

Abstract
Ischemia/reperfusion injury associated with kidney transplantation induces profound acute injury, influences early graft function and affects long-term graft outcomes. To determine whether renal dendritic cells play any role during initial innate ischemia/reperfusion injury and the subsequent development of adaptive immune responses, we studied the behavior and function of renal graft and host infiltrating dendritic cells during early and late phases of renal ischemia/reperfusion injury. Wild type to GFP-transgenic rat kidney transplantation was performed with and without 24 hours cold storage. Ischemia/reperfusion injury in cold stored grafts resulted in histopathological changes of interstitial fibrosis and tubular atrophy by 10 weeks accompanied by upregulation of mRNAs of mediators of interstitial fibrosis and inflammation. In normal rat kidneys we identified two populations of renal dendritic cells, predominant CD103−CD11b/c+ and minor CD103+CD11b/c+ cells. After transplantation without cold storage, grafts maintained CD103− but not CD103+ GFP-negative renal dendritic cells for 10 weeks. In contrast, both cell subsets disappeared from cold stored grafts, which associated with a significant GFP-expressing host CD11b/c+ cell infiltration that included CD103+ dendritic cells with a TNF-α producing phenotype. These changes in graft/host dendritic cell populations were associated with progressive infiltration of host CD4+ T cells with effector/effector-memory phenotypes and IFN-γ secretion. Thus, renal graft ischemia/reperfusion injury causes graft dendritic cell loss and was associated with progressive host dendritic cell and T cell recruitment. Renal resident dendritic cells might function as a protective regulatory network.
Keywords: CD103, IF/TA, effector CD4+ T cells, IFN-α, TNF-α
INTRODUCTION
Although significant improvements have been achieved in early transplant outcomes, progressive deterioration of renal allograft function and late allograft loss remain as major impediments to long-term successful kidney transplantation (KTx), with little clinical progress for many years (1, 2). Late graft loss typically associates with progressive renal dysfunction, proteinuria, and hypertension, and accompanied by histopathological findings of interstitial fibrosis, tubular atrophy, vascular occlusive changes, and glomerulosclerosis (2–5). Recently, these changes (formerly referred to chronic allograft nephropathy) have been renamed and redefined in histologic terms as interstitial fibrosis and tubular atrophy (IF/TA) without evidence of any specific etiology (6). The incidence of IF/TA is as high as 50% of kidney transplants at 1 year, 70% at 2 year, and nearly universal after 10 year of KTx (7–9). As there is no effective therapy, nor proven preventive strategies, IF/TA and late graft loss remain as a significant clinical problem in the field.
Ischemia/reperfusion injury (I/R) of the kidney graft is a leading cause of late graft loss and earlier development of IF/TA. Because of the current shortage of organs for transplantation, the donor pool has been expanded with the use of marginal donors (e.g. old donors, non-heart beating donors, grafts with prolonged cold storage), and grafts from these donors have a higher incidence of severe cold I/R injury and subsequent IF/TA development. During the acute phase, renal I/R injury presents a cascade of inflammatory events involving multiple interconnected factors, including peritubular capillary endothelial cell injury and disturbances of microvascular circulation, production and release of reactive oxygen species and inflammatory mediators, and extravasation of host inflammatory cells. However, the mechanisms of ongoing fibroinflammatory changes in the late phase of I/R injury are yet to be identified.
Dendritic cells (DC) function in and interact between the innate and adaptive arms of the immune system. In the normal kidney, DC together with macrophages are major constituents of the renal mononuclear phagocyte system, and widely distributed throughout the interstitium (10–12). These populations represent substantial heterogenicity and plasticity, and studies aiming the distinction of two populations based on cell surface markers have shown complicated pictures (13–16). They often co-express DC markers (DCSIGN, CD11c) and macrophage markers (CD68, F4/80), suggesting their overlapping cellular function in the kidney. Although renal DC have been shown to play key roles in many renal diseases (14, 17, 18), little is known about their function during KTx-induced I/R injury and the subsequent chronic inflammatory immune reactions leading to IF/TA. Several previous studies have aimed to determine in vivo functional roles of renal mononuclear phagocytes, in particular DC, in various kidney disease models by actively eliminating DC with liposome clodronate or using the CD11c-DTR mouse, and showed contradictory results. Renal DC are protective in cisplatin-induced acute kidney injury (19) and nephrotoxic nephritis (20). On the contrary, they are proinflammatory and detrimental in obstructive nephropathy (21, 22) and chronic glomerulonephritis (23–25). In the model of renal warm ischemia, cell population(s) that are depleted by liposome clodronate have protective roles (26), while the same liposome clodronate-depleted population(s) or CD11c+ cells depleted in CD11c-DRT mouse are shown to be detrimental (13, 27, 28). These conflicting outcomes might be due to different experimental settings or functions of non-DC populations, as these DC deletion methods are not completely specific for DC (21, 29, 30). However, contradictory results in these experiments could be explained by the different roles of kidney resident DC and locally recruited systemic DC and by the difficulty to differentiate two DC populations in these studies, as both populations could be influenced by the depletion methods. We hypothesized that renal graft resident DC and infiltrating host DC might have different functional roles in acute innate and subsequent chronic phases of renal I/R injury. Accordingly, using the GFP transgenic rat KTx model, the aim of this study was to understand the roles of kidney resident DC by examining the alteration of renal DC in the early and late phases of renal I/R injury and by characterizing recruited host DC and other host cells.
RESULTS
Two types of kidney resident DC are identified in rats
To characterized kidney resident DC, naïve rat kidney leukocytes were analyzed by flow cytometry (FCM) and immunohistochemistry (IHC). FCM of isolated renal CD45+ cells revealed that the major leukocyte population in normal kidneys was CD11b/c+ cells, followed by NK cells, T cells, and NKT cells (Fig. 1A). IHC of naïve kidney showed that CD11b/c+ cells form a contiguous network throughout the entire kidney interstitium (Fig. 1C, upper), suggesting that these cells were relevant to renal DC as previously reported in mice and humans (10–12, 14). Further analysis of CD11b/c+ renal DC showed that they expressed CD4, CD86, and MHC class II, but were negative for CD62L (Fig. 1B). Subpopulation (~10%) of CD11b/c+ renal DC also expressed CD103, which were scarcely found in the interstitium (Fig. 1C, lower). These results indicate the presence of two subsets of DC, predominant CD103−CD11b/c+ and minor 103+CD11b/c+ populations, in naïve rat kidneys.
Figure 1. Renal resident leukocytes in naïve rat kidney.

(A) Renal leukocytes were isolated from naïve rat kidneys with collagenase digestion and low-speed centrifugation, and analyzed by three to seven-color flow cytometry. CD45+ cells are gated, and kidney resident leukocyte composition was determined by surface marker expressions. Note that ~70% of renal CD45+ leukocytes were CD103−CD11b/c+ and ~10% CD103+CD11b/c+. (B) Expression of various surface molecules on CD45+CD11b/c+ renal DC. (C) Immunofluorescent stain of naïve kidney revealed networks of CD11b/c+ cells throughout the entire kidney interstitium (red, upper). CD103 expression was limited (red, lower). Representative images of n=5 experiments.
I/R injury results in fibroinflammatory changes in kidney grafts
Orthotopic syngenic KTx was conducted using GFP transgenic rats as recipients and WT as donors with static cold storage of kidney grafts for 24 hrs in UW solution (24h-CS group). Donor and recipient leukocytes in kidney grafts were analyzed with a comparison to control grafts that were transplanted immediately (no-CS group). Robust mRNA upregulation for TNF-α, IL-6, iNOS, and IL-10 at 3 hrs after transplantation were seen in 24h-CS grafts, as we have previously reported (31–33) (Fig. 2A). In contrast, no-CS grafts showed negligible mRNA levels for these mediators (Fig. 2A). In addition, host neutrophil infiltration was significantly increased in 24h-CS grafts, while they were only occasionally seen in no-CS grafts at 12 hrs after KTx (Fig. 2B). At 4 weeks after KTx, no-CS grafts showed CCr levels comparable with those of normal unoperated animals (1.65±0.3 mL/min), while 24h-CS grafts showed significantly reduced CCr levels (0.26±0.2 mL/min) with considerable proteinuria (33).
Figure 2. Early kidney graft injury caused by prolonged 24 hrs cold storage.

Kidney graft samples were obtained at 3 and 12 hrs after WT to GFP KTx with and without 24 hrs static cold storage in UW solution (24h-CS and no-CS groups). (A) Samples at 3 hrs were analyzed for mRNA levels for proinflammatory mediators with RT-PCR. (B) Neutrophil infiltration (yellow arrows) in kidney grafts at 12 hrs was evaluated in IHC with mAb for neutrophils (red, RP-1). N=3–4 for each group. *: p<.05 vs. no-CS controls. #: P<0.005 vs. no-CS control.
Histopathological analyses of 24h-CS grafts at 10W revealed patchy areas of chronic changes comprised of tubular atrophy, interstitial fibrosis, chronic inflammation, and lymphocytic infiltration, relevant to changes that described as IF/TA (Fig. 3A). No evidence of arteritis was seen. In contrast, in the control no-CS grafts, the glomeruli, tubules, and vessels in general appeared unremarkable. Masson’s Trichrome stain revealed significantly increased fibrotic area in 24h-CS grafts (Fig. 3A). Immunofluorescent stain also showed intense α-SMA expression in 24h-CS grafts at 10W, while control no-CS grafts showed marginal α-SMA expression.
Figure 3. Late kidney graft changes caused by I/R injury.


(A) Histopathological findings: WT to GFP KTx was performed with and without 24 hrs static cold storage in UW solution (24h-CS and no-CS groups). Representative histological images of kidneys grafts at 10 weeks are shown. (a) H&E stained section of 24h-CS grafts shows areas of interstitial fibrosis and tubular atrophy (arrow) with chronic interstitial inflammation. Tubular atrophy in the form of dilatation (“thyroidization”) is also seen (arrowhead) (H&E ×100). Glomeruli contained globular material suggestive of protein. (b) In control no-CS grafts, tubules are intact with no significant increase in interstitial fibrous tissue. Glomeruli are unremarkable (H&E ×100). (c) Masson’s trichrome stain reveals significant fibrosis (blue) in 24h-CS grafts. (d) Control no-CS grafts show minimum fibrosis. (e, f) Immunofluorescent stain for α-SMA (red) shows normal arterial α-SMA stain in no-CS grafts. 24h-CS graft shows area of α-SMA positive myofibroblasts with numerous GFP+ host infiltrates. Blue: nuclear stain. Representative images from no-CS (n=4) and 24h-CS (n=5) grafts. (B) mRNA levels for inflammatory and pro-fibrotic mediators in kidney grafts: RT-PCR was conducted using kidney graft samples obtained at 10 weeks. n=2–5 for each group. *: p <.05 vs. no-CS controls.
Active proinflammatory and profibrotic reactions in 24h-CS kidney grafts at 10W were confirmed with RT-PCR. mRNA levels for inflammatory cytokines, as well as RANTES and IL-10 were significantly increased in 24h-CS than in no-CS grafts (Fig. 3B). TGF-β, a key mediator in the progression of fibrosis, and collagen-1 also remarkably increased in 24h-CS grafts. These results indicated that acute innate I/R injury resulted in progressive fibroinflammatory changes in syngenic kidney grafts.
I/R injury associates with loss of graft renal DC and infiltration of host DC/macrophages
To investigate the roles of renal DC in acute I/R injury and the development of chronic fibroinflammatory changes in the WT to GFP KTx model, kidney resident GFP− DC were analyzed at early (3hrs, 12hrs) and late (4W, 10W) time points by FCM of graft CD45+ cells. Percentages of donor leukocytes (GFP−CD45+) started to decrease after KTx, and the reduction was significantly more in 24h-CS than in no-CS grafts. At 10W, only 11.3± 4.9% of leukocytes in 24h-CS grafts were donor phenotype, while 46.3 ± 10.2% in no-CS grafts (Fig. 4A). The reduction of donor leukocytes was mainly due to progressive decreases of GFP−CD11b/c+ renal DC, the major leukocyte population in native kidneys, and at 10W, frequencies of GFP−CD11b/c+ renal DC were markedly lower in 24h-CS than in no-CS grafts (Fig. 4B). These results indicate that I/R injury in 24h-CS grafts resulted in nearly total loss of graft renal DC, while GFP− renal DC were maintained in no-CS grafts.
Figure 4. CD11b/c+ cells in kidney grafts.

After WT to GFP KTx with or without 24 hrs cold storage in UW solution (24h-CS, no-CS groups), kidney grafts were obtained at 3 hrs, 12 hrs, 4 weeks, and 10 weeks (n=2–4 for each time point in no-CS and n=4–6 in 24h-CS groups) for flow cytometry of CD45+ leukocytes isolated from kidney grafts.
(A) Percentages of GFP− cells in graft CD45+ leukocytes. Significant decreases of GFP− graft cells n 24h-CS grafts compared to no-CS grafts. * p<0.05, # p<0.005
(B) Percentages of GFP−CD11b/c+ renal DC in graft CD45+ leukocytes. Graft resident GFP−CD11b/c+ DCsignificantly decreased due to I/R injury in 24h-CS than in no-CS grafts. * p<0.05, # p<0.005
(C) Flow cytometry of CD45+CD11b/c+ cells in kidney grafts. Donor GFP−CD103+ DC quickly disappeared from both 24h-CS and no-CS grafts within 1 week. In contrast, donor GFP−CD103− DC disappeared from 24h-CS, but not from no-CS, grafts. At the same time, host GFP+CD11b/c+ cells increased with time, and GFP+CD11b/c+CD103+ DC significantly increased in 24h-CS grafts. Representative histograms (upper) and bar graph showing percentages in CD45+CD11b/c+ gated cells. * p<0.05
(D) In 24h-CS grafts, both GFP−CD11b/c+ renal DC and GFP+CD11b/c+ infiltrates produced TNF-α at 12 hrs, but the frequencies were significantly higher in the host population. At 10 weeks, TNF-α was detected mostly in GFP+CD11b/c+ cells. * p<0.05
Interestingly, further analysis of GFP−CD11b/c+ renal DC revealed that CD103−CD11b/c+ DC subset disappeared from 24h-CS, but not no-CS, grafts. In contrast, CD103+CD11b/c+ renal DC disappeared from both 24h-CS and no-CS grafts, regardless of I/R injury (Fig. 4C), suggesting different functional roles of CD103+ and CD103− kidney DC populations. Concurrently, at 10W, significant numbers of host GFP+CD11b/c+ cells were found in 24h-CS grafts compared to no-CS grafts. Particularly, host CD103+CD11b/c+ DC were significantly higher in 24h-CS than in no-CS grafts (19.6 ± 2.0 vs. 6.7 ± 1.9%) (Fig. 4C).
IHC at 10W revealed that GFP−CD11b/c+ DC were maintained and homogenously distributed in the interstitium of no-CS grafts (Fig. 5A–C). In contrast, these renal DC were not detected in 24h-CS grafts, and abundant GFP+CD11b/c+ host cells were found among inflammatory infiltrates (Fig. 5D–F). Host CD11b/c+ cells could include monocytes, macrophages, DC, and neutrophils; however, neutrophils (naphthol+ or RP-1+) were rare at 10W (data not shown), indicating that infiltrating host CD11b/c+ cells in 24h-CS grafts at 10W were mostly DC and monocyte/macrophage populations. In IHC, host GFP+CD11b/c+ cells were found among inflammatory infiltrates and did not localize to peritubular area to replace the original renal DC (Fig. 5E), suggesting that functions of host GFP+CD11b/c+ cells were different from those of kidney resident DC.
Figure 5. Immunohistochemistry of CD11b/c+ cells in kidney grafts.
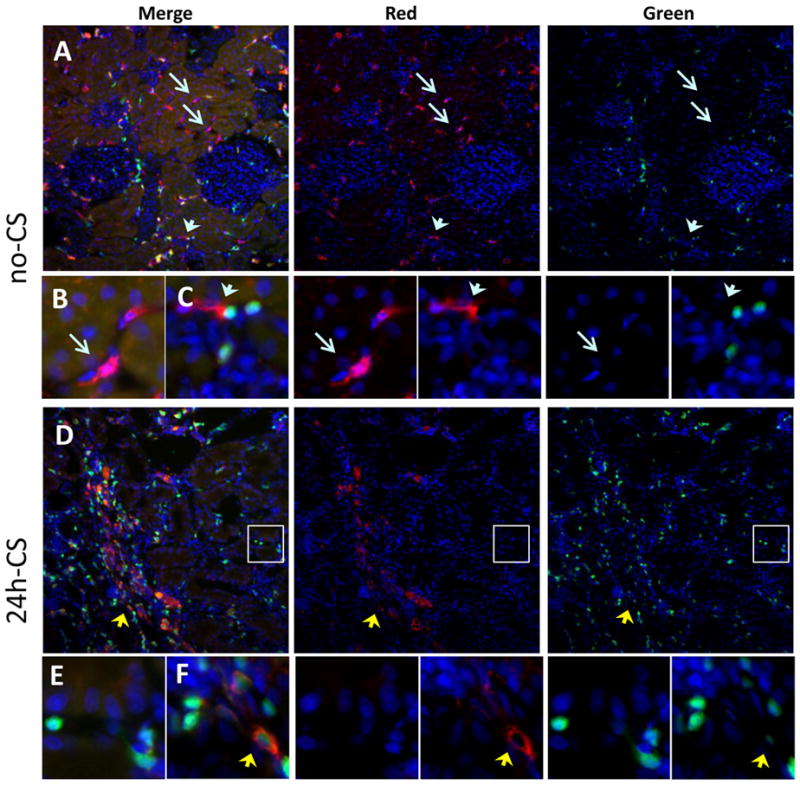
Representative images of CD11b/c stain (red) in kidney grafts at 10 weeks are shown as merged (left panels), red channel (middle panels), and green channel (right panels). In the peritubular area, no-CS grafts show numerous CD11b/c+GFP− renal DC (red), while 24h-CS grafts show GFP+ host infiltrates (green) without CD11b/c expressing renal DC. Instead, 24h-CS grafts show many GFP+CD11b/c+ host infiltrating DC/monocytes.
(A) No-CS grafts at 10 weeks show persistence of CD11b/c+GFP− renal DC (red, blue arrows). (B) High magnification image of CD11b/c+GFP− renal DC (blue arrow) in peritubular area. (C) High magnification image of CD11b/c+GFP− renal DC (blue arrowhead) in the area of GFP+ infiltrates.
(D) In contrast, 24h-CS grafts at 10 weeks had nearly no donor renal DC (GFP−CD11b/c+) but had numerous CD11b/c+ GFP+ infiltrates (double positive yellow, yellow arrowhead). (E) High magnification image of GFP+ infiltrates and loss of CD11b/c+ DC in peritubular area (white circled area in D). (F) High magnification image of GFP+CD11b/c+ cells among host infiltrates (yellow arrowhead).
Graft DC and host infiltrating CD11b/c+ cells produce TNF-α
As renal DC have been shown to produce TNF-α in the mouse warm I/R injury model (27), we examined whether renal resident graft DC and/or host infiltrating DC produced TNF-α in KTx-induced I/R injury. TNF-α production was exclusively seen on CD11b/c+ cells. At 12 hrs in 24h-CS grafts, both GFP− renal DC and GFP+CD11b/c+ host infiltrates produced TNF-α, and frequencies were higher in host GFP+CD11b/c+ cells (Fig. 4D). At 10W, 20.4 ± 2.8% of host CD11b/c+ cells produced TNF-α, while remaining small fractions (5.3 ± 1.2%) of donor renal DC were positive for TNF-α. TNF-α producing renal DC were CD103− subtype.
Renal I/R injury results in continual host cell infiltration
In conjunction with the alteration in DC populations, GFP+ host leukocytes infiltrated kidney grafts after KTx, and percentages of host cells were significantly higher in 24h-CS than in control no-CS grafts as early as 3 hrs after KTx. GFP+ host cells in 24h-CS grafts further increased with time, and nearly all of graft leukocytes became host phenotype by 10W, while no-CS grafts showed significantly less GFP+ host cells (88.7 ± 4.9 vs. 53.7 ± 10.2%) (Fig. 6A). Increases of GFP+ host infiltration into kidney grafts were confirmed in tissue sections, and 24h-CS grafts showed abundant GFP+ cells at 10W (Fig. 6B).
Figure 6. Host (GFP+) cell infiltrations into kidney grafts.

WT to GFP KTx was performed with or without 24 hrs cold storage in UW solution (24h-CS, no-CS groups). Kidney grafts were obtained at 3 hrs, 12 hrs, 4 weeks, and 10 weeks after KTx (n=2–4 for each time point in no-CS and n=4–6 in 24h-CS groups) for immunohistochemistry and flow cytometry.
(A) Percentages of GFP+ host cells in isolated CD45+ leukocytes from kidney grafts. Significantly more GFP+ cells were seen in 24h-CS than in no-CS grafts. * p<0.05, # p<0.005
(B) Kidney graft sections are examined for GFP+ cells with nuclear Hoechst stain (blue). GFP+ host cells increased with time and numerous GFP+ infiltrates were seen in 24h-CS grafts at 10 weeks.
(C) Percentages of CD3+ T cells in graft CD45+ leukocytes. T cells increased with time and were significantly more in 24h-CS than in no-CS grafts. * p<0.05, # p<0.005
(D) Immunohistochemistry for CD3 (red) with nuclear Hoechst stain (blue). Significant host (GFP+) T cell infiltration in 24h-CS grafts at 10 weeks after KTx. Control no-CS grafts showed few CD3+ T cells. Lower panels show images of green GFP channel and red CD3 channel.
Effector-memory type CD4+ T cells accumulate in kidney grafts with I/R injury
FCM analysis of host infiltrates during early and late phases of I/R injury revealed significant increases of CD3+ T cell frequencies in 24h-CS grafts at 4W and 10W compared to control no-CS kidney grafts (Fig. 6C). IHC with anti-CD3 mAb also confirmed numerous GFP+CD3+ cells among infiltrates in 24 h-CS grafts at 10W (Fig. 6D).
Characterization of CD3+ cells in kidney grafts revealed gradual increases of CD4+ T cells with I/R injury. At 10W, infiltrating GFP+ host T cells in 24h-CS grafts were dominated by CD4+ T cells with significantly higher CD4/CD8 ratios, compared to those in no-CS grafts (1.8 ± 0.4 vs. 0.7 ± 0.2) (Fig. 7A, B). Majority of GFP+CD3+ cells were CD62L negative and expressed low molecular weight CD45 isoforms, indicating that they were effector or effector-memory phenotype T cells (Fig. 7A). Function of T cells was analyzed by their capability to produce IFN-γ. CD3+ cells producing IFN-γ were mostly GFP−donor cells at 3 hrs of I/R injury, but quickly shifted to GFP+ host T cells at 12 hrs. At 10W, one third of GFP+ host T cells were able to produce IFN-γ. Frequencies of IFN-γ producing host T cells were similar among CD4+ and CD8+ T cells (Fig. 7C).
Figure 7. Characterization of host (GFP+) T cells in kidney grafts with I/R injury.

(A) Analysis of host infiltrating T cells (GFP+CD45+CD3+) for CD4/CD8 and CD62L/CD45RC. CD4+ T cells gradually increased in 24h-CS grafts. Host T cells were mainly CD4+ CD62L−CD45RC− effector-memory phenotype T cells.
(B) CD4/CD8 ratios were significantly higher in 24h-CS than in no-CS grafts at 4 and 10 weeks (n=2–4 for each time point in no-CS and n=4–6 in 24h-CS groups). * p<0.05
(C) Intracellular IFN-γ expression on CD45+ leukocytes from kidney grafts. Early after KTx at 3 hrs, IFN-γ was detected in GFP−CD3+ graft T cells. At 10 weeks, GFP+ host T cells produced IFN-γ. In GFP+CD3+ cells in 24h-CS grafts at 10 weeks, both CD4+ and CD8+ T cells were able to produce IFN-γ. N=2–4 for each time point in no-CS and n=4–6 in 24h-CS groups.
DISCUSSION
Renal I/R injury, which occurs in some degree in every transplant graft, is a significant deleterious factor responsible for early poor graft function as well as for late graft loss due to chronic inflammation and fibrosis, characterized as IF/TA. To understand the mechanisms by which acute I/R injury results in continual adaptive immune responses, the current study focused on graft renal DC alteration and host DC infiltration during acute and late phases of KTx-induced renal I/R injury. DC are known to play crucial roles in regulating normal and abnormal immune functions. Numerous studies have described the roles of DC in lymphoid tissues during steady-state as well as various disease conditions; however, relatively little is known about roles of tissue resident DC, particularly in the kidney. Previous histopathological studies show that normal kidney DC form a contiguous network throughout the entire interstitium and create an anatomic surveillance network to sense and respond to substances diffusing into the kidney (11, 12, 14). In vitro analyses of isolated renal DC have shown that they are in immature status (low costimulatory function) and possess full phagocytic ability (10). These studies certainly have established the location and phenotypes of renal DC; however, in vivo functional roles of renal DC are not defined.
Using the WT to GFP-transgenic rat KTx model, which enabled us to differentiate donor kidney resident DC from recruited host DC, we demonstrate here an early loss of GFP− donor renal DC and progressive GFP+ host DC infiltration into kidney grafts with I/R injury. At 4–10 weeks after initial I/R injury, the majority of donor renal DC disappear from 24h-CS grafts, and CD11b/c+ cells in these kidney grafts become nearly totally GFP+ host phenotype, which are located among inflammatory infiltrates, and do not form the interstitial network. In contrast, control grafts without I/R injury maintain the interstitial network of donor phenotype renal DC for 10 weeks with significantly less host DC infiltration. These results suggest that kidney resident DC and recruited systemic DC have different functional roles. Infiltrating host DC promote fibroinflammatory reactions, while renal resident DC might have protective roles in regulating chronic inflammation and infiltration of host DC. Although further investigation is needed, renal resident DC could be involved in control of renal microenvironment and regulation of adaptive immune responses.
We have identified two subpopulations of renal DC in normal rat kidneys, predominant CD103−CD11b/c+ and minor CD103+CD11b/c+ DC, and they behave differently in responding to KTx-induced renal I/R injury. GFP−CD103+ DC quickly disappear from both 24h-CS and control no-CS grafts, regardless of injury. In contrast, GFP−CD103− DC are maintained in no-CS grafts, but disappear from 24h-CS grafts with I/R injury. Recent studies show that DC exhibit distinct functional roles depending on CD103 (integrin αE) expression; CD103 DCs are highly qualified to perform innate mechanisms such as antigen clearance and chemokine-mediated attraction of leukocytes. In contrast, CD103+DC are primarily involved in cross-presentation of self or foreign antigens after migrating to draining lymph nodes (34, 35). E-cadherin, the ligand for CD103, mediates calcium-dependent cell-cell adhesion, and DC maturation, function, and migration are suggested to be connected to E-cadherin (36). In Langerhans cell cultures, IL-1, TNF-α, and LPS decrease E-cadherin expression and dissociate DC aggregates with increased expressions of MHC class II, CD40, and CD86, suggesting that in vivo DC migration with cytokines/LPS might be caused by a loss of E-cadherin-mediated adhesion (37–39). It is long known that MHC class II+ DC disappear from non-lymphoid tissues as early as 4 hrs and maximum of 48 hrs after LPS, IL-1 or TNF-α injection (40). It is also shown that renal DC migrate to T cell areas of draining lymph nodes to promote antigen-specific T-cell proliferation (41). In organ transplantation, the migration of donor DC from allografts to recipient lymphoid tissues has been well documented (42, 43). Although these studies do not differentiate CD103+ and CD103− DC, the migration from the peripheral tissues to the lymphoid organs after antigen/pathogen exposure is an essential feature oftissue resident DC. The disappearance of renal CD103+ DC with migratory features in both no-CS and 24h-CS grafts in this study could be caused by KTx and might be involved in T cell activation. In contrast, the mechanisms of CD103− renal DC loss in 24h-CS, but not in no-CS, grafts remain to be determined. As we find negligible frequencies of donor cells in host spleens after I/R injury (unpublished observation), massive loss of this population may not be caused by migration. It is tempting to speculate that CD103− DCs might die after being involved in innate responses to clean antigens and produce cytokines/chemokines, such as TNF-α, as shown in this study.
Although the information on the turnover of DCs in non-lymphoid tissues is limited, several studies have shown that DC homeostasis in the non-lymphoid tissues requires constant replacement with new cells. Similar to findings in the lymphoid tissues (44, 45), kidney DC turnover was trucked with BrdU labeling, with bone marrow transplantation into irradiated recipients, and with parabiosis (35, 46, 47). In the steady state, both CD103+ and CD103− DC undergo limited numbers of divisions and are entirely replaced within 2–4 weeks by DC precursors most likely from blood (35). However, in the current study, donor type graft CD103− DCs were maintained for 10 weeks in syngenic recipients with minimum graft injury. We also observed the same maintenance of kidney graft resident CD103− DC in allogenic recipients under the control of rejection with tacrolimus (unpublished data). Epidermal Langerhans cells were shown to remain as host type for at least 18 months in bone marrow chimera, and in parabiotic mice with shared blood circulation, there was no mixing of skin Langerhans cells (48), suggesting that under steady-state conditions, Langerhans cells can be locally maintained. Further study is warranted to identify the mechanisms in maintaining kidney resident CD103− DC. It is intriguing to investigate the possibility that kidney CD103− DC are maintained in part by local self-renewing progenitors that divide in situ.
Progressive T cell infiltration in 24h-CS grafts is caused by accumulation of CD62L−CD45RC− effector-memory phenotype host CD4 T cells. These GFP+ infiltrating T cells most likely include both antigen-specific (e.g. injury-associated antigens, self antigens) and non-specifically activated T cells. Previous mouse studies report continual presence of effector/memory T cells in the kidney with renal warm ischemia (49–51). These T cells could play roles in promoting IF/TA development; however, T cell depletion starting 3 days after mouse warm I/R injury increased IL-1 and did not show long-term protective effects (49, 50, 52). Further studies to understand roles of CD4+ memory T cells in kidney grafts, their specificity, and activation signals will be important to advance our understanding of late graft loss with IF/TA and to develop therapeutic strategies to prevent/treat IF/TA and other renal fibroinflammatory conditions.
The current study uses the syngenic kidney transplantation model to exclusively study the effects of ischemic injury without any additional factor influencing the endpoints, such as immunosuppression and alloimmunity. These factors are unavoidably involved in clinical transplantation and could significantly influence I/R injury and graft outcomes. In accordance with previous studies (53–56), current ongoing study in the allotransplantation model suggests augmented graft injury in allogenic than in syngenic kidney grafts. Renal resident CD103− DC were not detected in allografts with I/R injury, while they were maintained in those without injury (unpublished data). Further studies identifying the contributing factors and mechanisms of I/R injury in allografts would be beneficial to improve outcomes of clinical kidney transplantation.
In summary, using the WT to GFP rat KTx model, the study demonstrates that renal cold I/R injury resulted in the early loss of kidney graft resident CD103−CD11b/c+ and CD103+CD11b/c+ DC, while CD103− renal DC were maintained in grafts without I/R injury. Progressive infiltration of host DC and CD4+ T cells with effector/effector-memory phenotypes (CD62L−CD45RC−) occurred during the following 10W after I/R injury and associated with an upregulation of fibroinflammatory mediators and histopathological IF/TA development. As the loss of renal graft resident DC due to acute innate I/R injury associates with a shift to adaptive immunity and chronic inflammation, renal resident DC might function as a protective regulatory network against profibroinflammatory changes and host DC infiltration. The current experimental study uses young animals, and the impact of I/R injury on kidney grafts obtained from healthy animals cannot be extended to that of cold ischemic injury on a damaged elderly human kidney. However, the study provides new insight into mechanisms of persistent renal I/R injury and roles of renal resident DC.
MATERIALS & METHODS
Reagents
Phycoerythrin (PE), Allophycocyanin (APC), Pacific Blue (PB), Alexa Fluor 647, Alexa Fluor 700, and PE/Cy7-conjugated monoclonal antibodies (mAbs) used in this study included anti-rat CD3, CD4, CD8, CD11b/c, CD45, CD45R, CD45RC, CD62L, CD86, CD103, CD161a, IFN-γ, and TNF-α obtained from eBioscience (San Diego, CA), Serotec (Oxford, UK), and BD Pharmingen (San Diego, CA). RPMI 1640, L-glutamine, PBS, and FBS were from Cellgro Mediatech, Inc. (Manassas, VA). 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES) buffer and gentamicin were from Life Technologies (Grand Island, NY). Collagenase (Type IV), phorbol 12-myristate 13-acetate (PMA), calcium ionomycin, paraformaldehyde (PFA), and saponin were obtained from Sigma (St. Louis, MO). Golgi Plug™ (BD Pharmingen) and sodium azide (Fisher Scientific, Fair Lawn, NJ) also were used.
Animals and surgical procedures
A breeding colony of GFP-transgenic and WT Sprague–Dawley (SD) rats, originally generated by Dr. Masaru Okabe (University of Osaka, Osaka, Japan) (57), was maintained at the University of Pittsburgh. All procedures in this experiment were performed according to the guidelines of the National Research Council's Guide for the Humane Care and Use of Laboratory Animals and approved by the Council on Animal Care at the University of Pittsburgh.
Orthotopic KTx was performed using GFP transgenic rats as recipients and WT as donors using the technique described previously (31). The donor left kidney was flushed with 3 ml UW solution (Du Pont, Wilmington, DE, USA) and transplanted either immediately or after 24 hrs preservation in UW at 4°C into the recipient by end-to-side anastomoses to recipient infrarenal abdominal aorta and infrarenal vena cava. The recipient left native kidney was removed, and end-to-end ureteral anastomosis was performed. Graft samples were obtained during the early phase at 3 and 12 hrs, and late phase at 4 and 10W.
Renal leukocyte isolation
Renal leukocytes were isolated from the kidney by the collagenase digestion method. Briefly, the kidney was perfused in situ with 25 mL PBS, excised, then cut into small pieces in RPMI 1640 containing 5% FBS, L-glutamine, 25 mM HEPES, 4 μg/ml gentamycin, and 0.05% collagenase. After incubation for 30 mins at 37°C, cells were filtrated through a 70 μm filter, and leukocytes were separated by low-speed centrifugation (five times at 500 rpm/5 mins/4°C) and obtained by high-speed centrifugation (1500 rpm/10 mins/4°C).
Flow cytometry
Isolated renal leukocytes were incubated with mAbs at 4°C for 30 mins and analyzed on an LSR (BD Biosciences). Data were analyzed using Flowjo 7.5 (Tree Star Inc, Oregon Corporation, Ashland, OR). TNF-α producing cells were detected after culturing isolated leukocytes (106 cells/100 μL) with Golgi Plug™ for 6 hrs at 37°C. For the detection of IFN-γ, leukocytes were incubated with PMA, calcium ionomycin and Golgi Plug™ for 6 hrs at 37°C. At the end of 6 hrs, cells were washed, incubated with mAbs for surface markers, fixed with 2% PFA, permialized with 0.1% Saponin, and incubated with anti-TNF-α or IFN-γ mAbs at 4°C.
PCR
After total RNA was extracted from the graft tissues and cDNA was prepared, mRNA expression was quantified by SYBR Green two-step, real-time RT-PCR previously described (58). The expression of each gene was normalized to GAPDH mRNA content and calculated relative to normal control using DD Ct method (59, 60).
Routine pathology and IHC
The kidney samples were stored in 2% PFA at 4°C, then in 2.3 M sucrose in PBS overnight, embedded in optimal cutting temperature compound, and frozen in liquid nitrogen-cooled isopentane. Samples were cut into 6 μm sections, blocked with superblock (ScyTek Lab, Logan, UT), and stained with mAbs for αSMA, CD3, CD11b/c and neutrophils (RP-1). Sections were nuclear DNA stained with Hoechst dye (Bisbenzimide), coverslipped with Gelvatol, and visualized with an Olympus BX51 epifluorescence microscope or a Fluoview 1000 confocal microscope (Olympus, Center Valley, PA).
Data analysis
Results are expressed as means ± SD. Statistical analysis was performed using Student's t-test or one-way ANOVA and Fisher's protected least significant difference test where appropriate. A probability level of P < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by the National Institutes of Health Grant DK071753.
We thank Lisa Chedwick for excellent technical support and Carla Forsythe for the preparation and organization of the manuscript.
Footnotes
DISCLOSURE
The authors do not have any financial relationships with any company that may have a financial interest in the information contained in this manuscript.
References
- 1.Meier-Kriesche HU, Schold JD, Srinivas TR, Kaplan B. Lack of improvement in renal allograft survival despite a marked decrease in acute rejection rates over the most recent era. Am J Transplant. 2004 Mar;4(3):378–83. doi: 10.1111/j.1600-6143.2004.00332.x. [DOI] [PubMed] [Google Scholar]
- 2.Kasiske BL, Gaston RS, Gourishankar S, Halloran PF, Matas AJ, Jeffery J, et al. Long-term deterioration of kidney allograft function. Am J Transplant. 2005 Jun;5(6):1405–14. doi: 10.1111/j.1600-6143.2005.00853.x. [DOI] [PubMed] [Google Scholar]
- 3.Joosten SA, Sijpkens YW, van Kooten C, Paul LC. Chronic renal allograft rejection: pathophysiologic considerations. Kidney Int. 2005 Jul;68(1):1–13. doi: 10.1111/j.1523-1755.2005.00376.x. [DOI] [PubMed] [Google Scholar]
- 4.Demetris AJ, Murase N, Starzl TE, Fung JJ. Pathology of Chronic Rejection: An Overview of Common Findings and Observations About Pathogenic Mechanisms and Possible Prevention. Graft (Georget Tex) 1998 May;1(2):52–9. [PMC free article] [PubMed] [Google Scholar]
- 5.Paul LC. Chronic renal transplant loss. Kidney Int. 1995 Jun;47(6):1491–9. doi: 10.1038/ki.1995.211. [DOI] [PubMed] [Google Scholar]
- 6.Solez K. Making global transplantation pathology standards truly global. Am J Transplant. 2007 Dec;7(12):2834. doi: 10.1111/j.1600-6143.2007.02001.x. [DOI] [PubMed] [Google Scholar]
- 7.Nankivell BJ, Borrows RJ, Fung CL, O'Connell PJ, Allen RD, Chapman JR. The natural history of chronic allograft nephropathy. N Engl J Med. 2003 Dec 11;349(24):2326–33. doi: 10.1056/NEJMoa020009. [DOI] [PubMed] [Google Scholar]
- 8.Bosmans JL, Ysebaert DK, Verpooten GA. Chronic allograft nephropathy: what have we learned from protocol biopsies? Transplantation. 2008 Apr 15;85(7 Suppl):S38–41. doi: 10.1097/TP.0b013e318169c5d0. [DOI] [PubMed] [Google Scholar]
- 9.Seron D, Moreso F. Protocol biopsies in renal transplantation: prognostic value of structural monitoring. Kidney Int. 2007 Sep;72(6):690–7. doi: 10.1038/sj.ki.5002396. [DOI] [PubMed] [Google Scholar]
- 10.Steptoe RJ, Patel RK, Subbotin VM, Thomson AW. Comparative analysis of dendritic cell density and total number in commonly transplanted organs: morphometric estimation in normal mice. Transpl Immunol. 2000 Mar;8(1):49–56. doi: 10.1016/s0966-3274(00)00010-1. [DOI] [PubMed] [Google Scholar]
- 11.Soos TJ, Sims TN, Barisoni L, Lin K, Littman DR, Dustin ML, et al. CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. 2006 Aug;70(3):591–6. doi: 10.1038/sj.ki.5001567. [DOI] [PubMed] [Google Scholar]
- 12.Kaissling B, Le Hir M. The renal cortical interstitium: morphological and functional aspects. Histochem Cell Biol. 2008 Aug;130(2):247–62. doi: 10.1007/s00418-008-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li L, Okusa MD. Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin Nephrol. 2010 May;30(3):268–77. doi: 10.1016/j.semnephrol.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woltman AM, de Fijter JW, Zuidwijk K, Vlug AG, Bajema IM, van der Kooij SW, et al. Quantification of dendritic cell subsets in human renal tissue under normal and pathological conditions. Kidney Int. 2007 May;71(10):1001–8. doi: 10.1038/sj.ki.5002187. [DOI] [PubMed] [Google Scholar]
- 15.Segerer S, Heller F, Lindenmeyer MT, Schmid H, Cohen CD, Draganovici D, et al. Compartment specific expression of dendritic cell markers in human glomerulonephritis. Kidney Int. 2008 Jul;74(1):37–46. doi: 10.1038/ki.2008.99. [DOI] [PubMed] [Google Scholar]
- 16.Ferenbach D, Hughes J. Macrophages and dendritic cells: what is the difference? Kidney Int. 2008 Jul;74(1):5–7. doi: 10.1038/ki.2008.189. [DOI] [PubMed] [Google Scholar]
- 17.John R, Nelson PJ. Dendritic cells in the kidney. J Am Soc Nephrol. 2007 Oct;18(10):2628–35. doi: 10.1681/ASN.2007030273. [DOI] [PubMed] [Google Scholar]
- 18.Hochheiser K, Tittel A, Kurts C. Kidney dendritic cells in acute and chronic renal disease. Int J Exp Pathol. 2011 Jun;92(3):193–201. doi: 10.1111/j.1365-2613.2010.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tadagavadi RK, Reeves WB. Renal dendritic cells ameliorate nephrotoxic acute kidney injury. J Am Soc Nephrol. 2010 Jan;21(1):53–63. doi: 10.1681/ASN.2009040407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scholz J, Lukacs-Kornek V, Engel DR, Specht S, Kiss E, Eitner F, et al. Renal dendritic cells stimulate IL-10 production and attenuate nephrotoxic nephritis. J Am Soc Nephrol. 2008 Mar;19(3):527–37. doi: 10.1681/ASN.2007060684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitamoto K, Machida Y, Uchida J, Izumi Y, Shiota M, Nakao T, et al. Effects of liposome clodronate on renal leukocyte populations and renal fibrosis in murine obstructive nephropathy. J Pharmacol Sci. 2009 Nov;111(3):285–92. doi: 10.1254/jphs.09227fp. [DOI] [PubMed] [Google Scholar]
- 22.Dong X, Bachman LA, Miller MN, Nath KA, Griffin MD. Dendritic cells facilitate accumulation of IL-17 T cells in the kidney following acute renal obstruction. Kidney Int. 2008 Nov;74(10):1294–309. doi: 10.1038/ki.2008.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holdsworth SR, Neale TJ, Wilson CB. Abrogation of macrophage-dependent injury in experimental glomerulonephritis in the rabbit. Use of an antimacrophage serum. J Clin Invest. 1981 Sep;68(3):686–98. doi: 10.1172/JCI110304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heymann F, Meyer-Schwesinger C, Hamilton-Williams EE, Hammerich L, Panzer U, Kaden S, et al. Kidney dendritic cell activation is required for progression of renal disease in a mouse model of glomerular injury. J Clin Invest. 2009 May;119(5):1286–97. doi: 10.1172/JCI38399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hochheiser K, Engel DR, Hammerich L, Heymann F, Knolle PA, Panzer U, et al. Kidney Dendritic Cells Become Pathogenic during Crescentic Glomerulonephritis with Proteinuria. J Am Soc Nephrol. 2011 Feb;22(2):306–16. doi: 10.1681/ASN.2010050548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim MG, Boo CS, Ko YS, Lee HY, Cho WY, Kim HK, et al. Depletion of kidney CD11c+ F4/80+ cells impairs the recovery process in ischaemia/reperfusion-induced acute kidney injury. Nephrol Dial Transplant. 2010 Sep;25(9):2908–21. doi: 10.1093/ndt/gfq183. [DOI] [PubMed] [Google Scholar]
- 27.Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. 2007 Apr;71(7):619–28. doi: 10.1038/sj.ki.5002132. [DOI] [PubMed] [Google Scholar]
- 28.Day YJ, Huang L, Ye H, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: role of macrophages. Am J Physiol Renal Physiol. 2005 Apr;288(4):F722–31. doi: 10.1152/ajprenal.00378.2004. [DOI] [PubMed] [Google Scholar]
- 29.Probst HC, Tschannen K, Odermatt B, Schwendener R, Zinkernagel RM, Van Den Broek M. Histological analysis of CD11c-DTR/GFP mice after in vivo depletion of dendritic cells. Clin Exp Immunol. 2005 Sep;141(3):398–404. doi: 10.1111/j.1365-2249.2005.02868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bennett CL, Clausen BE. DC ablation in mice: promises, pitfalls, and challenges. Trends Immunol. 2007 Dec;28(12):525–31. doi: 10.1016/j.it.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 31.Neto JS, Nakao A, Kimizuka K, Romanosky AJ, Stolz DB, Uchiyama T, et al. Protection of transplant-induced renal ischemia-reperfusion injury with carbon monoxide. Am J Physio Renal Physiol. 2004 Nov;287(5):F979–89. doi: 10.1152/ajprenal.00158.2004. [DOI] [PubMed] [Google Scholar]
- 32.Nakao A, Faleo G, Shimizu H, Nakahira K, Kohmoto J, Sugimoto R, et al. Ex vivo carbon monoxide prevents cytochrome P450 degradation and ischemia/reperfusion injury of kidney grafts. Kidney Int. 2008 Oct;74(8):1009–16. doi: 10.1038/ki.2008.342. [DOI] [PubMed] [Google Scholar]
- 33.Faleo G, Neto JS, Kohmoto J, Tomiyama K, Shimizu H, Takahashi T, et al. Carbon monoxide ameliorates renal cold ischemia-reperfusion injury with an upregulation of vascular endothelial growth factor by activation of hypoxia-inducible factor. Transplantation. 2008 Jun 27;85(12):1833–40. doi: 10.1097/TP.0b013e31817c6f63. [DOI] [PubMed] [Google Scholar]
- 34.del Rio ML, Bernhardt G, Rodriguez-Barbosa JI, Forster R. Development and functional specialization of CD103+ dendritic cells. Immunol Rev. 2010 Mar;234(1):268–81. doi: 10.1111/j.0105-2896.2009.00874.x. [DOI] [PubMed] [Google Scholar]
- 35.Ginhoux F, Liu K, Helft J, Bogunovic M, Greter M, Hashimoto D, et al. The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med. 2009 Dec 21;206(13):3115–30. doi: 10.1084/jem.20091756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang A, Bloom O, Ono S, Cui W, Unternaehrer J, Jiang S, et al. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity. 2007 Oct;27(4):610–24. doi: 10.1016/j.immuni.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jakob T, Udey MC. Regulation of E-cadherin-mediated adhesion in Langerhans cell-like dendritic cells by inflammatory mediators that mobilize Langerhans cells in vivo. J Immunol. 1998 Apr 15;160(8):4067–73. [PubMed] [Google Scholar]
- 38.Riedl E, Stockl J, Majdic O, Scheinecker C, Knapp W, Strobl H. Ligation of E-cadherin on in vitro-generated immature Langerhans-type dendritic cells inhibits their maturation. Blood. 2000 Dec 15;96(13):4276–84. [PubMed] [Google Scholar]
- 39.Jakob T, Brown MJ, Udey MC. Characterization of E-cadherin-containing junctions involving skin-derived dendritic cells. J Invest Dermatol. 1999 Jan;112(1):102–8. doi: 10.1046/j.1523-1747.1999.00475.x. [DOI] [PubMed] [Google Scholar]
- 40.Roake JA, Rao AS, Morris PJ, Larsen CP, Hankins DF, Austyn JM. Dendritic cell loss from nonlymphoid tissues after systemic administration of lipopolysaccharide, tumor necrosis factor, and interleukin 1. J Exp Med. 1995 Jun 1;181(6):2237–47. doi: 10.1084/jem.181.6.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong X, Swaminathan S, Bachman LA, Croatt AJ, Nath KA, Griffin MD. Antigen presentation by dendritic cells in renal lymph nodes is linked to systemic and local injury to the kidney. Kidney Int. 2005 Sep;68(3):1096–108. doi: 10.1111/j.1523-1755.2005.00502.x. [DOI] [PubMed] [Google Scholar]
- 42.Larsen CP, Morris PJ, Austyn JM. Migration of dendritic leukocytes from cardiac allografts into host spleens. A novel pathway for initiation of rejection. J Exp Med. 1990 Jan 1;171(1):307–14. doi: 10.1084/jem.171.1.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murase N, Starzl TE, Tanabe M, Fujisaki S, Miyazawa H, Ye Q, et al. Variable chimerism, graft-versus-host disease, and tolerance after different kinds of cell and whole organ transplantation from Lewis to brown Norway rats. Transplantation. 1995 Jul 27;60(2):158–71. doi: 10.1097/00007890-199507000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kabashima K, Banks TA, Ansel KM, Lu TT, Ware CF, Cyster JG. Intrinsic lymphotoxin-beta receptor requirement for homeostasis of lymphoid tissue dendritic cells. Immunity. 2005 Apr;22(4):439–50. doi: 10.1016/j.immuni.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 45.Liu K, Waskow C, Liu X, Yao K, Hoh J, Nussenzweig M. Origin of dendritic cells in peripheral lymphoid organs of mice. Nat Immunol. 2007 Jun;8(6):578–83. doi: 10.1038/ni1462. [DOI] [PubMed] [Google Scholar]
- 46.Leszczynski D, Renkonen R, Hayry P. Localization and turnover rate of rat renal 'dendritic' cells. Scand J Immunol. 1985 Apr;21(4):355–60. doi: 10.1111/j.1365-3083.1985.tb01441.x. [DOI] [PubMed] [Google Scholar]
- 47.Helft J, Ginhoux F, Bogunovic M, Merad M. Origin and functional heterogeneity of non- lymphoid tissue dendritic cells in mice. Immunol Rev. 2010 Mar;234(1):55–75. doi: 10.1111/j.0105-2896.2009.00885.x. [DOI] [PubMed] [Google Scholar]
- 48.Merad M, Manz MG, Karsunky H, Wagers A, Peters W, Charo I, et al. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol. 2002 Dec;3(12):1135–41. doi: 10.1038/ni852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ascon M, Ascon DB, Liu M, Cheadle C, Sarkar C, Racusen L, et al. Renal ischemia-reperfusion leads to long term infiltration of activated and effector-memory T lymphocytes. Kidney Int. 2009 Mar;75(5):526–35. doi: 10.1038/ki.2008.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burne-Taney MJ, Yokota N, Rabb H. Persistent renal and extrarenal immune changes after severe ischemic injury. Kidney Int. 2005 Mar;67(3):1002–9. doi: 10.1111/j.1523-1755.2005.00163.x. [DOI] [PubMed] [Google Scholar]
- 51.Satpute SR, Park JM, Jang HR, Agreda P, Liu M, Gandolfo MT, et al. The role for T cell repertoire/antigen-specific interactions in experimental kidney ischemia reperfusion injury. J Immunol. 2009 Jul 15;183(2):984–92. doi: 10.4049/jimmunol.0801928. [DOI] [PubMed] [Google Scholar]
- 52.Hochegger K, Schatz T, Eller P, Tagwerker A, Heininger D, Mayer G, et al. Role of alpha/beta and gamma/delta T cells in renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2007 Sep;293(3):F741–7. doi: 10.1152/ajprenal.00486.2006. [DOI] [PubMed] [Google Scholar]
- 53.Coulson MT, Jablonski P, Howden BO, Thomson NM, Stein AN. Beyond operational tolerance: effect of ischemic injury on development of chronic damage in renal grafts. Transplantation. 2005 Aug 15;80(3):353–61. doi: 10.1097/01.tp.0000168214.84417.7d. [DOI] [PubMed] [Google Scholar]
- 54.Herrero-Fresneda I, Torras J, Cruzado JM, Condom E, Vidal A, Riera M, et al. Do alloreactivity and prolonged cold ischemia cause different elementary lesions in chronic allograft nephropathy? Am J Pathol. 2003 Jan;162(1):127–37. doi: 10.1016/S0002-9440(10)63804-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kouwenhoven EA, de Bruin RW, Heemann UW, Marquet RL, Ijzermans JN. Late graft dysfunction after prolonged cold ischemia of the donor kidney: inhibition by cyclosporine. Transplantation. 1999 Oct 15;68(7):1004–10. doi: 10.1097/00007890-199910150-00018. [DOI] [PubMed] [Google Scholar]
- 56.Kouwenhoven EA, de Bruin RW, Bajema IM, Marquet RL, Ijzermans JN. Cold ischemia augments allogeneic-mediated injury in rat kidney allografts. Kidney Int. 2001 Mar;59(3):1142–8. doi: 10.1046/j.1523-1755.2001.0590031142.x. [DOI] [PubMed] [Google Scholar]
- 57.Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. 'Green mice' as a source of ubiquitous green cells. FEBS Lett. 1997 May 5;407(3):313–9. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 58.Neto JS, Nakao A, Toyokawa H, Nalesnik MA, Romanosky AJ, Kimizuka K, et al. Low-dose carbon monoxide inhalation prevents development of chronic allograft nephropathy. Am J Physiol Renal Physiol. 2006 Feb;290(2):F324–34. doi: 10.1152/ajprenal.00026.2005. [DOI] [PubMed] [Google Scholar]
- 59.Cikos S, Bukovska A, Koppel J. Relative quantification of mRNA: comparison of methods currently used for real-time PCR data analysis. BMC Mol Biol. 2007;8:113. doi: 10.1186/1471-2199-8-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]