

Abstract
As a persistent pump, the mammalian heart demands a high-capacity mitochondrial system. Significant progress has been made in delineating the gene regulatory networks that control mitochondrial biogenesis and function in striated muscle. The PPARγ coactivator-1 (PGC-1) coactivators serve as inducible boosters of downstream transcription factors that control the expression of genes involved in mitochondrial energy transduction, ATP synthesis, and biogenesis. PGC-1 gain-of-function and loss-of-function studies targeting two PGC-1 family members, PGC-1α and PGC-1β, have provided solid evidence that these factors are both necessary and sufficient for perinatal mitochondrial biogenesis and maintenance of high-capacity mitochondrial function in postnatal heart. In humans, during the development of heart failure owing to hypertension or obesity-related diabetes, the activity of the PGC-1 coactivators, and several downstream target transcription factors, is altered. Gene targeting studies in mice have demonstrated that loss of PGC-1α and PGC-1β in heart leads to heart failure. Interestingly, the pattern of dysregulation within the PGC-1 transcriptional regulatory circuit distinguishes the heart disease caused by hypertension from that caused by diabetes. This transcriptional regulatory cascade and downstream metabolic pathways should be considered as targets for novel etiology-specific therapeutics aimed at the early stages of heart failure.
As a pump, the heart serves a critical role throughout the life of higher organisms. The energy demands of the heart are immense, demanding kilogram quantities of ATP daily in humans. The high energy requirements of the heart are met through a specialized, high-capacity mitochondrial system. During the past decade, significant advances have been made in delineating the gene regulatory circuitry controlling mitochondrial biogenesis and function in heart and other mitochondrial-rich organs. This work has defined many of the key transcriptional regulators of muscle mitochondrial fuel metabolism and energy transduction pathways, including the peroxisome proliferator-activated receptors (PPARs), estrogen-related receptors (ERRs), and nuclear respiratory factors 1 and 2 (NRF-1 and -2; Puigserver and Spiegelman 2003; Kelly and Scarpulla 2004). PPARs regulate the mitochondrial fatty acid oxidation (FAO) pathway (Finck and Kelly 2002). ERRs and NRFs regulate genes involved in FAO, the tricarboxylic acid cycle, electron transport chain, and oxidative phosphorylation (OXPHOS). In addition, NRF-1, a nuclearly encoded transcription factor, activates mitochondrial transcription factor A (Tfam), which is involved in mitochondrial DNA replication, a key component of the mitochondrial biogenic response.
A major breakthrough in the understanding of how these transcription factors are dynamically coregulated in a temporally orchestrated manner came with the discovery of PGC-1α, initially described as a coactivator of the adipocyte-enriched nuclear receptor PPARγ in brown adipose tissue (Puigserver et al. 1998). This work revealed that PGC-1α activated transcription of the NRF-1 gene and the expression of many downstream enzymes and proteins involved in mitochondrial biogenesis and function including respiratory uncoupling, a key process for thermogenesis in the brown adipocyte (Puigserver et al. 1998). Subsequently, it was shown that PGC-1α is expressed in heart, where mitochondria are responsible for high levels of ATP production, serving as coactivator for PPARα and ERRα in addition to NRF-1 (Vega et al. 2000; Huss et al. 2004). PGC-1α enhances gene transcription by interacting directly with its target transcription factors, serving as a platform to recruit molecules that mediate chromatin remodeling via histone acetyltransferase (HAT) activity and by recruiting components of the transcriptional machinery, including RNA polymerase II, to activate transcription of its target genes (Ge et al. 2008). Interestingly, PGC-1α expression is induced by a variety of upstream cellular signaling pathways, thus serving as a transducer of cellular energy stores and physiological stimuli that demand increased energy requirements such as exercise, cold exposure, and fasting (Kelly and Scarpulla 2004; Lin et al. 2005; Finck and Kelly 2006; Handschin and Spiegelman 2006). Thus, PGC-1α orchestrates the activity of multiple transcription factors in response to physiological stimuli to control mitochondrial functional capacity (Fig. 1). PGC-1α is a member of a family that includes PGC-1α (Lin et al. 2002) and PGC-1-related coactivator (PRC; Andersson and Scarpulla 2001; Kressler et al. 2002; Puigserver and Spiegelman 2003). The PGC-1β molecule has significant homology with PGC-1α and, as described below, shares many if not all of its functions in striated muscle (Kelly and Scarpulla 2004; Lai et al. 2008; Zechner et al. 2010).
Figure 1.
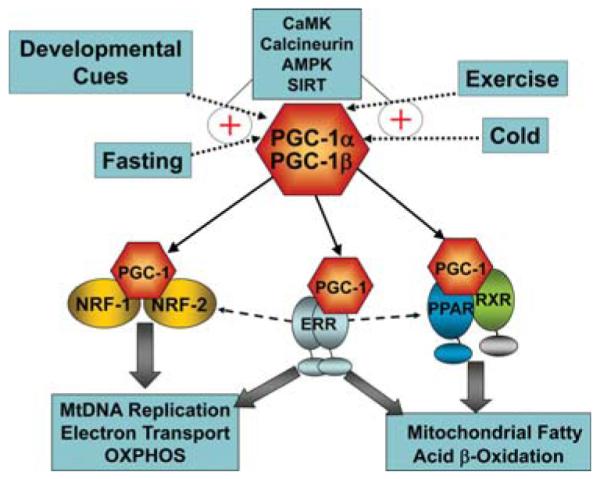
The PGC-1 gene regulatory circuit. PGC-1, PPARγ coactivator-1; NRF, nuclear respiratory factor; ERR, estrogen-related receptor; PPAR, peroxisome proliferator-activated receptor; OXPHOS, oxidative phosphorylation; RXR, retinoid X receptor.
THE ROLE OF THE PGC-1 GENE REGULATORY CASCADE IN CARDIAC METABOLIC MATURATION
Perinatal Fuel Shifts
At birth, the heart undergoes a dramatic shift in fuel preference from relying on glucose and lactate during the fetal period to the use of fatty acids (FA) oxidized in mitochondria after birth. This change in cardiac energy substrate utilization is accompanied by a surge in the expression of PGC-1α and β mRNA and downstream targets initiating a burst of mitochondrial biogenesis (Lehman et al. 2000; Lai et al. 2008). The importance of this perinatal metabolic maturation for postnatal cardiac function is best exemplified by the dramatic phenotype of mice with combined PGC-1αβ gene disruption (PGC-1αβ-/- mice) that die several days after birth from heart failure (Lai et al. 2008; Fig. 2). In addition, mice deficient for ERRγ, which is coactivated by PGC-1α (as shown in Fig. 1), fail to switch to using FA as a fuel source, leading to an early postnatal death (Alaynick et al. 2007).
Figure 2.

Lethal early postnatal death due to heart failure in PGC-1αβ-deficient hearts. (A) High-resolution echocardiography was performed within a few hours after birth. Bars represent left ventricular (LV) performance (cardiac output) among the four genotypes measured in microliters per minute; *, p ≤ 0.5 compared with other groups. (B) Mortality curve depicting the percent of survival of male and female PGC-1α-/- (diamonds, n = 55) and PGC-1αβ-deficient (PGC-1αβ-/-; squares, n = 31) pups 28 days after birth. (Figure modified from Lai et al. 2008.)
Mitochondrial Biogenesis
Early studies in cultured cardiac myocytes and in cardiac-specific transgenic mice demonstrated that forced expression of PGC-1α is capable of activating virtually all aspects of mitochondrial biogenesis in heart, including increased expression of OXPHOS and FAO genes, increased mitochondrial number, and augmentation of mitochondrial respiratory capacity (Lehman et al. 2000). Chronic transgenic overexpression of PGC-1α in mice triggers uncontrolled cardiac mitochondrial proliferation, leading to a loss of sarcomeric structure and a fatal cardiomyopathy at an early age. To assess a more physiologically relevant induction pattern of PGC-1α in heart, a tissue-specific, doxycycline-inducible transgenic mouse expression system was developed (Russell et al. 2004). Induction of PGC-1α expression beginning immediately after birth resulted in a dramatic increase in number and size of mitochondria. However, cardiac overexpression of PGC-1α in the adult resulted in a minimal increase in mitochondrial number and mitochondrial ultrastructural derangements (Russell et al. 2004). Taken together, these results indicate that PGC-1α is sufficient to drive mitochondrial biogenesis in the postnatal heart. However, it appears that the adult heart may not always be permissive for this program.
Complimentary loss-of-function studies in mice have confirmed the necessity of PGC-1α and the related coactivator, PGC-1β, for perinatal mitochondrial biogenesis and cardiac function (Lai et al. 2008). Interestingly, mice with targeted ablation of the genes encoding either PGC-1α or PGC-1β have a minimal phenotype until faced with a physiological stress that increases demands on the mitochondrion for fuel utilization and ATP production (Lin et al. 2004; Leone et al. 2005; Lelliott et al. 2006; Sonoda et al. 2007). For example, PGC-1α-/- mice are unable to exercise rigorously on a treadmill compared with wild-type counterparts (Leone et al. 2005), and fasting leads to hepatic steatosis (Leone et al. 2005). The mild phenotypes of the single PGC-1α and PGC-1β mouse “knockout” lines strongly suggested that the two related transcriptional coactivators serve redundant functions, at least in tissues with high mitochondrial content such as heart and skeletal muscle. To formally test this hypothesis, mice that were doubly deficient in both PGC-1α and PGC-1β were generated. As predicted, the phenotype of PGC-1αβ-/- mice was dramatic; they died early after birth of severe heart failure (Fig. 2B; Lai et al. 2008). Electron microscopy studies of PGC-1α/β null heart sections over a time period ranging from the late fetal stages into the first 48 h after birth revealed that the normal perinatal mitochondrial biogenic response was completely absent in the PGC-1α/β-deficient mice in heart (Fig. 3; Lai et al. 2008). Mitochondrial ultrastructural abnormalities were also noted in the PGC-1α/β-deficient heart (Fig. 3). These results demonstrated that the PGC-1 gene regulatory circuit is necessary for the surge of perinatal mitochondrial biogenesis in heart and for postnatal heart function and survival.
Figure 3.

Perinatal mitochondrial biogenesis is blocked in PGC-1αβ hearts. Representative electron micrographs of cardiac muscle sections (left ventricular free wall) at day 1 after birth. Arrows denote vacuoles within the mitochondria. Quantitative measurements of the cellular volume density for the mitochondrial fractions based on analysis of electron micrographs reveal that the perinatal increase in mitochondrial density is completely blocked in the PGC-1αβ-/- mice. In the graph, bars represent mean ± S.E.M.; *, p ≤ 0.5 compared with other groups. (Figure modified from Lai et al. 2008.)
FUEL METABOLIC AND ENERGETIC DERANGEMENTS IN MYOCARDIAL DISEASES
Fuel Switches in the Hypertrophied and Failing Heart
Numerous studies have demonstrated that myocardial fuel utilization patterns are reprogrammed in the hypertrophied and failing heart (Taegtmeyer et al. 2004). The direction of the “fuel shifts” varies with the etiology and severity of ventricular dysfunction. Pathologic cardiac hypertrophy and heart failure caused by chronic pressure overload, such as occurs with hypertension, results in the myocardium shifting away from mitochondrial FAO, relying predominantly on glucose as the chief energy substrate (van Bilsen et al. 2004). This metabolic reprogramming is commonly referred to as a “fetal” shift owing to the fact that, as described above, during fetal development, myocardial ATP is mainly generated largely by glycolysis (Neely et al. 1972). Consistent with these observations in animal models, cardiac positron emission tomography studies in humans have shown a shift away from FA utilization with hypertensive cardiac hypertrophy (Davila-Roman et al. 2002; de las Fuentes et al. 2003). Conversely, the diabetic myocardium relies largely and, at times exclusively, on FA as the main energy substrate, with the contribution of glucose to ATP production being minimal (Paulson and Crass 1982; Rodrigues et al. 1995; Stanley et al. 1997; Belke et al. 2000; Carley and Severson 2005).
The central question of whether cardiac fuel metabolic shifts are “bystanders” of the underlying disease process or actually contribute to the etiology of pathologic cardiac remodeling is the focus of intense investigation. The best evidence to support a causal role for metabolic derangements in the development of cardiac dysfunction is the observation that children with genetic defects in FAO enzymes, which force the heart to rely on glucose, often develop cardiomyopathy (Kelly and Strauss 1994). However, partial shifts away from mitochondrial FAO may serve an early compensatory role to maintain contractile function while minimizing oxygen consumption costs.
Mitochondrial Derangements
The adult heart undergoes distinct forms of hypertrophic growth in response to physiological and pathophysiological loading. Discrete structural, metabolic, and molecular signatures distinguish pathologic from physiologic forms of cardiac hypertrophic growth (Iemitsu et al. 2001). Whereas pathologic hypertrophy is an independent risk factor for future heart failure and death (Vakili et al. 2001), physiologic hypertrophy may be cardio-protective. Physiologic hypertrophy, as occurs during postnatal development or with endurance training (“runner’s heart”), enhances functional capacity through coordinated increases in cardiac mass and oxygen consumption, thereby matching mitochondrial respiratory capacity with energetic demand (Strom et al. 2005). Pathophysiologic stressors, such as chronic pressure overload or following an ischemic insult, induce pathologic hypertrophic growth accompanied by dissociation of increased cardiac mass and workload from available energy reserves (Anversa et al. 1978; Kayar and Weiss 1992). Initial metabolic and structural growth responses during pathologic hypertrophic growth are likely to be adaptive. Over time, however, ATP synthetic capacity becomes diminished in the pathologically hypertrophied heart. Magnetic resonance spectroscopy has shown reduced “high-energy” phosphate stores (phosphocreatine or PCr) in pressure-overload-induced ventricular hypertrophy in animal models and in humans that progressively declines during the transition to heart failure (Ingwall et al. 1985; de Roos et al. 1992; Tian et al. 1996; Ingwall and Weiss 2004). Creatine-kinase-mediated flux between PCr and ATP is reduced in failing human hearts (Weiss et al. 2005). The [PCr]/[ATP] ratio correlates with heart failure severity and is a strong predictor of cardiovascular mortality (Neubauer et al. 1997; Neubauer 2007). Such observations support the notion that, regardless of primary etiology, functional deterioration of the hypertrophied heart probably has an energetic basis.
Genetic evidence for a link between mitochondrial dysfunction and heart failure is also compelling. Mutations in both nuclear- and mitochondrial-encoded genes account for heritable respiratory chain defects that cause cardiomyopathy in humans (Wallace 1999; DiMauro and Schon 2001; Larsson and Oldfors 2001; Kirby et al. 2004). Respiratory chain defects typically present as multisystem dysfunction, disproportionately affecting organs with high ATP demand, such as heart, skeletal muscle, and the central nervous system. Cardiomyopathy may develop during childhood or at later ages. Mouse models of mitochondrial dysfunction have also provided important information. Tfam is a nuclear-encoded mitochondrial transcription factor necessary for mitochondrial biogenesis and gene expression. Cardiac-specific Tfam-/- mice exhibit reduced respiratory capacity and mitochondrial DNA in the heart before birth, coincident with a high neonatal mortality (Li et al. 2000). Surviving animals develop compensated cardiac hypertrophy, which progresses to dilated cardiomyopathy, leading to death by 4 months of age (Hansson et al. 2004). Collectively, these observations have led to the hypothesis that the cardiac remodeling en route to heart failure is driven, in part, by a relative mitochondrial insufficiency.
Derangements of the PGC-1 Gene Regulatory Circuit in the Hypertrophied and Failing Heart
The alterations in mitochondrial function and fuel metabolic abnormalities that occur in the hypertrophied and failing heart are due, at least in part, to dysregulation of the PGC-1 regulatory circuit. The expression of PGC-1α and its downstream targets, ERR and PPAR, has been shown to be down-regulated in animal models of pressure-overload-induced cardiac hypertrophy and heart failure and in humans (Sack et al. 1996; Lehman and Kelly 2002; Sihag et al. 2009). The down-regulation of PPARα and its downstream target genes occurs early during pathologic hypertrophic growth, indicating that this is likely to be a fundamental event in the fuel shifts that are known to accompany cardiac hypertrophy (Sack et al. 1997).
ETIOLOGY-SPECIFIC METABOLIC DERANGEMENTS IN HEART FAILURE: LESSONS LEARNED FROM TARGETING THE PGC-1 CASCADE IN MICE
Hypertensive Heart Disease
Long-standing hypertension (pressure overload) is an important cause of heart failure in humans. Several lines of evidence, based on modeling these diseases in wild-type and genetically modified mice, suggest that the deactivation of components of the PGC-1 gene regulatory network contributes to heart failure. For example, mice with targeted deletion of the gene encoding ERRα, a key PGC-1α target, develop heart failure in association with PCr depletion and reduced maximal rates of ATP synthesis following an ischemia-reperfusion insult—metabolic signatures of human heart failure (Huss et al. 2007). Transverse aortic constriction, which models the chronic pressure overload of hypertension, caused robust heart failure in both PGC-1α-/- and PGC-1β-/- but not in wild-type mice (Arany et al. 2006; Lu et al. 2010; Riehle et al. 2011). Interestingly, cardiac-specific overexpression of Cdk9 in mice, which results in decreased PGC-1α expression, also leads to severe heart failure in mice associated with myocyte death (Sano et al. 2004). Taken together, these recent findings suggest that, in the context of a mechanical stress such as chronic pressure overload, chronically reduced levels of PGC-1α predispose the heart to pathological remodeling related to energy starvation caused by mitochondrial dysfunction. It is likely that mitochondrial dysfunction in this context also predisposes to the generation of high levels of reactive oxygen species and cell death. The established link between reduced PGC-1α expression and pathologic cardiac remodeling makes the PGC-1 cascade an appealing therapeutic target for heart failure due to chronic hypertension (and other diseases).
Heart Disease Caused by Obesity and Diabetes
We are witnessing a virtual pandemic of obesity that is driving a dramatic increase in type 2 diabetes. The leading cause of death in the diabetic population is cardiovascular disease, including a unique form of heart failure. The metabolic derangements that characterize cardiac dysfunction occurring in the obese and diabetic population probably involve derangements in the PGC-1 transcriptional regulatory network, but in ways that is distinct from the early events driving pathologic cardiac remodeling owing to hypertension. First, it is important to note that, in contrast to the “fetal” switch in fuel utilization that occurs in response to chronic pressure overload, the insulin-resistant and type 2 diabetic heart has reduced capacity for glucose utilization and increased reliance on FA, a substrate that is catabolized in the mitochondrial FAO pathway. Whereas the normal heart is equipped for high-capacity FAO, the loss of flexibility to switch back and forth between glucose and fats, together with the extraordinary high rates of delivery of FA to the heart, leads to a scenario referred to as “lipotoxic cardiomyopathy.” Lipotoxic cardiomyopathy manifests when high rates of FAO are associated with accumulation of neutral lipid in the myocyte, predominately in the form of triglyceride in lipid droplets. The mechanistic basis for the development of heart failure relevant to the accumulation of lipid in high rates of FAO is not clear. However, as described below, accumulation of triglyceride is a key signature in the vast majority of mouse models of lipotoxic cardiomyopathy (Chiu et al. 2001; Finck et al. 2002, 2003; Yagyu et al. 2003; Haemmerle et al. 2006).
Early studies focused on animal models revealed that reprogramming of the heart to oxidize and store lipid in the insulin-resistant and diabetic state involved chronic activation of the nuclear receptor PPARα, which, as described above, drives almost every step of FA uptake and oxidation. Interestingly, activation of PPARα in the lipotoxic heart is directionally opposite of the dysregulation that occurs in response to chronic pressure overload and provides a signature to distinguish these types of heart failure based on etiology and metabolic signatures. Mice with cardiac-specific overexpression of PPARα (MHC-PPARα mice) were developed to model the metabolic derangements known to occur in the diabetic heart (Finck et al. 2002, 2003). MHC-PPARα, mice exhibit many, if not all, of the signatures of the diabetic heart, including high rates of FAO, low rates of glucose oxidation, triglyceride accumulation in cardiac myocytes, and the development of a cardiomyopathy that is worsened by a high-fat diet. The importance of the lipid accumulation signature in signifying the development of cardiomyopathy was demonstrated by the rescue of MHC-PPARα mice when bred into a CD36-null background (Yang et al. 2007). CD36 is one of the major FA transporters for the cardiac myocyte, and its deficiency blocked the FA overload of the diabetic and MHC-PPARα heart. Similarly, MHC-PPARα mice bred into mice that lack lipoprotein lipase (LPL) in heart were also rescued, presumably because most of the extra FA that accumulates in the lipotoxic heart is derived from the VLDL particle (Duncan et al. 2010).
How does chronic activation of PPARα, and corresponding lipid metabolic derangements, lead to cardiac dysfunction? Although this question has not been completely resolved, several lines of evidence suggest that this ultimately relates to reduced mitochondrial function owing, in part, to the loss of PGC-1-mediated control. Studies conducted in insulin-resistant and diabetic mice have demonstrated that in the early stages of insulin resistance, the activity of both PGC-1α and PPARα is high—an adaptive response of the heart to increased FA delivery, increasing the capacity for mitochondrial FAO (Duncan et al. 2007). However, it is believed that in the later stages of this process, when diabetes sets in, PGC-1α levels fall and mitochondrial dysfunction ensues, driving the development of heart failure in a final common pathway that is not dissimilar to that described for hypertensive cardiomyopathy (Fig. 4).
Figure 4.
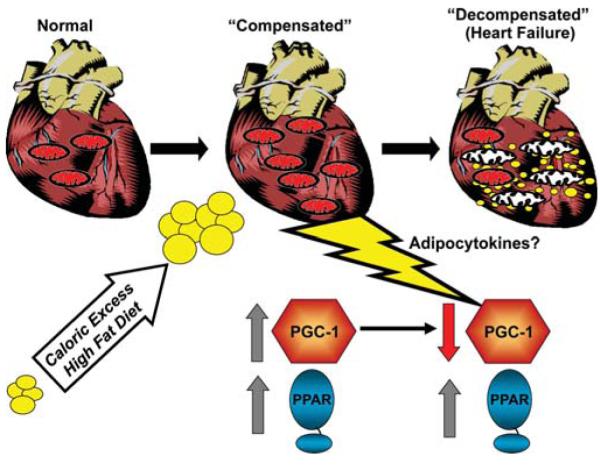
Mechanistic model for the development of lipotoxic cardiomyopathy. Caloric excess outstrips the capacity for adipose storage, increasing delivery of circulating FA to the heart, which leads to an adaptive induction of PPARα and PGC-1α and an increase in capacity for fat oxidation and mitochondrial biogenesis. Over time, PGC-1α levels fall and mitochondrial dysfunction ensues, leading to a vicious cycle of myocyte lipid accumulation and mitochondrial dysfunction.
A key question for this mechanistic model relates to the mechanism whereby PGC-1α levels fall. Recent evidence suggests that this late-stage fall in PGC-1α levels may be due to the effects of innate immune signaling. It is well known that, in the obese state, adipose tissue elaborates a number of cytokines referred to as “adipocytokines.” Some of these factors activate the cellular innate immune response through the Toll-like receptor 4 (TLR4). We hypothesized that activation of the innate immune system of cardiac myocytes could alter mitochondrial function and PGC-1α. To this end, lipopolysaccharide (LPS), a potent activator of TLR4, was administered to mice (Schilling et al. 2011). As expected, the wild-type mice developed reduced cardiac function. Interestingly, levels of both PGC-1α and PGC-1β were markedly diminished in response to LPS in vivo and in cardiac myocytes in culture. In addition, LPS increased myocardial triglyceride levels. The mechanism whereby LPS reduces PGC-1 gene expression was shown to be NF-κB dependent, at least in the cardiac myocytes. Taken together, these results suggest that the lipotoxic cardiomyopathy that develops in states of obesity and diabetes relates, in part, to a fall in the coordinated activation of PGC-1 and PPARα, as shown in Figure 4. Whereas PPARα activation persists, the fall of PGC-1 levels, possibly in response to inflammatory signals, results in an inability to maintain high mitochondrial respiratory capacity in the context of continued FA uptake, leading to a vicious cycle of further accumulation of lipid and mitochondrial dysfunction.
SUMMARY
In summary, the pathologic cardiac remodeling that is known to occur in common forms of heart failure probably has a significant metabolic component. Interestingly, the corresponding derangements in fuel metabolism signify different pathogenic pathways for hypertensive (and probably ischemic) versus diabetic forms of heart failure. However, the final common pathway is likely to be mitochondrial dysfunction. Significant evidence indicates that the fuel metabolic derangements and mitochondrial defects of the hypertrophied and failing heart involve alterations in PGC-1 signaling. Given that the events driving the metabolic derangements differ based on etiology, there is great hope for the development of therapeutics that are targeted at etiology rather than a “one size fits all” treatment strategy for heart failure. An important future area of investigation relates to the identification and validation of biomarkers that will accurately define the heart failure disease phenotype, leading to metabolic modulator therapy aimed at the appropriate etiology in the very early stages.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants RO1-HL058493 and HL101189. We thank Shonna Hyde for assistance with manuscript preparation.
REFERENCES
- Alaynick WA, Kondo RP, Xie W, He W, Dufour CR, Downes M, Jonker JW, Giles W, Naviaux RK, Giguere V, Evans RM. ERRγ directs and maintains the transition to oxidative metabolism in the postnatal heart. Cell Metab. 2007;6:13–24. doi: 10.1016/j.cmet.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Andersson U, Scarpulla RC. Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol Cell Biol. 2001;21:3738–3749. doi: 10.1128/MCB.21.11.3738-3749.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anversa P, Loud AV, Giacomelli F, Wiener J. Absolute morphometric study of myocardial hypertrophy in experimental hypertension. II. Ultrastructure of myocytes and interstitium. Lab Invest. 1978;38:597–609. [PubMed] [Google Scholar]
- Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-γ coactivator 1α. Proc Natl Acad Sci. 2006;103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2000;279:E1104–E1113. doi: 10.1152/ajpendo.2000.279.5.E1104. [DOI] [PubMed] [Google Scholar]
- Carley AN, Severson DL. Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochim Biophys Acta. 2005;1734:112–126. doi: 10.1016/j.bbalip.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila-Roman VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, Gropler RJ. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2002;40:271–277. doi: 10.1016/s0735-1097(02)01967-8. [DOI] [PubMed] [Google Scholar]
- de las Fuentes L, Herrero P, Peterson LR, Kelly DP, Gropler RJ, Davila-Roman VG. Myocardial fatty acid metabolism: Independent predictor of left ventricular mass in hypertensive heart disease. Hypertension. 2003;41:83–87. doi: 10.1161/01.hyp.0000047668.48494.39. [DOI] [PubMed] [Google Scholar]
- de Roos A, Doornbos J, Luyten PR, Oosterwaal LJ, van der Wall EE, den Hollander JA. Cardiac metabolism in patients with dilated and hypertrophic cardiomyopathy: Assessment with proton-decoupled P-31 MR spectroscopy. J Magn Reson Imaging. 1992;2:711–719. doi: 10.1002/jmri.1880020616. [DOI] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. Mitochondrial DNA mutations in human disease. Am J Med Genet. 2001;106:18–26. doi: 10.1002/ajmg.1392. [DOI] [PubMed] [Google Scholar]
- Duncan JG, Fong JL, Medeiros DM, Finck BN, Kelly DP. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-α/PGC-1α gene regulatory pathway. Circulation. 2007;115:909–917. doi: 10.1161/CIRCULATIONAHA.106.662296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JG, Bharadwaj KG, Fong JL, Mitra R, Sambandam N, Courtois MR, Lavine KJ, Goldberg IJ, Kelly DP. Rescue of cardiomyopathy in peroxisome proliferator-activated receptor-α transgenic mice by deletion of lipoprotein lipase identifies sources of cardiac lipids and peroxisome proliferator-activated receptor-α activators. Circulation. 2010;121:426–435. doi: 10.1161/CIRCULATIONAHA.109.888735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Kelly DP. Peroxisome proliferator-activated receptor α (PPARα) signaling in the gene regulatory control of energy metabolism in the normal and diseased heart. J Mol Cell Cardiol. 2002;34:1249–1257. doi: 10.1006/jmcc.2002.2061. [DOI] [PubMed] [Google Scholar]
- Finck BN, Kelly DP. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac phenotype induced by PPARα overex-pression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, Gross RW, Kelly DP. A critical role for PPARα-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: Modulation by dietary fat content. Proc Natl Acad Sci. 2003;100:1226–1231. doi: 10.1073/pnas.0336724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge K, Cho YW, Guo H, Hong TB, Guermah M, Ito M, Yu H, Kalkum M, Roeder RG. Alternative mechanisms by which mediator subunit MED1/TRAP220 regulates peroxisome proliferator-activated receptor γ-stimulated adipogenesis and target gene expression. Mol Cell Biol. 2008;28:1081–1091. doi: 10.1128/MCB.00967-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312:734–737. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor γ coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27:728–735. doi: 10.1210/er.2006-0037. [DOI] [PubMed] [Google Scholar]
- Hansson A, Hance N, Dufour E, Rantanen A, Hultenby K, Clayton DA, Wibom R, Larsson NG. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc Natl Acad Sci. 2004;101:3136–3141. doi: 10.1073/pnas.0308710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huss JM, Torra IP, Staels B, Giguere V, Kelly DP. Estrogen-related receptor α directs peroxisome proliferator-activated receptor α signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol Cell Biol. 2004;24:9079–9091. doi: 10.1128/MCB.24.20.9079-9091.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, Giguere V, Murphy E, Kelly DP. The nuclear receptor ERRα is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007;6:25–37. doi: 10.1016/j.cmet.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Iemitsu M, Miyauchi T, Maeda S, Sakai S, Kobayashi T, Fujii N, Miyazaki H, Matsuda M, Yamaguchi I. Physiological and pathological cardiac hypertrophy induce different molecular phenotypes in the rat. Am J Physiol Regul Integr Comp Physiol. 2001;281:R2029–R2036. doi: 10.1152/ajpregu.2001.281.6.R2029. [DOI] [PubMed] [Google Scholar]
- Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circul Res. 2004;95:135–145. doi: 10.1161/01.RES.0000137170.41939.d9. [DOI] [PubMed] [Google Scholar]
- Ingwall JS, Kramer MF, Fifer MA, Lorell BH, Shemin R, Grossman W, Allen PD. The creatine kinase system in normal and diseased human myocardium. New Engl J Med. 1985;313:1050–1054. doi: 10.1056/NEJM198510243131704. [DOI] [PubMed] [Google Scholar]
- Kayar SR, Weiss HR. Diffusion distances, total capillary length and mitochondrial volume in pressure-overload myocardial hypertrophy. J Mol Cell Cardiol. 1992;24:1155–1166. doi: 10.1016/0022-2828(92)93179-n. [DOI] [PubMed] [Google Scholar]
- Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18:357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- Kelly DP, Strauss AW. Inherited cardiomyopathies. New Engl J Med. 1994;330:913–919. doi: 10.1056/NEJM199403313301308. [DOI] [PubMed] [Google Scholar]
- Kirby DM, Salemi R, Sugiana C, Ohtake A, Parry L, Bell KM, Kirk EP, Boneh A, Taylor RW, Dahl HH, et al. NDUFS6 mutations are a novel cause of lethal neonatal mitochondrial complex I deficiency. J Clin Invest. 2004;114:837–845. doi: 10.1172/JCI20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kressler D, Schreiber SN, Knutti D, Kralli A. The PGC-1-related protein PERC is a selective coactivator of estrogen receptor α. J Biol Chem. 2002;277:13918–13925. doi: 10.1074/jbc.M201134200. [DOI] [PubMed] [Google Scholar]
- Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A, Kelly DP. Transcriptional coactivators PGC-1α and PGC-lβ control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22:1948–1961. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson NG, Oldfors A. Mitochondrial myopathies. Acta Physiol Scand. 2001;171:385–393. doi: 10.1046/j.1365-201x.2001.00842.x. [DOI] [PubMed] [Google Scholar]
- Lehman JJ, Kelly DP. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin Exp Pharmacol Physiol. 2002;29:339–345. doi: 10.1046/j.1440-1681.2002.03655.x. [DOI] [PubMed] [Google Scholar]
- Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor γ coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelliott CJ, Medina-Gomez G, Petrovic N, Kis A, Feldmann HM, Bjursell M, Parker N, Curtis K, Campbell M, Hu P, et al. Ablation of PGC-1β results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol. 2006;4:e369. doi: 10.1371/journal.pbio.0040369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, Courtois M, Wozniak DF, Sambandam N, Bernal-Mizrachi C, et al. PGC-1α deficiency causes multisystem energy metabolic derangements: Muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005;3:e101. doi: 10.1371/journal.pbio.0030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang J, Wilhelmsson H, Hansson A, Thoren P, Duffy J, Rustin P, Larsson NG. Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proc Natl Acad Sci. 2000;97:3467–3472. doi: 10.1073/pnas.97.7.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor γ coactivator 1β (PGC-1β), a novel PGC-1-related transcription coactivator associated with host cell factor. J Biol Chem. 2002;277:1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]
- Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Lu Z, Xu X, Hu X, Fassett J, Zhu G, Tao Y, Li J, Huang Y, Zhang P, Zhao B, Chen Y. PGC-1 α regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid Redox Signal. 2010;13:1011–1022. doi: 10.1089/ars.2009.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely JR, Rovetto MJ, Oram JF. Myocardial utilization of carbohydrate and lipids. Prog Cardiovasc Dis. 1972;15:289–329. doi: 10.1016/0033-0620(72)90029-1. [DOI] [PubMed] [Google Scholar]
- Neubauer S. The failing heart—An engine out of fuel. New Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, Pabst T, Ertl G, Hahn D, Ingwall JS, Kochsiek K. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 1997;96:2190–2196. doi: 10.1161/01.cir.96.7.2190. [DOI] [PubMed] [Google Scholar]
- Paulson DJ, Crass MF., 3rd Endogenous triacylglycerol metabolism in diabetic heart. Am J Physiol. 1982;242:H1084–H1094. doi: 10.1152/ajpheart.1982.242.6.H1084. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-Γ coactivator 1 α (PGC-1 α): Transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Riehle C, Wende AR, Zaha VG, Pires KM, Wayment B, Olsen C, Bugger H, Buchanan J, Wang X, Moreira AB, et al. PGC-1Β deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circul Res. 2011 doi: 10.1161/CIRCRESAHA.111.243964. doi:101161/CircResaha.III.243964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues B, Cam MC, McNeill JH. Myocardial substrate metabolism: Implications for diabetic cardiomyopathy. J Mol Cell Cardiol. 1995;27:169–179. doi: 10.1016/s0022-2828(08)80016-8. [DOI] [PubMed] [Google Scholar]
- Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, Medeiros DM, Valencik ML, McDonald JA, Kelly DP. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor γ coactivator-1α promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circul Res. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- Sack MN, Rader TA, Park S, Bastin J, McCune SA, Kelly DP. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation. 1996;94:2837–2842. doi: 10.1161/01.cir.94.11.2837. [DOI] [PubMed] [Google Scholar]
- Sack MN, Disch DL, Rockman HA, Kelly DP. A role for Sp and nuclear receptor transcription factors in a cardiac hypertrophic growth program. Proc Natl Acad Sci. 1997;94:6438–6443. doi: 10.1073/pnas.94.12.6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Wang SC, Shirai M, Scaglia F, Xie M, Sakai S, Tanaka T, Kulkarni PA, Barger PM, Youker KA, et al. Activation of cardiac Cdk9 represses PGC-1 and confers a predisposition to heart failure. EMBO J. 2004;23:3559–3569. doi: 10.1038/sj.emboj.7600351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling J, Lai L, Sambandam N, Dey CE, Leone TC, Kelly DP. Toll-like receptor-mediated inflammatory signaling reprograms cardiac energy metabolism by repressing peroxisome proliferator-activated receptor {γ} coactivator-1 signaling. Circul Heart Fail. 2011;4:474–482. doi: 10.1161/CIRCHEARTFAILURE.110.959833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sihag S, Cresci S, Li AY, Sucharov CC, Lehman JJ. PGC-1α and ERRα target gene downregulation is a signature of the failing human heart. J Mol Cell Cardiol. 2009;46:201–212. doi: 10.1016/j.yjmcc.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda J, Mehl IR, Chong LW, Nofsinger RR, Evans RM. PGC-1β controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc Natl Acad Sci. 2007;104:5223–5228. doi: 10.1073/pnas.0611623104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley WC, Lopaschuk GD, McCormack JG. Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc Res. 1997;34:25–33. doi: 10.1016/s0008-6363(97)00047-3. [DOI] [PubMed] [Google Scholar]
- Strom CC, Aplin M, Ploug T, Christoffersen TE, Langfort J, Viese M, Galbo H, Haunso S, Sheikh SP. Expression profiling reveals differences in metabolic gene expression between exercise-induced cardiac effects and maladaptive cardiac hypertrophy. FEBS J. 2005;272:2684–2695. doi: 10.1111/j.1742-4658.2005.04684.x. [DOI] [PubMed] [Google Scholar]
- Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M. Linking gene expression to function: Metabolic flexibility in the normal and diseased heart. Ann N Y Acad Sci. 2004;1015:202–213. doi: 10.1196/annals.1302.017. [DOI] [PubMed] [Google Scholar]
- Tian R, Nascimben L, Kaddurah-Daouk R, Ingwall JS. Depletion of energy reserve via the creatine kinase reaction during the evolution of heart failure in cardiomyopathic hamsters. J Mol Cell Cardiol. 1996;28:755–765. doi: 10.1006/jmcc.1996.0070. [DOI] [PubMed] [Google Scholar]
- Vakili BA, Okin PM, Devereux RB. Prognostic implications of left ventricular hypertrophy. Am Heart J. 2001;141:334–341. doi: 10.1067/mhj.2001.113218. [DOI] [PubMed] [Google Scholar]
- van Bilsen M, Smeets PJ, Gilde AJ, van der Vusse GJ. Metabolic remodelling of the failing heart: The cardiac burnout syndrome? Cardiovasc Res. 2004;61:218–226. doi: 10.1016/j.cardiores.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol Cell Biol. 2000;20:1868–1876. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- Weiss RG, Gerstenblith G, Bottomley PA. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci. 2005;102:808–813. doi: 10.1073/pnas.0408962102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y, Seo T, Hu Y, Lutz EP, Merkel M, et al. Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest. 2003;111:419–426. doi: 10.1172/JCI16751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN, Kelly DP. CD36 deficiency rescues lipotoxic cardiomyopathy. Circul Res. 2007;100:1208–1217. doi: 10.1161/01.RES.0000264104.25265.b6. [DOI] [PubMed] [Google Scholar]
- Zechner C, Lai L, Zechner JF, Geng T, Yan Z, Rumsey JW, Collia D, Chen Z, Wozniak DF, Leone TC, Kelly DP. Total skeletal muscle PGC-1 deficiency uncouples mitochondrial derangements from fiber type determination and insulin sensitivity. Cell Metab. 2010;12:633–642. doi: 10.1016/j.cmet.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]