


Abstract
Next-generation sequencing data will soon become routinely available for association studies between complex traits and rare variants. Sequencing data, however, are characterized by the presence of sequencing errors at each individual genotype. This makes it especially challenging to perform association studies of rare variants, which, due to their low minor allele frequencies, can be easily perturbed by genotype errors. In this article, we develop the quality-weighted multivariate score association test (qMSAT), a new procedure that allows powerful association tests between complex traits and multiple rare variants under the presence of sequencing errors. Simulation results based on quality scores from real data show that the qMSAT often dominates over current methods, that do not utilize quality information. In particular, the qMSAT can dramatically increase power over existing methods under moderate sample sizes and relatively low coverage. Moreover, in an obesity data study, we identified using the qMSAT two functional regions (MGLL promoter and MGLL 3′-untranslated region) where rare variants are associated with extreme obesity. Due to the high cost of sequencing data, the qMSAT is especially valuable for large-scale studies involving rare variants, as it can potentially increase power without additional experimental cost. qMSAT is freely available at http://qmsat.sourceforge.net/.
INTRODUCTION
Analysis of common variants through genome-wide association (GWA) studies has enjoyed much success over the last few years. More than a thousand genetic loci associated with more than 200 complex traits have been identified (1). However, these variants typically explain only a small fraction of the inheritable variability for common diseases (2). This may, in part, be explained by the genetic theory that implicates natural selection in pruning out disease-causing variants efficiently before they may become common among the population (3). On the other hand, rare variants, having minor allele frequencies (MAFs) between 0.1 and 1%, are often evolved from more recent mutations and less subjected to natural selection. Thus, association studies of rare variants may hold the potential for identifying genetic components that are functionally relevant and causal for a larger proportion of inheritable variability (4–6). Several studies have already demonstrated the relevance of rare variants on complex traits (7–13). The availability of emerging sequencing technologies will soon allow association studies to be routinely performed between complex traits and rare variants, making the development of robust and powerful procedures for rare variants association especially relevant.
Recent studies reveal several important characteristics of rare variants association. The rare variant hypothesis states that a large proportion of common diseases may be due to the aggregate effects of multiple rare variants, acting independently and with detectable effects on disease risks (4,6,14). Due to low allele frequencies, rare variants often do not exhibit strong linkage disequilibrium (LD) with either rare or common SNPs (6,15). Hence, GWA studies through indirect LD-mapping with tagged SNPs are usually ineffective in detecting rare variants. Rare variants identified in recent literatures usually have strong effects with a mean odds ratio (OR) of 3.74 and most variants having ORs greater than 2 (4). This is quite different from the effects of identified common variants, which usually have ORs between 1.1 and 1.4. Recent studies also present evidences of multiple rare variants acting collectively on disease risks (8,11,16).
Impelled by growing interests, several association tests have been proposed for rare variants. These tests often employ the idea of pooling or collapsing multiple rare variants. This is partly due to the need to aggregate effects of low-frequency variants in order to increase power and further due to recent evidences suggesting that multiple rare variants often act collectively upon disease risks (8,11,16,17). For example, the combined multivariate and collapsing (CMC) test (18) extends the cohort allelic sum test (CAST) (17) by collapsing variants in subgroups according to allele frequencies and combining these subgroups using Hotelling's T2 test. The step-up procedure determines whether each variant is protective, deleterious or non-causal and aggregates them accordingly (19). The C-alpha test is a homogeneity test for case-control data via binomial proportions (20). The sequence kernel association test (SKAT) extends kernel machine-based tests for rare variants with more accurate asymptotic approximations in the tail distribution (21). Moreover, several popular multivariate tests for GWA studies have been evaluated for rare variants association, including the mininum of univariate P-values (UminP) (22), sum score (23), sum of squared score (SSU) (24) and weighted sum of squared score (SSUw) tests (25).
These existing methods can sometimes be limited for rare variants association as they do not incorporate sequencing quality information. Associating rare variants to complex traits can be challenging due to the presence of sequencing errors. Under low MAFs, a few minor alleles mistaken as major or a few major alleles mistaken as minor can both result in significant loss of power in rare variants association. The quality of individual genotype estimate can vary significantly across both variants and samples, even when the mean sequencing coverage is high. Nevertheless, accurate measures of sequencing qualities can be readily obtained for each genotype using variant calling toolkits (26,27). Genotype quality scores (GQ) are often provided in Phred scale with GQ = −10 log10(1 − Pr), where Pr is the probability of a genotype being correctly called (28). Thus, a GQ value of 10 indicates a 10% sequencing error, whereas a value of 30 indicates 0.1% error. Incorporating sequencing qualities in association tests may allow us to increase power and to fully explore the potential of emerging sequencing technologies for identifying causal rare variants.
Some existing methods allow the incorporation of prior weights on each variant (21,24,29), which can potentially incorporate average quality scores across variants. However, sequencing qualities also vary significantly across samples, in addition to variants. For example, a variant may consist of some proportion of high-quality genotype samples but others that are sequenced at low qualities, with the extreme case of having missing values. Utilizing these genotypes without considering varying sequencing qualities across samples can significantly decrease power in rare variants association studies due to large perturbations from low-quality samples. Thus, a new procedure is needed that can address the incorporation of sequencing qualities at each individual genotype.
To achieve a robust and powerful test for rare variants association studies, we propose a quality-weighted multivariate score association test (qMSAT) that directly incorporates sequencing qualities in association tests with multiple rare variants. The qMSAT utilizes a quality-weighted multivariate regression model to incorporate sequencing qualities at each individual genotype. It is robust towards the inclusion of noncausal variants and variants having effects with different magnitudes and directions. Furthermore, it can coherently account for missing genotypes and conjoin in a principled way individuals and variants sequenced at varying coverage depths. When no quality information is utilized, the qMSAT is equivalent to the C-alpha, SSU and SKAT using the linear kernel. The qMSAT is therefore expected to perform equivalently or better than established methods when quality information is utilized.
We present extensive simulation studies based upon sequencing qualities from real data, obtained from the 1000 Genomes Project (30). To guide investigators, we examine the performances of the qMSAT with current methods across a spectrum of MAFs, odds ratios, coverage depths, sample sizes and LD structures. We also provide scenarios of having non-causal variants and variants having effects with different magnitudes and directions. These results suggest that the qMSAT can often dominate over current methods, that do not utilize quality information. In particular, the qMSAT often demonstrates significant improvements in power under moderate sample sizes and relatively low coverage for rare variants association. Furthermore, we apply the qMSAT in an application to the UCSD obesity data study, where we identified two functional regions, the MGLL promoter and MGLL 3′-untranslated region (3′-UTR), that exhibit significant multiple rare variants association with extreme obesity. Most importantly, our results suggest the qMSAT to be a potentially valuable tool for increasing power without incurring additional experimental costs, such as by sequencing at higher coverage depths or larger sample sizes (31).
MATERIALS AND METHODS
Quality-Weighted Multivariate Score Association Test (qMSAT)
In this article, we consider case-control association studies having d multiple rare variants and n individuals. For the i-th individual, yi denotes a dichotomous trait with yi = 1 for cases and yi = 0 for controls and Gi = (Gi1,…, Gid) are variant genotypes. We assume, in this article, that genotypes are represented by the additive genetic model where Gij = 0, 1 or 2 are the numbers of minor alleles at a locus. However, our procedure can be used with other genetic models, such as the dominant or recessive model, and our conclusions regarding the inclusion of quality information will remain the same using another genotypic assignment.
Consider the logistic regression model,
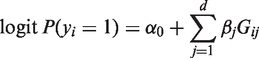 |
(1) |
where α0 is an intercept term and βj is the effect of the j-th variant on trait. The rare variants hypothesis suggests that rare variants often act independently on disease risks. Further, having low MAFs, rare variants usually do not exhibit strong LD, even at close proximity (6,11,15). Hence, we consider a quality-weighted log-likelihood,
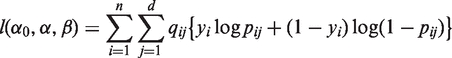 |
(2) |
where
and qij are quality weights on each pair (i, j) of individuals and variants. The quality-weighted log-likelihood (2) utilizes relative independence among rare variants by aggregating over weighted marginal likelihoods. It allows effects of individual genotypes to be evaluated based upon sequencing qualities, down-weighting those having relatively low qualities. The weighted multivariate log-likelihood approach has been utilized successfully in several other statistical contexts (32,33). It is often employed for the incorporation of quality-related constituents (34). The approach enables conjoined analysis of sequencing data over a spectrum of qualities and read depths. Moreover, problems associated with imputing rare variants for traditional association tests can be circumvented by assigning qij = 0 at missing genotypes in Equation (2). The quality-weighted likelihood naturally allows missing genotypes without the need for imputation, which can be quite challenging under low MAFs.
For simplicity, we utilize the genotype quality scores directly as weights qij in this article. The quality-based weights qij can also be assigned using the number of reads and monotonically non-decreasing functions of quality scores, such as 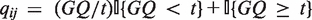 where
where  {·} is an indicator function and t is some threshold above which qualities are believed to be equally acceptable. A myriad of quality-based weights can be applied based upon an investigator's preferences and prior knowledge.
{·} is an indicator function and t is some threshold above which qualities are believed to be equally acceptable. A myriad of quality-based weights can be applied based upon an investigator's preferences and prior knowledge.
To achieve a robust and powerful test for multiple rare variants association, we utilize a quality-weighted Rao's score statistic (35) in testing the multivariate null hypothesis,
| (3) |
Computing for the slope of the log-likelihood Equation (2) with genotype coefficients βj restricted at the null hypothesis, we obtain the quality-weighted score vector S = (S1, S2,…, Sd)T where
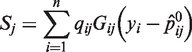 |
(4) |
and
are the predicted proportions of cases at the j-th variant.
We define the qMSAT statistic as the norm squared of the quality-weighted score vector
| (5) |
If no quality information is utilized (qij = 1), the qMSAT is reduced to the SSU and linear SKAT and is also equivalent to the C-alpha test. The qMSAT provides a computationally efficient means to estimate expected improvements in model fit according to sequencing qualities when the null hypothesis in Equation (3) may be violated. It admits a simple form as explicit computations of genotype coefficients βj are not required. Utilizing a quadratic form, the qMSAT is robust towards the inclusion of variants having effects with opposing directions. However, with prior weights imposed upon the responses yi in Equation (2), the qMSAT statistic does not assume a readily approximable asymptotic distribution. Thus, we utilize a permutation-based strategy, where the distribution of the qMSAT statistic is approximated using statistics computed with randomly re-sampled responses 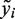 in Equation (4) (36). As asymptotic-based tests are not guaranteed to be reliable under low MAFs and limited sample sizes, permutation-based procedures are often preferred in rare variants association studies (20,29). Having a simple form, the qMSAT can be computed efficiently even when large numbers of permutations are required. Indeed, the qMSAT is equivalent in computational speed to C-alpha and other score-based procedures, that have been successfully employed in several earlier studies (20,25,29).
in Equation (4) (36). As asymptotic-based tests are not guaranteed to be reliable under low MAFs and limited sample sizes, permutation-based procedures are often preferred in rare variants association studies (20,29). Having a simple form, the qMSAT can be computed efficiently even when large numbers of permutations are required. Indeed, the qMSAT is equivalent in computational speed to C-alpha and other score-based procedures, that have been successfully employed in several earlier studies (20,25,29).
Simulations
We present extensive simulations based upon sequencing qualities from the Pilot Phase 3 study of the 1000 Genomes project (30). The study provides sequencing data for 90 Caucasian individuals over 2385 rare variant sites, amounting to 214 650 genotype quality scores across a spectrum of read depths. In order to compare performances of various methods under realistic error distributions, we sampled directly from these empirical quality scores in generating simulated data.
We generated simulated data over a spectrum of MAFs, odds ratios, coverage depths, sample sizes and LD structures. We also provided scenarios where non-causal variants and variants having effects with different magnitudes and directions are included. As in previous studies (25,37), we first generated d latent variables from the multivariate normal distribution with the autoregressive covariance structure Σ = ρ|i−j|, where ρ = 0 was used to generate independent variants and ρ close to 1 was used to generate variants in strong LD with its neighbors. The latent variables were then used to produce a haplotype at a given MAF by dichotomizing variables at a specified quantile. Two independent haplotypes thus generated were combined to obtain the underlying genotypes 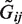 , with which we generated dichotomous phenotypes for a case–control study under the logistic regression model
, with which we generated dichotomous phenotypes for a case–control study under the logistic regression model
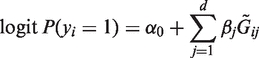 |
(6) |
at given effect sizes βj or odds ratios exp(βj). The intercept term α0 was set to attain a disease rate of 5%. As in conventional case–control design, we randomly sampled n/2 cases and n/2 controls along with their underlying genotypes 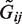 . Next, we generated observed genotypes Gij by perturbing the underlying ones
. Next, we generated observed genotypes Gij by perturbing the underlying ones 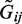 with sequencing qualities sampled from the 1000 Genomes project. At a given mean coverage depth (dp), we simulated the number of reads for each genotype from the gamma distribution with shape and scale parameters at 6.3 and dp/6.3, respectively. The gamma distribution with the specified parameters have been found to approximate observed numbers of reads well from mean coverage in previous studies (38,39). The quality score GQ was obtained for each genotype by sampling from quality scores of the 214 650 Pilot Phase 3 study genotypes with an observed number of reads equal to or closest to the number of reads simulated from mean coverage. Finally, we generated genotypes Gij by randomly perturbing
with sequencing qualities sampled from the 1000 Genomes project. At a given mean coverage depth (dp), we simulated the number of reads for each genotype from the gamma distribution with shape and scale parameters at 6.3 and dp/6.3, respectively. The gamma distribution with the specified parameters have been found to approximate observed numbers of reads well from mean coverage in previous studies (38,39). The quality score GQ was obtained for each genotype by sampling from quality scores of the 214 650 Pilot Phase 3 study genotypes with an observed number of reads equal to or closest to the number of reads simulated from mean coverage. Finally, we generated genotypes Gij by randomly perturbing 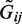 using the Bernoulli distribution with error probabilities 10(−GQ/10). Each procedure was applied by associating phenotypes yi with perturbed genotypes Gij. Sampled quality scores at each genotype were utilized as weights qij in the qMSAT.
using the Bernoulli distribution with error probabilities 10(−GQ/10). Each procedure was applied by associating phenotypes yi with perturbed genotypes Gij. Sampled quality scores at each genotype were utilized as weights qij in the qMSAT.
We compared the qMSAT with 10 other methods that have been proposed recently for rare variants association. The SSU, SSUw, Score, SKAT with linear and quadratic kernels, and UminP are score-based procedures similar to the qMSAT, whereas the Sum and CMC tests are derived from estimates in a logistic regression with collapsed variants. The step-up procedure also performs variable selection to determine causality and directions of effects in addition to collapsing variants together. The C-alpha procedure is a homogeneity test based upon binomial proportions. In a case–control study, the C-alpha is equivalent to the SSU and linear SKAT (21,40). Moreover, the C-alpha, SSU and linear SKAT are, in turn, equivalent to the qMSAT if no quality information is utilized (qij = 1). We implemented the qMSAT in R, available online at http://qmsat.sourceforge.net/. The SSU, SSUw and Score tests were computed using R codes from an earlier study (24), whereas the SKAT was computed using the R package from the original paper (21). The SKAT package also includes options for incorporating MAF-based weights on each variant. Nonetheless, in order to clearly demonstrate the effects of MAF on association tests, our simulations were performed at a spectrum of values at fixed MAFs. Incorporating MAF-based weight on each variant is only beneficial if both rare and common variants are included. Thus, we do not incorporate MAF-based weights for the SKAT in this study. The thgenetics R package was used for the step-up procedure (19). All other procedures were computed using available codes in a recent study (40). P-values for the qMSAT, C-alpha, and step-up procedures were computed using 500 permutations. Simulated data for type I errors were generated using the restricted logistic regression model instead of (6),
where ORs were set to 1 for each variant. Both empirical powers and type I errors for each procedure were estimated using 1000 repetitions as the proportion of P-values < 0.05.
Application to UCSD obesity data set
We analyzed a targeted re-sequencing data of candidate genes at extreme obesity levels from the UCSD obesity study (41,42). The data set consisted of 289 individuals of European ancestry; among which, 73 men and 70 women having body mass index (BMI) ≥40 kg/m2 were selected as cases, and 74 men and 72 women with BMI ≤ 30 kg/m2 were assigned as controls. Two intervals were sequenced over 188 kb that encompassed regions encoding the endocannabinoid metabolic enzymes, fatty-acid amide hydrolase (FAAH) and monoglyceride lipase (MGLL). Earlier studies had found genes in the endocannabinoid system that regulates obesity levels (43,44), making the FAAH and MGLL prime candidates for association analysis of variants to extreme obesity. We mapped short reads using the Burrows-Wheeler Alignment (BWA) algorithm (45). We employed the most recent state-of-the-art variant calling toolkit GATK (26) to perform variant calls and extract corresponding quality scores at each genotype. An earlier algorithm MAQ (46) was used in the original study (42) for variant calling. We obtained 977 variants with a mean coverage of 101×. These variants were then annotated using SeattleSeq (http://gvs.gs.washington.edu/SeattleSeqAnnotation/). Given the annotations, we focused on the 455 rare variants that were not cataloged in the dbSNP database (47); these variants were then divided based on their genomic positions into groups of promoters, 5′-UTRs, exon regions, intron regions and 3′-UTRs, for performing association tests. For each gene, we combined all exon variants together in a group and considered only non-synonymous variants. The FAAH promoter was sequenced over the 11.5 kb upstream region, whereas the MGLL promoter encompassed the 12.5 kb upstream region. Short functional regions with less than two variants were not included in this study. Missing genotypes were imputed by sampling randomly from non-missing ones in the same variant, and their associated quality scores were set to be 0. Number of permutations in statistical tests was increased to 10 000 for increased precision in P-values.
RESULTS
Presence of sequencing errors
Figure 1 presents sequencing qualities of the data from the Pilot Phase 3 study of the 1000 Genomes project at each genotype in terms of genotype quality scores (GQ) and probability of sequencing errors (1 − Pr) over varying coverage depths. The quantities are related through the conversion formula Pr = 1 − 10−GQ/10. We observe that sequencing qualities tend to increase with increasing coverage depths or numbers of reads at a genotype. Nonetheless, even at relatively high coverage depths, a significant proportion of genotypes still assumes low sequencing qualities. For example, the 99% interval includes genotypes with GQ = 15 (or 1 − Pr = 0.03) at a coverage depth of 20×. In association analysis of rare variants where MAFs are low, even a small proportion of unaccounted sequencing errors can influence results unexpectedly. In the following studies, we evaluate the effects of having sequencing errors under realistic settings by sampling sequencing qualities directly from the Pilot Phase 3 study as presented in Figure 1 (see ‘Material and Methods’ section).
Figure 1.

Genotype sequencing qualities generated in Pilot Study 3 of the 1000 Genomes Project. Sequencing qualities in terms of genotype quality scores (GQ) and probability of sequencing errors (1 − Pr) are displayed, where 1 − Pr = 10−GQ/10. A total of 2385 rare variants are called, amounting to 214 650 genotypes at various coverage depths. (a) Plot of sequencing qualities at all genotypes, with coverage depths spaced logarithmically. (b) Modified box plot of genotypes at coverage depths of 5×, 10×, 20×, 30×, 50× and 100×. Short, bold lines are drawn at the median. Each whisker ends at the 1 and 99% quantiles, and each box encloses the 5–95% interval.
Evaluation of the effects of coverage depth and sample size
We evaluated empirical powers and type I errors over mean coverage depths of 5×, 10×, 20×, 30×, 50× and 100× and moderate sample sizes of 200, 400, 600, 800 and 1000, each having balanced numbers of cases and controls. We considered 20 rare variants (d = 20) consisting of five causal ones with odds ratios of 2, 3, 4, 0.5 and 0.5 for power computations. No LD was assumed among the variants. In each case, we compared the qMSAT with 10 other methods that do not utilize quality information (see ‘Material and Methods’ section).
Figure 2 presents type I errors at an MAF of 0.5%. Type I errors are generally well controlled. However, score-based procedures using asymptotic distributions (SSU, SSUw, Score, SKAT and UminP) tend to be conservative at moderate or high coverage, especially when sample sizes are small. On the other hand, permutation-based methods (qMSAT, C-alpha and step-up) are usually well controlled around the 0.05 error rate. This may suggest that permutation methods can sometimes be advantageous under finite samples. Supplementary Figures S1 and S3 further depict type I errors at MAFs of 0.1% and 1%, respectively.
Figure 2.

Type I errors over varying coverage depths and sample sizes at MAF=0.5%. There are n/2 numbers of cases and controls each. Twenty non-causal rare variants (d = 20) are considered with odds ratios at 1 for all variants. No LD is assumed among the variants. Reference lines (in red) are drawn at the 0.05 error rate.
Empirical powers are provided in Figure 3 at an MAF of 0.5%. We observe that the qMSAT often dominates over other procedures, that do not utilize quality information. Improvements in power are most significant at relatively low coverage. Consider the permutation-based C-alpha, which is related to the qMSAT when no quality information is utilized (see ‘Material and Methods’ section). At n = 600, the qMSAT outperforms C-alpha with 78.0, 158, 61.2, 27.4, 21.0 and 14.8% increases in power at mean coverage depths of 5×, 10×, 20×, 30×, 50×, and 100×, respectively (Figure 3). Moreover, the qMSAT often achieves fairly high powers even at relatively low coverage. At n = 1000, the qMSAT attains powers of 0.530, 0.739 and 0.773 at mean coverage depths of 10×, 20× and 30×, respectively, whereas a similar level of power is attained at 0.780 with a much higher mean coverage depth of 100× (Figure 3). This suggests the qMSAT as a valuable tool for achieving powerful results without incurring additional costs, such as by sequencing at higher coverage depths. The SSU usually tends to have higher powers than SSUw and score tests. Previous studies have suggested that the SSUw and score tests that depend upon accurate estimations of variances and the covariance matrix, respectively, may be limited for multivariate analysis when the dimension d is large (24,48,49). The SKAT using linear kernels often has higher powers than the SKAT using quadratic kernels as epistatic effects were not included in this analysis. Due to the inclusion of causal variants having opposing directions, the sum test often has limited powers. Supplementary Figures S2 and S4 further provide power evaluations at MAFs of 0.1 and 1%, respectively.
Figure 3.

Power evaluation of the effects of coverage depths and sample sizes at MAF=0.5%. There are n/2 numbers of cases and controls each. Twenty rare variants (d = 20) are considered, of which five are causal with odds ratios at 2, 3, 4, 0.5 and 0.5. No LD is assumed among the variants.
Evaluation of the effects of MAF and odds ratio
We compared empirical powers and type I errors over varying MAFs of 0.1, 0.5, 1, 5 and 10% and ORs of 1–6 for each of the five variants among the 20 (d = 20) considered, in which the remaining are non-causal with ORs of 1. Identified rare variants in recent literatures have a mean OR of 3.74, and most have ORs greater than 2 (4). To focus on evaluating effects of MAF and OR, we considered n = 500 observations with balanced numbers of cases and controls and assumed no LD among the variants.
Figure 4 presents empirical powers and type I errors (ORs = 1) at a mean coverage depth of 30×. Power increases for all methods with increasing MAF and OR. The qMSAT often outperforms other procedures at low MAFs for rare variants, whereas all procedures do well at high MAFs for common variants. For low MAFs, power for the qMSAT improves dramatically for increasing ORs. For example, at an MAF of 0.1%, the qMSAT outperforms C-alpha with 66.0, 84.4, 113, 124 and 127% increases in power at causal variants' ORs of 2, 3, 4, 5 and 6, respectively (Figure 4). This may suggest the qMSAT as a potentially useful procedure for identifying rare variants (with MAFs between 0.1 and 1%) under moderate sample sizes and, possibly, for identifying novel variants (with MAFs <0.1%) having strong effect sizes in large-scale studies. The sum test performs competitively with related methods in this example, as all causal variants have the same direction. Supplementary Figures S5–S9 present empirical powers and type I errors at mean coverage depths of 5×, 10×, 20×, 50× and 100×, respectively. Improvements of the qMSAT tend to be more pronounced at lower coverage depths in this example.
Figure 4.

Effects of MAFs and odds ratios at a mean coverage depth of 30×. Empirical powers and type I errors are presented at a mean coverage depth of 30× with varying MAFs and odds ratios for five variants amongst twenty (d = 20) considered altogether. We use n = 500 observations, where there are n/2 = 250 numbers of cases and controls each. No LD is assumed among the variants. Reference lines (in red) are drawn at the 0.05 error rate.
Evaluation of the effects of LD
We evaluated the effects of LD at a mean coverage depth of 30× and sample sizes of n = 500, with equal numbers of cases and controls. A spectrum of LD structures was evaluated with latent variables having autoregressive covariance matrices Σ = ρ|i−j| with ρ at 0, 0.25, 0.5, 0.75 and 0.9 (Figure 5). We employed 20 variants (d = 20) with ORs of (3, 2, 1, 1, 4, 1, 1, 1, 0.5, 0.5, 1,…,1). Variants generated with ρ = 0 are independent, whereas those generated with large values of ρ are in LD with its neighbors.
Figure 5.

Power evaluation of the effects of linkage disequilibrium. Empirical powers are presented at a mean coverage depth of 30× and sample sizes n = 500 with equal numbers of cases and controls for a spectrum of LD structures generated from latent variables having autoregressive covariance matrices Σ = ρ|i−j|. Variants generated with ρ = 0 have no LD, whereas variants generated with large values of ρ are in high LD. Twenty variants (d = 20) are considered having odds ratios of (3, 2, 1, 1, 4, 1, 1, 1, 0.5, 0.5, 1,…,1).
Figure 5 presents empirical powers for evaluating LD effects. Power increases for all methods with increasing ρ or degree of LD among neighboring variants. This is largely because variants in LD tend to have effect sizes augmented by aggregation. The qMSAT tends to dominate other methods at relatively low MAFs, even at high LD. Rare variants usually do not exhibit strong LD (6,11,15). However, these results suggest that the qMSAT can also be effective when LD effects are strong. Supplementary Figure S10 further provides type I errors.
Application to UCSD obesity data set
We analyzed the UCSD obsesity data from a previous study that suggested multiple rare variants to be associated with the FAAH promoter, the MGLL promoter, and a region of MGLL introns (42). We performed multiple rare variants association tests using all statistical approaches as above. As in the original study (42), none of these methods found significant rare variants in the FAAH and MGLL exon regions (Supplementary Table S1); several methods confirmed associations of multiple rare variants in the FAAH promoter and MGLL promoter regions (Table 1). Specifically, the qMSAT attained P-values of 0.061 and 0.037 for the FAAH promoter and MGLL promoter regions, respectively. No intron regions were found to be significant by any of these methods (Supplementary Table S1). Interestingly, several methods, including the qMSAT (Table 1), identified the MGLL 3′-UTR to be significant, which was missed in the previous study (42). Promoters are well known to be gene transcription regulators that specify binding of transcription factors to DNA sequences. Moreover, protein binding to 3′-UTR is known to function in some situations by protecting mRNA from nuclease degradation (50). These results suggest that recent mutations in the FAAH promoter, MGLL promoter, and MGLL 3′-UTR may have led to extreme obesity levels in some individuals by altering gene transcription mechanisms. Further studies, such as by associating rare variants to gene expression levels, may help to verify these results. On the other hand, recent mutations in MGLL introns might not have played as significant a role as previous study (42) suggested in extreme obesity. Differences between our results from those of the previous study might be due to using more recent variant calling algorithm in our analysis (see ‘Material and Methods’ section). Among the procedures evaluated, the qMSAT has the smallest P-values for association of rare variants in the MGLL promoter and MGLL 3′-UTR with extreme obesity levels (Table 1). The C-alpha, SSU and linear SKAT are equivalent to the qMSAT when no quality information is utilized. Both C-alpha and qMSAT utilize a permutation-based estimation of P-values in order to better control type I errors under moderate samples. In all three regions where at least one method indicates significant association, the qMSAT has significantly smaller P-values than the C-alpha (Table 1). These results confirm our simulation studies suggesting that the incorporation of quality information can often improve power in rare variants association analysis. We observe that the SKAT using linear kernels has smaller P-values than the SKAT using quadratic kernels (Table 1). This may suggest that rare variants, having low MAFs, might not be able to exhibit strong interaction and higher-order effects. In addition, we performed marginal association analysis of rare variants in the FAAH promoter, MGLL promoter and MGLL 3′-UTR (Supplementary Table S2). The MGLL promoter contains two variants with marginal associations significant at the 0.05 P-value level but with opposing effects on extreme obesity (Supplementary Table S2). In this region at the MGLL promoter, the multivariate sum test has the largest P-value of 0.578 (Table 1). This may support previous studies asserting the sum test to be limited when association of significant variants in a region are in opposite directions, though it might be powerful when directions are the same (25). Due to low MAFs, marginal association tests for rare variants often have low power and can be subjected to perturbations from genotype errors. Both the FAAH promoter and MGLL 3′-UTR, indicated to be significant by several multivariate tests, do not have rare variants exhibiting marginal associations significant at the 0.05 P-value level (Supplementary Table S2). This result further suggests the benefit of pooling multiple variants together in identifying rare variants associated with common diseases.
Table 1.
Multiple rare variants association analysis of the UCSD obesity data
| FAAH promoter (d=17) | MGLL promoter (d=25) | MGLL 3′-UTR (d=6) | |
|---|---|---|---|
| qMSAT | 0.061 | 0.037 | 0.002 |
| C-alpha | 0.077 | 0.049 | 0.044 |
| SSU | 0.080 | 0.038 | 0.246 |
| SSUw | 0.145 | 0.234 | 0.308 |
| Score | 0.146 | 0.123 | 0.303 |
| SKAT (Linear) | 0.079 | 0.038 | 0.246 |
| SKAT (Quad) | 0.107 | 0.050 | 0.250 |
| UminP | 0.631 | 0.211 | 0.627 |
| Step up | 0.022 | 0.063 | 0.015 |
| Sum | 0.002 | 0.578 | 0.052 |
| CMC | 0.015 | 0.109 | 0.052 |
P-values are provided for association tests performed over d multiple rare variants. FAAH promoter, MGLL promoter and MGLL 3′-UTR are regions exhibiting significant association of multiple rare variants with extreme BMI at the 0.05 level by one or more methods. P-values significant at the 0.05 level are boldfaced.
In this application, the qMSAT P-values were computed in 25.039 s for all 12 regions considered using 10 000 permutations with the R programming language. In large-scale rare variants association studies involving hundreds of sequenced genes, it is expected to take only a couple of hours. Computing speed can also be greatly improved by using more efficient programming languages, such as C or Fortran. Among the methods evaluated, the step-up procedure has comparatively much slower computational speed, as it has to ascertain whether each variant is protective, deleterious or non-causal.
DISCUSSION
We have proposed a novel procedure that allows the incorporation of sequencing qualities directly in association tests of complex traits and multiple rare variants. Extensive simulation studies with sequencing qualities sampled directly from real data have been used to evaluate our method across a spectrum of factors of interest. Results have suggested the qMSAT as a potentially very useful method for improving power, especially under moderate sample sizes and relatively low coverage. Moreover, our analysis of the UCSD obesity data set using the qMSAT has identified two regions (MGLL promoter and MGLL 3′-UTR), where multiple rare variants are significantly associated with extreme obesity.
We have not considered the inclusion of additional covariates in order to provide a concise presentation. It is clear that our conclusions regarding the incorporation of sequencing qualities will remain the same whether additional covariates are included or not. The qMSAT can be easily extended to include additional covariates, such as relevant common variants and environmental effects. For dichotomous traits yi and Xi a dc-vector of additional covariates, the qMSAT statistic qMSAT = ST S is defined using scores in Equation (4) with
where 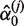 and
and 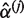 are estimated under the null hypothesis by maximizing the log-likelihood in Equation (2) with
are estimated under the null hypothesis by maximizing the log-likelihood in Equation (2) with 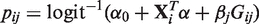 using only the covariates Xi and quality-based weights q1j, q2j,…, qnj at the j-th variant, i.e. qij′ = 0 for all i = 1,…, n and j′ ≠ j.
using only the covariates Xi and quality-based weights q1j, q2j,…, qnj at the j-th variant, i.e. qij′ = 0 for all i = 1,…, n and j′ ≠ j.
Moreover, we focused on association tests with the logistic regression for case–control studies. This is partly due to the prevalence of case–control data in biomedical researches and the fact that effects of rare variants, having extremely low MAFs, can be more readily modeled with dichotomous traits than continuous ones. However, with the advent of large-scale rare variants association studies in the near future, quantitative traits may also be considered. For continuous traits yi, the qMSAT statistic is defined with scores
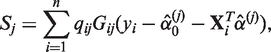 |
for which the quality-weighted log-likelihood under the linear model
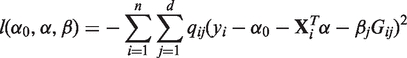 |
is employed in place of Equation (2). The coefficients 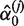 and
and 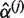 are estimated under the null hypothesis by weighted least squares regression of yi on additional covariates Xi only with quality-based weights q1j, q2j,…, qnj at the j-th variant. The same benefits demonstrated for the qMSAT for case–control data are expected to extend to applications involving quantitative traits.
are estimated under the null hypothesis by weighted least squares regression of yi on additional covariates Xi only with quality-based weights q1j, q2j,…, qnj at the j-th variant. The same benefits demonstrated for the qMSAT for case–control data are expected to extend to applications involving quantitative traits.
We have not considered association studies simultaneously involving both rare and common variants. A common strategy proposed in the literature is to re-weight each variant according to its MAF, augmenting effects of rare variants relative to common ones. However, it is not clear which is the best way to re-weight variants objectively. For example, re-weighting by standard deviations, control MAFs (51), and beta transformations of MAFs (21) have been proposed. As rare variants usually act on disease risks independently of common ones (6,15), a natural approach is to first identify common variants that may be causal, such as by routine GWA studies, and then incorporate them as additional covariates Xi as in the qMSAT.
In this article, we have utilized genotype quality scores as weights in the qMSAT. These scores can account for errors, such as those arising from base calling in sequencer traces and mapping of short sequencing reads, and can quantify genotype uncertainty accurately at most loci. However, variant calling at some loci may suffer due to artifacts, such as from strand bias, that cannot be easily accounted for and may lead to less accurate genotype quality scores. Nonetheless, current variant calling programs usually can filter out the variants at these suspicious loci (26,27,52). In the situation when genotype qualities are less accurate, the qMSAT may be less powerful while type I errors are expected to be controlled due to permutation (36). Nevertheless, our results demonstrate that the incorporation of sequencing qualities can consistently improve power over methods that do not incorporate quality information. Moreover, the development of variant calling methods is an active area (26,27,52,53), and our procedure will continue to benefit from future improvements in variant calling programs.
On the other hand, the qMSAT is quite flexible and can incorporate other forms of weights, in addition to quality information. For example, if there is evidence to believe that some variants are more likely to be functional, such as by prior knowledge or evolutionary models (54), or some individuals are more representative of the effected population, the qMSAT weights qij can be redefined accordingly in order to incorporate additional information at each variant or individual. Indeed, the qMSAT extends earlier methods with weights on each variant (24,51,55) to allow varying weights on both individuals and variants.
Statistical analysis of sequencing data is ultimately limited by inadequacies in the underlying technology employed. With the qMSAT, we approached this challenge by incorporating sequencing quality, a technology-derived constituent, directly in a statistical procedure. The qMSAT also offers a coherent strategy for integrating missing genotypes and individual genotypes sequenced at varying coverage depths and quality scores. Improvements in performances that we observed for the qMSAT in both empirical and real-data studies may suggest the importance of considering the limitations of the technology at hand in order to improve statistical analysis of genomic data sets.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables S1–S2 and Supplementary Figures S1–S10.
FUNDING
National Institutes of Health (grant numbers CA127334, ES009911). Funding for open access charge: Waived by Oxford University Press.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank the two anonymous referees for their very generous comments and improvements made in the article based on their suggestions.
REFERENCES
- 1.Hindorff LA, Junkins HA, Hall PN, Mehta JP, Manolio TA. A catalog of published genome-wide association studies. 2011 Available at: www.genome.gov/gwastudies. Accessed July 15, 2011. [Google Scholar]
- 2.Maher B. Personal genomes: the case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 3.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am. J. Hum. Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? Am. J. Hum. Genet. 2001;69:124–137. doi: 10.1086/321272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahituv N, Kavaslar N, Schackwitz W, Ustaszewska A, Martin J, Hebert S, Doelle H, Ersoy B, Kryukov G, Schmidt S, et al. Medical sequencing at the extremes of human body mass. Am. J. Hum. Genet. 2007;80:779–791. doi: 10.1086/513471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen JC, Kiss RS, Pertsemlidis A, Marcel YL, McPherson R, Hobbs HH. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. doi: 10.1126/science.1099870. [DOI] [PubMed] [Google Scholar]
- 9.Cohen JC, Boerwinkle E, Jr, THM, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006;354:1264–1272. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 10.Ji W, Foo JN, O'Roak BJ, Zhao H, Larson MG, Simon DB, Newton-Cheh C, State MW, Levy D, Lifton RP. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008;40:592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romeo S, Pennacchio LA, Fu Y, Boerwinkle E, Tybjaerg-Hansen A, Hobbs HH, Cohen JC. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat. Genet. 2007;39:513–516. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romeo S, Yin W, Kozlitina J, Pennacchio LA, Boerwinkle E, Hobbs HH, Cohen JC. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J. Clin. Invest. 2009;119:70–79. doi: 10.1172/JCI37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorlov IP, Gorlova OY, Sunyaev SR, Spitz MR, Amos CI. Shifting paradigm of association studies: Value of rare single-nucleotide polymorphisms. Am. J. Hum. Genet. 2008;82:100–112. doi: 10.1016/j.ajhg.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pritchard JK, Cox NJ. The allelic architecture of human disease genes: common disease-common variant… or not? Hum. Mol. Genet. 2002;11:2417–2423. doi: 10.1093/hmg/11.20.2417. [DOI] [PubMed] [Google Scholar]
- 16.Fearnhead NS, Wilding JL, Winney B, Tonks S, Bartlett S, Bicknell DC, Tomlinson IP, Mortensen NJ, Bodmer WF. Multiple rare variants in different genes account for multifactorial inherited susceptibility to colorectal adenomas. Proc. Natl Acad. Sci. USA. 2004;101:15992–15997. doi: 10.1073/pnas.0407187101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Morgenthaler S, Thilly WG. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST) Mutat. Res. 2007;615:28–56. doi: 10.1016/j.mrfmmm.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffmann TJ, Marini NJ, Witte JS. Comprehensive approach to analyzing rare genetic variants. PLoS One. 2010;5:e1001289. doi: 10.1371/journal.pone.0013584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neale BM, Rivas MA, Voight BF, Altshuler D, Devlin B, Orho-Melander M, Kathiresan S, Purcell SM, Roeder K, Daly MJ. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7:e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare variant association testing for sequencing data using the sequence kernel association test (SKAT) Am. J. Hum. Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conneely KN, Boehnke M. So many correlated tests, so little time! rapid adjustment of p values for multiple correlated tests. Am. J. Hum. Genet. 2007;81:1158–1168. doi: 10.1086/522036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chapman J, Whittaker J. Analysis of multiple SNPs in a candidate gene or region. Genet. Epidemiol. 2008;32:560–566. doi: 10.1002/gepi.20330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan W. Asymptotic tests of association with multiple SNPs in linkage disequilibrium. Genet. Epidemiol. 2009;33:497–507. doi: 10.1002/gepi.20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basu S, Pan W. Comparison of statistical tests for disease association with rare variants. Genet. Epidemiol. 2011;35:606–619. doi: 10.1002/gepi.20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei Z, Wang W, Hu P, Lyon GJ, Hakonarson H. SNVer: a statistical tool for variant calling in analysis of pooled or individual next-generation sequencing data. Nucleic Acids Res. 2011;39:e132. doi: 10.1093/nar/gkr599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- 29.Lin DY, Tang ZZ. A general framework for detecting disease associations with rare variants in sequencing studies. Am. J. Hum. Genet. 2011;89:354–367. doi: 10.1016/j.ajhg.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. The 1000 Genomes Project Consortium (2010) A map of human genome variation from population-scale sequencing. Nature, 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Sidore C, Kang HM, Boehnke M, Abecasis GR. Low-coverage sequencing: Implications for design of complex trait association studies. Genome Res. 2011;21:940–951. doi: 10.1101/gr.117259.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruppert D, Wand MP. Multivariate locally weighted least squares regression. Ann. Statist. 1994;22:1346–1370. [Google Scholar]
- 33.Zellner A. An efficient method of estimating seemingly unrelated regressions and tests for aggregation bias. J. Amer. Statist. Assoc. 1962;57:348–368. [Google Scholar]
- 34.Carroll RJ, Ruppert D. Transformation and Weighting in Regression. New York: Chapman and Hall/CRC; 1988. [Google Scholar]
- 35.Rao CR. Large sample tests of statistical hypotheses concerning several parameters with applications to problems of estimation. Proc. Camb. Phil. Soc. 1948;44:50–57. [Google Scholar]
- 36.Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138:963–971. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang T, Elston RC. Improved power by use of a weighted score test for linkage disequilibrium mapping. Am. J. Hum. Genet. 2007;80:353–360. doi: 10.1086/511312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prabhu S, Pe'er I. Overlapping pools for high-throughput targeted resequencing. Genome Res. 2009;19:1254–1261. doi: 10.1101/gr.088559.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarin S, Prabhu S, O'Meara MM, Pe'er I, Hobert O. Caenorhabditis elegans mutant allele identification by whole-genome sequencing. Nat. Methods. 2008;5:865–867. doi: 10.1038/nmeth.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pan W. Relationship between genomic distance-based regression and kernel machine regression for multi-marker association testing. Genet. Epidemiol. 2011;35:211–216. doi: 10.1002/gepi.20567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhatia G, Bansal O, Harismendy O, Schork NJ, Topol EJ, Frazer K, Bafna V. A covering method for detecting genetic associations between rare variants and common phenotypes. PLoS Comput. Biol. 2010;6:e1000954. doi: 10.1371/journal.pcbi.1000954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harismendy O, Bansal V, Bhatia G, Nakano M, Scott M, Wang X, Dib C, Turlotte E, Sipe JC, Murray SS, et al. Population sequencing of two endocannabinoid metabolic genes identifies rare and common regulatory variants associated with extreme obesity and metabolite level. Genome Biol. 2010;11:R118. doi: 10.1186/gb-2010-11-11-r118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodriguez de Fonseca F, Del Arco I, Bermudez-Silva FJ, Bilbao A, Cippitelli A, Navarro M. The endocannabinoid system: physiology and parmacology. Alcohol Alcohol. 2005;40:2–14. doi: 10.1093/alcalc/agh110. [DOI] [PubMed] [Google Scholar]
- 44.Walker JM, Krey JF, Chu CJ, Huang SM. Endocannabinoids and related fatty acid derivatives in pain modulation. Chem. Phys. Lipids. 2002;121:159–172. doi: 10.1016/s0009-3084(02)00152-4. [DOI] [PubMed] [Google Scholar]
- 45.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. Mar. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sherry ST, Ward M, Sirotkin K. dbSNP - database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999;8:677–679. [PubMed] [Google Scholar]
- 48.Bai Z, Saranadasa H. Effect of high dimension: by an example of a two sample problem. Statistica Sinica. 1996;6:311–329. [Google Scholar]
- 49.Chen SX, Qin YL. A two-sample test for high-dimensional data with applications to gene-set testing. Ann. Statist. 2010;38:808–835. [Google Scholar]
- 50.Cooper GM, Hausman RE. The Cell A Molecular Approach. 4th edn. Washington, DC: Sinauer Associates Inc.; 2007. p. 326. [Google Scholar]
- 51.Madsen BE, Browning SR. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meacham F, Boffelli D, Dhahbi J, Martin DIK, Singer M, Pachter L. Identification and correction of systematic error in high-throughput sequence data. BMC Bioinformatics. 2011;12 doi: 10.1186/1471-2105-12-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.King CR, Rathouz PJ, Nicolae DL. An evolutionary framework for association testing in resequencing studies. PLoS Genet. 2010;6:e1001202. doi: 10.1371/journal.pgen.1001202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kwee LC, Liu D, Lin X, Ghosh D, Epstein MP. A powerful and flexible multilocus association test for quantitative traits. Am. J. Hum. Genet. 2008;82:386–397. doi: 10.1016/j.ajhg.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.