

SUMMARY
Balanced chromosomal abnormalities (BCAs) represent a reservoir of single gene disruptions in neurodevelopmental disorders (NDD). We sequenced BCAs in autism and related NDDs, revealing disruption of 33 loci in four general categories: 1) genes associated with abnormal neurodevelopment (e.g., AUTS2, FOXP1, CDKL5), 2) single gene contributors to microdeletion syndromes (MBD5, SATB2, EHMT1, SNURF-SNRPN), 3) novel risk loci (e.g., CHD8, KIRREL3, ZNF507), and 4) genes associated with later onset psychiatric disorders (e.g., TCF4, ZNF804A, PDE10A, GRIN2B, ANK3). We also discovered profoundly increased burden of copy number variants among 19,556 neurodevelopmental cases compared to 13,991 controls (p = 2.07×10−47) and enrichment of polygenic risk alleles from autism and schizophrenia genome-wide association studies (p = 0.0018 and 0.0009, respectively). Our findings suggest a polygenic risk model of autism incorporating loci of strong effect and indicate that some neurodevelopmental genes are sensitive to perturbation by multiple mutational mechanisms, leading to variable phenotypic outcomes that manifest at different life stages.
INTRODUCTION
Mounting evidence indicates that genomic structural variants (SV) collectively play a substantial role in susceptibility to autism spectrum disorders (ASD) and other neurodevelopmental disorders (NDD). However, the near ubiquitous use of chromosomal microarrays in research and clinical diagnostics has limited the assessment of those classes of SV that do not involve large gains and losses of genetic material, such as inversions, excision/insertions, and translocations, which together constitute balanced chromosomal abnormalities (BCAs). These events are typically defined clinically at karyotypic resolution, implicating only broad chromosomal regions, without the gene-level sequence specificity that would permit informative interpretation. Even in the research setting, there is often a failure to consider or to assess BCAs, consequently bypassing a meaningful proportion of subjects with genetic events that may mark a single locus of potentially large effect size. These balanced events offer a unique route, complementary to conventional approaches, for identifying individual genes or functional sequences that contribute to otherwise genetically complex human disorders. At cytogenetic resolution, the estimated frequency of BCAs in ASD is 1.3% (Marshall et al., 2008), an approximately six-fold increase over that observed in more than 10,000 reproductively normal controls (Ravel et al., 2006). This ratio is almost certainly a lower bound for relative risk, given the resolution of available techniques and the inability to survey submicroscopic balanced alterations. Thus, BCAs clearly have a meaningful impact in ASD and represent a fertile area for high resolution study to identify functional sequences that contribute to human neurodevelopment.
We previously described innovations in the molecular approach to massively parallel sequencing and tailored bioinformatics applicable to the rapid, high resolution discovery of chromosomal rearrangement breakpoints (Talkowski et al., 2011a). These and conceptually similar methods were recently used to sequence B lymphocytes to derive potential mechanisms of rearrangement (Chiarle et al., 2011; Klein et al., 2011), as they have been previously to delineate complex chromosomal rearrangements and chromothripsis in cancer cells (Stephens et al., 2011), as well as balanced chromothripsis and predominant non-homologous repair in the human germline and transgenic animals (Chiang et al., 2012). Here, we precisely characterize to nucleotide resolution karyotypically-defined human constitutional chromosomal rearrangements: 36 de novo BCAs and two inherited rearrangements that were transmitted from an affected parent. The results of our sequencing analyses, coupled with extensive secondary orthogonal genomics support, indicate that disruption of genes from a wide range of biological pathways can contribute to ASD, but that in many instances the same genes also confer risk, sometimes via different mutational mechanisms, to other NDDs or to a range of psychiatric disorders in both children and adults.
RESULTS
Identification of genes contributing to neurodevelopmental abnormalities
Using a series of previously developed next-generation sequencing techniques ranging from high-depth whole-genome sequencing to a targeted capture of breakpoints approach (Talkowski et al., 2011a), we delineated BCAs in 38 subjects with neurodevelopmental abnormalities, including two monozygotic twin pairs (36 independent probands). All harbored a BCA that appeared balanced at karyotypic resolution and was interpreted as pathogenic by the clinical geneticist; 36 aberrations arose de novo while two alterations were transmitted from an affected parent and thus segregated with the phenotype. Extensive clinical data were collected for all subjects and affected parents, as described in the Supplemental Information (SI). If a structured diagnostic interview was performed, or a patient was formally diagnosed with autism or an ASD by the referring clinician using DSM-IV criteria, they are referred to herein as ASD (50% of subjects); otherwise the disorder was classified as NDD, although many such subjects also displayed clinical features consistent with ASD. Complete clinical information for each subject and each BCA breakpoint, confirmed by PCR and capillary sequencing, are provided in SI. Previously performed genetic testing was also obtained and all results were unremarkable with 244,000 or one million feature aCGH unless otherwise described in the SI.
This BCA sequencing approach uncovered genes that conformed to four general classifications, with meaningful overlap between categories: (1) genes implicated previously in ASD or NDD and confirmed here by their heterozygous inactivation, (2) genes discovered to be single locus contributors to previously recognized microdeletion syndromes, (3) completely novel genes not previously implicated individually in ASD or NDD, and (4) genes that had been associated with adolescent and adult onset psychiatric disorders by common variant genome-wide association studies (GWAS) or other approaches, all but three of which represent novel ASD or NDD loci. Alterations in gene expression were also assessed for all subjects where a lymphoblastoid cell line could be obtained (33 of 38 subjects; Fig. S1). If a gene was not directly disrupted, positional effects on expression were evaluated for genes in proximity to the breakpoint. All genes and sequences disrupted by BCAs are presented in Table S1, along with results of mRNA expression studies, while the subset of genes supported by secondary analyses is presented below and in Table 1.
Table 1.
Genes disrupted by chromosomal rearrangements*
| Cat | ID | Dx | ChrA | ChrB | Disrupted | Fisher’s Exact P‡ | Function |
|---|---|---|---|---|---|---|---|
| 1 | DGAP201 | ASD | 7q11.22 | 7q36.3 | AUTS2 | 5.6E-04 | Unknown |
| 1&4 | NDR27031 | NDD | 3q13.32 | 18q21.2 | TCF4 | 6.2E-04 | Transcription factor |
| 1 | DGAP093 | NDD | Xp22.13 | 19p13.3 | CDKL5 | 7.2E-02 | Protein kinase |
| 1 | DGAP157 | NDD | 3p13 | 10q21.2 | FOXP1 | 4.5E-02 | Transcription factor |
| 1&4 | NDR25941 | ASD | 12p13.1 | 12q21.31 | GRIN2B | 7.9E-02 | Glutamate receptor |
| 1 | DGAP189 | NDD | 11p13 | 12p12.1 | SOX5 | 8.4E-02 | Transcription factor in embryonic development |
| 2 | DGAP232 | ASD | 9p11.2 | 15q11.2 | SNURF-SNRPN | 1.1E-13 | Genomic Imprinting in Angelman – PWS region |
| 2&4 | DGAP155 | ASD | 9q34.3 | 11p11.2 | EHMT1 | 3.3E-07 | Histone Methyltransferase |
| 2 | DGAP142 | ASD | 2q23.1 | 22q13 | MBD5 | 3.1E-05 | Methylation binding |
| 2 | DGAP211 | ASD | 2q33.1 | 6q16.3 | SATB2 | 1.1E-03 | Transcriptional regulation and Chromatin remodeling |
| 3 | DGAP148 | NDD | Xp11.4 | 11q24.2 | KIRREL3 | 1.6E-04 | Cell adhesion |
| 3 | DGAP154 | NDD | Xq22 | 17p13.3 | SMG6 | 5.9E-04 | Nonsense mediated decay |
| 3 | NDR26867 | ASD | 3q25.31 | 14q11.2 | CHD8 | 2.4E-02 | Chromatin remodeling |
| 3 | DGAP125 | NDD | 7q32.1 | 19q13.11 | ZNF507 | 8.0E-02 | Zinc Finger |
| 3 | DGAP132** | NDD | 5q12.2 | 7q21.3 | PON3 | 1.5E-01 | Lactonase |
| 3 | AC02-0053 | ASD | 6q16.1 | 9q21.13 | GNA14 | 2.7E-01 | G-protein signaling |
| 3 | DGAP131 | NDD | 1p22.3 | 5q33 | ZNHIT6 | 2.7E-01 | Zinc finger protein |
| 3 | DGAP193 | ASD | 2p22.3 | 2q31.3 | SPAST | 2.7E-01 | Membrane trafficking |
| 3&4 | DGAP143 | NDD | 6q22.1 | 6q27 | PDE10A | 5.2E-03 | Phosphodiesterase |
| 3&4 | DGAP171 | NDD | 17p13.2 | 18p11.21 | C18orf1 | 3.2E-02 | Unknown |
| 3&4 | DGAP180** | NDD | 2q32 | 11q14 | ZNF804A | 4.7E-02 | Zinc finger protein |
BCA-disrupted genes individually implicated by case-control CNV burden at p < 0.10 or by a minimum of 3 CNVs in cases with none in controls are provided. See Table S1 and SI for all subjects and phenotypes and Table S2 for CNV counts on all subjects.
Fisher’s exact test p-value from comparison of CNV burden between NDD cases and controls.
Cat = disruption category, Dx = diagnosis
ASD = autism spectrum disorder
NDD = other neurodevelopmental disorders
ChrA and ChrB = sequenced chromosomal sub-band containing the BCA
BCA inherited from similarly affected parent.
Convergent data from molecular diagnostics
Although the presence of a BCA is conservatively associated with an ~6 fold increased risk of ASD, any individual BCA is rare and generally non-recurrent, precluding assessment of false discovery by replication; consequently, we sought secondary support, by analysis of copy number variants (CNVs), for the hypothesis that specific locus hemizygosity contributes to genetic risk of neurodevelopmental abnormalities. We curated and analyzed a large collection of 33,573 cases from molecular diagnostic facilities and identified that subset of cases referred for a variety of neurodevelopmental abnormalities (n = 19,556), as well as that comprising cases without a referral indication of NDD (n = 14,017). We also surveyed CNV data from 13,991 controls screened for absence of a reported developmental or psychiatric phenotype. Although almost 25% of the 19,556 cases with neurodevelopmental abnormalities were referred for testing with an indication of autism or ASD, we did not have direct access to clinical records to confirm this diagnosis so we conservatively describe these cases using the broader term NDD (see SI). Our findings indicate that genes disrupted by BCAs in ASD individuals frequently show increased CNV burden across the diagnostic cases referred for ASD as well as those referred for other neurodevelopmental abnormalities, suggesting that the genes discovered can contribute to phenotypic outcomes that are not constrained by ASD diagnostic criteria.
An example of the convergent genomic information collected in this study is depicted in Figure 1, where sequencing of monozygotic twins with extensive clinical information revealed a translocation disrupting TCF4, a gene previously implicated in both Pitt-Hopkins syndrome and schizophrenia, with concomitantly decreased mRNA expression in both subjects and a significant impact of CNV burden across the clinical diagnostic samples (14 case CNVs, 0 control CNVs; p = 0.0006). Additional follow-up analyses revealed a significant GWAS signal from common SNPs at TCF4 in both autism and schizophrenia (see below).
Figure 1. Convergent genomic evidence implicates TCF4 in neurodevelopment.

(a) A t(3;18)(q13.32;q21.2) translocation was sequenced using our custom jumping library protocol in monozygotic twin boys with multiple developmental abnormalities, directly disrupting TCF4 in intron 8, a gene previously associated with Pitt-Hopkins syndrome (PHS) by mutation analysis and with schizophrenia by GWAS. (b) Analysis of CNVs revealed disruption of coding and noncoding exons in 14 independent NDD among 19,556 NDD cases compared to no CNVs observed in 13,991 controls (p = 0.0006), implicating hemizygosity of TCF4 as a highly penetrant locus in NDD, PHS, and ASD (see also Table S2 and Rosenfeld et al., 2009b). (c) Reduced mRNA expression of TCF4 was detected in both cases from EBV-transformed lymphoblastoid cell lines compared to two gender-matched controls using two independent primers spanning exons 16–17 and exons 18–19.
Across all loci disrupted by BCAs, a dramatic increase in overall CNV burden was observed in cases compared to controls (CNV burden across 33 loci: p = 2.07×10−47, OR = 5.12, 95% CI = 3.92–6.79) and this result remained robust to subset analyses and one million random simulations to assess empirical significance (see SI for all CNV analyses and results). Comparison of the NDD cases to the 14,017 diagnostic cases referred for a primary indication other than NDD and analyzed on identical platforms also showed an increased CNV burden across these genes (p = 1.8×10−5), a result that again exceeded the significance of 1 million random simulations despite the previously established enrichment of large CNVs in the latter cases, which we replicate here. Notably, restricting CNV analyses to only those genes disrupted by a BCA in an individual with a confirmed diagnosis of ASD was also highly significant (p = 2.76×10−28; Table 1). Individually, increased CNV burden was nominally significant for 14 of the genes in this study, with three non-significant trends (p < 0.10), while five additional genes were disrupted by CNVs in three or more independent cases but never altered in controls. For the latter, the rarity of dosage alteration in cases and absence of alterations in controls limit statistical power but are consistent with a strong deleterious effect. In each category discussed further below, those genes supported by secondary CNVs are briefly mentioned and presented in Table 1, while the full list of genes disrupted by BCAs is delineated in Table S1 and discussed in the SI.
Category 1. Balanced alterations confirming loci previously implicated in neurodevelopment
The power of sequencing BCAs as a discovery tool for loci contributing to disease etiology is illustrated in our independent identification of several genes previously suggested as candidates in ASD or NDD. We found direct disruption of AUTS2 and CDKL5, two established neurodevelopmental loci (Bakkaloglu et al., 2008; Sultana et al., 2002). We also found disruption of genes implicated during completion of this study by de novo mutations from an exome-sequencing study of 20 ASD families (the fork-head transcription factor FOXP1 and the glutamate receptor GRIN2B) (O’Roak et al., 2011), or by mutations in Pitt-Hopkins syndrome (TCF4). Other genes have been implicated by CNV analysis from an autism cohort (the transcription factor SOX5 and the dystrophin regulator SNTG2) (Rosenfeld et al., 2010). Consistent with their status as previously recognized contributors to neurodevelopment, their collective CNV burden was significant (p = 7.74×10−20; OR = 3.6) (Table 2).
Table 2.
Analysis of copy number variants in independent samples
| Gene Categories | NDD Count* | Control Count* | P-Value | OR | 95% CI |
|---|---|---|---|---|---|
| All Genes | 443 | 63 | 2.1E-47 | 5.1 | 3.9–6.8 |
| Category 1 | 114 | 23 | 7.7E-20 | 3.6 | 2.3–5.9 |
| Category 2 | 169 | 12 | 1.6E-26 | 10.2 | 5.7–20.1 |
| Category 3 | 160 | 28 | 2.2E-15 | 4.1 | 2.7–6.4 |
| Category 4 | 111 | 12 | 5.1E-15 | 6.7 | 3.7–13.3 |
| Categories 1, 3, 4 | 274 | 51 | 5.2E-24 | 3.9 | 2.9–5.3 |
Counts of CNVs from 19,556 NDD cases and 13,991 controls.
All Genes = all 33 loci disrupted by BCAs and included in the CNV analyses
Categories 1, 3, 4 = analyses of all genes not localized to microdeletion syndromes known to be associated with increased CNV burden. See Table S2 for all genes in each category.
Category 2. Individual genes contributing to microdeletion syndromes
Microdeletion syndromes, which are recognized contributors to neurodevelopmental and psychiatric disorders, typically involve hemizygosity of large genomic regions where the difficulty of defining specific genes responsible for core phenotypes has been an obstacle for clinical genetics, predictive diagnostics, and the study of disease pathogenesis. Here, BCA sequencing in three ASD subjects pinpointed three individual gene contributors to microdeletion syndromes, each of which is involved in transcriptional and/or epigenetic regulation, in the 2q23.1, 2q33.1, and 9q34.3 microdeletion syndrome regions, respectively. In 2q23.1, a translocation disrupted MBD5, a member of the methyl-CpG binding domain protein family defined by a highly conserved methyl binding domain and including MeCP2, a causal locus in Rett syndrome. We recently completed an international consortium follow-up study that found 65 structural variations spanning the 2q23.1 microdeletion region in cases with syndromic features including ASD, seizures, and intellectual disability (including another translocation that disrupted MBD5), establishing MBD5 as a necessary, sufficient, and predictive locus contributing to a majority of the phenotypic features of the 2q23.1 microdeletion syndrome (Talkowski et al., 2011b). In the 2q33.1 and 9q34.3 regions, we respectively identified disruption of SATB2, a gene involved in transcriptional regulation and chromatin remodeling (Rosenfeld et al., 2009a), and of EHMT1, encoding a histone methyltransferase (Kleefstra et al., 2006). In a fourth region (15q11-13), the nested genes SNURF-SNRPN were disrupted at 15q11.2 in a subject with ASD and multiple developmental abnormalities, including sensory integration disorder, but without Angelman or Prader-Willi syndromes, both of which result from imprinting within the region. Our data argue for increased resolution and interpretation from molecular diagnostic testing of regional disorders, particularly for loci in which individual gene disruptions can yield phenotypes that are similar to or indistinguishable from that defining the syndrome. Given their localization to syndromic regions, it was not surprising that CNV analysis of genes in this category reflected a strong impact in neurodevelopment (p = 1.64×10−26; OR = 10.2) (Table 2, Fig. 3).
Figure 3. Disruption of individual genes in microdeletion syndromes regions.

Gene-specific view of sequenced BCAs and CNVs implicating individual genes involved in transcriptional or epigenetic regulation and localized to previously described microdeletion syndromes, each of which implicates a single gene by the combined BCA and CNV analyses. Haploinsufficiency of mRNA was confirmed for each gene (Figure S1). A breakpoint disrupting a two gene locus (SNURF and SNRPN) was detected in a fourth microdeletion syndrome, 15q11-13 (not shown). Red bars indicate copy loss, blue bars are copy gains, and striped bars are gene-specific alterations. Green bars represent “Other” alterations (balanced or unbalanced translocation, inversion, or complex alteration involving both deletion and duplication). The bar at the top provides the count of gains and losses that extend beyond the window and is color coded to reflect the relative proportion of each variant type. Confidence intervals provide the distance to the next probe without a detected dosage imbalance. A single RefSeq transcript is provided. Breakpoint sequence from each derivative is provided with text color coded by originating chromosome or non-templated inserted sequence in gray. A subset of the SATB2 patients included here were previously reported by Rosenfeld et al. (Rosenfeld et al., 2009), and all cases reported here for MBD5, as well as 44 additional structural variants, are reported in a follow-up consortium study (Talkowski et al., 2011b).
Category 3. Novel genes conferring risk to autism and neurodevelopment
BCA sequencing yielded 22 novel ASD/NDD candidate genes that, like the genes in Categories 1 and 2, are also likely to contribute to the neurodevelopmental phenotype in the corresponding subjects. The collective increase in CNV burden for these novel candidates was highly significant (p = 2.21×10−15, OR = 4.1) (Table 2).
The most significant individual genes were two loci localized to previously identified genomic disorder regions (KIRREL3, SMG6) and one encoding a novel DNA helicase (CHD8). Similar to the Category 2 genes, we observed dysregulation by BCA and secondary CNV support for novel genes in regions associated with classic terminal deletion disorders: KIRREL3 in 11q24.1 (Jacobsen syndrome) and SMG6 in 17p13.3 (Miller-Dieker syndrome). KIRREL3 encodes a cell adhesion molecule of the immunoglobulin family expressed in developing and adult brain of mouse and developing sensory pathways (Morikawa et al., 2007; Serizawa et al., 2006; Tamura et al., 2005). The locus was disrupted by a BCA 39.6 kb upstream of the mRNA coding region that altered both mRNA and protein levels (Table 1 and Fig. S2). Disruption of SMG6 nominates nonsense mediated decay as another novel pathway in ASD and NDD, but not necessarily in the lissencephaly phenotype commonly seen in Miller-Dieker syndrome (see SI and Fig. S3). Like many of the genes described above that are involved in transcriptional regulation, epigenetic modification, and methylation patterning, CHD8 encodes a DNA helicase that remodels chromatin structure (Thompson et al., 2008). It has never been individually linked to a human disorder but was disrupted by a BCA in a subject diagnosed with ASD, was supported by our CNV analyses, was among the loci within the minimal region of overlap from previous analyses of de novo microdeletions, is an important transcriptional repressor, and interacts with genes involved in NDD such as CHD7, a causal locus in CHARGE syndrome, thus representing a strong autism and NDD candidate locus (Batsukh et al., 2010; Nishiyama et al., 2009; Rodriguez-Paredes et al., 2009).
Additionally, in seven subjects no annotated gene was disrupted, but several expressed sequence tags (ESTs), conserved sequences, and regions with predicted regulatory effects were impacted by breakpoints (Table S1), suggesting that BCAs may also provide a novel entrée into such regions. In these subjects, we considered both loci in proximity to the breakpoint for positional effects (e.g., KIRREL3), as well as disrupted but functionally unannotated sequences themselves, such as the highly conserved 6q16.3 sequence of unknown function disrupted by a BCA in an ASD subject (denoted as “High Cons” in Table S1) and the noncoding RNA LOC401324.
Category 4. Neurodevelopmental loci associated with risk that crosses traditional diagnostic boundaries
A remarkable number of genes implicated in ASD or NDD by single gene disruption from BCAs in our study have also been recently associated with a spectrum of developmental, psychiatric, and behavioral phenotypes by other strategies, such as GWAS and mutation screening, including TCF4 (Pitt-Hopkins syndrome, intellectual disability, schizophrenia) (Amiel et al., 2007; Blake et al., 2010; Rosenfeld et al., 2009b), GRIN2B (schizophrenia, bipolar disorder, neurodevelopment) (Endele et al., 2010), EHMT1 (schizophrenia) (Kirov et al., 2012) and four novel neurodevelopmental genes that overlap with Category 3: ZNF804A (schizophrenia, psychosis, cognitive function) (O’Donovan et al., 2008; Walters et al., 2010), ANK3 (bipolar disorder, schizophrenia) (Ferreira et al., 2008; Williams et al., 2011), C18orf1 (schizophrenia) (Meerabux et al., 2009) and PDE10A (schizophrenia) (Kehler, 2011). All loci with the exception of ANK3 are supported by secondary CNV analyses (Table 1). Of these novel ASD or NDD candidates, only PDE10A has an established function, encoding a phosphodiesterase suggested as a biological candidate in schizophrenia due to its high tissue specific expression in the caudate nucleus. Specific PDE10A inhibitors provide a potential therapeutic approach to schizophrenia due to their regulation of cAMP and cGMP, thereby altering dopamine D1 and D2 receptor activity (Kehler, 2011; Lakics et al., 2010).
Genes in this category are apparently capable of contributing to pleiotropic effects ranging from early onset autism and intellectual disability to adult onset psychosis, often through different mutational mechanisms. Several were previously associated with psychiatric disorders by unbiased GWAS and/or by candidate gene studies of common variants, which are thought to reflect a more subtle effect on gene regulation than the outright inactivation caused by the BCA disruption. Perhaps the most compelling example of different mutational mechanisms is TCF4, where rare mutations are recognized as causing NDD, sometimes with a diagnosis of Pitt-Hopkins syndrome, but common variation has recently emerged as a significant risk factor for schizophrenia. Taken together, our findings support the long-hypothesized notion of a neurodevelopmental component to adult onset neuropsychiatric disorders like schizophrenia (Murray and Lewis, 1987; Owen et al., 2011; Weinberger, 1986). However, our data extend this hypothesis to suggest that differing mutational impact on the same sets of genes constitutes a significant overlap in the genetic etiology of autism, schizophrenia, psychosis, bipolar disorder, and intellectual disability, comprising at least a subset of the total genetic variance for each of these disorders. The collective CNV burden for these genes decisively supports the fundamental hypothesis that some psychiatric disease-associated genes are important in neurodevelopment (p = 5.1×10−15; OR = 6.7) (Table 2).
Polygenic risk from genome-wide association studies
Our identification of genes disrupted by BCA in ASD or NDD with an increased CNV burden among diagnostic cases with neurodevelopmental abnormalities suggests that these are relatively penetrant alterations in human development, consistent with oligogenic risk factors of modest to large effect. However, the discovery of the genes in Category 4 suggests that for some loci, an accumulation of subtle genetic effects associated with common polymorphisms could have a pleiotropic impact across a spectrum of early childhood and adult onset psychiatric disorders. To test this hypothesis, we performed gene-set enrichment analyses in autism and schizophrenia GWAS datasets (Ripke et al., 2011; Wang et al., 2009; Weiss et al., 2009) using an established method in which each linkage disequilibrium block across the genome is scored with the maximum Z-score achieved in the block (Rossin et al., 2011). Analysis of an initial autism study revealed a highly significant enrichment of risk alleles across the BCA disrupted gene-set (empirical p = 0.0018), a result that persisted in the second autism GWAS study (p = 0.068). Moreover, we discovered a significant enrichment of associated alleles from the largest schizophrenia GWAS meta-analysis to date (empirical p = 0.0009). Struck by these results, we evaluated the potential for any unforeseen confounding variables by performing identical enrichment analyses in phenotype-permuted datasets from the schizophrenia and autism studies meta-analysis (p = 0.444 and p = 0.518, respectively), in a well-powered GWAS study of Crohn’s disease (p = 0.819) (Franke et al., 2010), and from GWAS data for seven other unrelated traits (p-values ranged from p = 0.06 to p = 0.917, fitting nicely to the expected null distribution). These data indicate an unusually strong enrichment of subtle effects from common polygenic risk loci in autism and schizophrenia among the genes identified by our BCA sequencing, and further support the hypothesis that diverse mutational mechanisms at these loci can confer pleiotropic effects across conventional diagnostic classifications (Fig. 2).
Figure 2. Genes disrupted by chromosomal abnormalities confer risk across diagnostic groups.
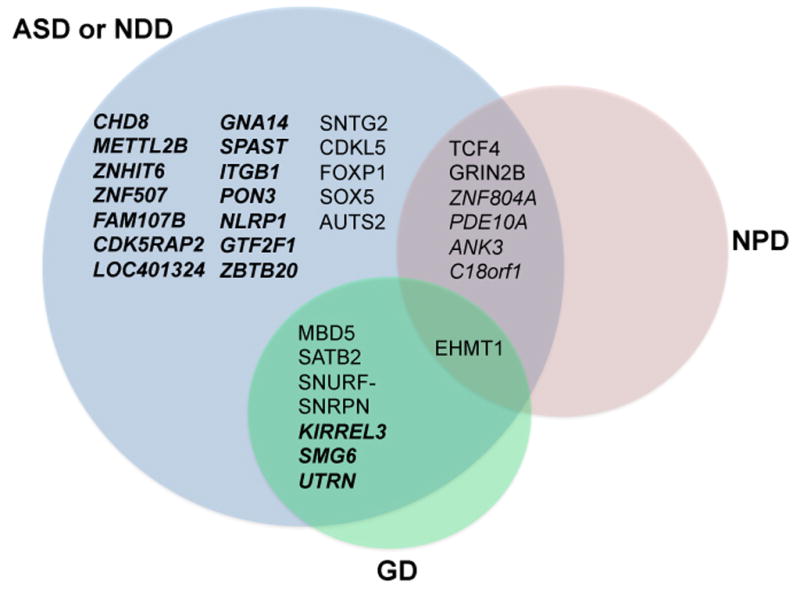
All genes disrupted by a BCA and analyzed in the CNV analyses are shown. While all genes are implicated in autism spectrum disorder (ASD) or other neurodevelopmental disorder (NDD) by BCA disruption in this study, some loci also represented single gene contributors to previously recognized genomic disorder (GD) regions (three microdeletion syndromes, two terminal deletion syndromes, and one duplication syndrome), and genes discovered in ASD or NDD in this study that had been previously linked to adolescent or adult onset neuropsychiatric disorders (NPD) by common variation association studies. Genes in italics are novel loci implicated for the first time in ASD or NDD by this study (Category 3), bold italics represents novel loci not previously implicated in any ASD, NDD, or NPD.
Genes sensitive to dosage dysregulation
The finding that some genes associated with ASD or NDD due to inactivation of one allele may also contribute to abnormal phenotypes when more subtly disrupted suggests that some ASD or NDD genes require tight control of their expression for appropriate neurodevelopmental function. In such circumstances, disruption by increased dosage might also produce an NDD phenotype. This is highlighted in more detailed examination of the CNV analyses presented in Figures 1, 3, and 4 which indicate that for some genes, the CNV data predict that both deletion and duplication are risk factors for abnormal neurodevelopment, whereas for other loci the mechanism of disruption appears to be dosage specific. For example, previously established NDD risk loci (TCF4, Fig. 1; SATB2, MBD5, Fig. 3) almost exclusively display deletion among the CNV cases, clearly supporting a similar mechanism of dysregulation to that seen for the BCA disruption. This was also true for novel NDD candidates such as PDE10A and KIRREL3. However, in Figure 4a are three instances, including two well-established loci (AUTS2, CDKL5), for which CNV analysis supported genetic risk from both deletion and duplication, approaching a near 50:50 balance. Interestingly, Figure 4b shows a group of genes where the CNV support is primarily from duplication at the locus (CHD8, GRIN2B, FOXP1), suggesting similar phenotypic outcomes from both dosage increase and heterozygous inactivation by BCA disruption, as in this study, or de novo mutations in a previous study (for GRIN2B and FOXP1) (O’Roak et al., 2011). These data build upon previous findings in recurrent rearrangement regions, such the common recurrent 16p11.2 microdeletion/microduplication (Weiss et al., 2008), where both deletion and duplication increase risk for autism to different degrees and have disparate impact (including reciprocal phenotypes) for other disorders and physiological traits. The clear distinction between neurodevelopmental loci associated primarily with deletion or duplication, and those displaying similarly abnormal neurodevelopment from either event emphasizes the need for detailed experimental annotation of the genome with respect to dosage sensitive loci and phenotypic prediction.
Figure 4. Genes sensitive to dosage dysregulation.

Provided is an example of genes illustrated by this study that appear to be sensitive to a spectrum of mutational mechanisms reciprocally altering their gene dosage. In Figures 1 and 2 are examples of genes such as TCF4 and SATB2 which are disrupted exclusively by deletions. (a) Genes sensitive to dosage dysregulation from which both deletion and duplication appear to confer risk for similar phenotypes (AUTS2, CDKL5, C18orf1). (b) Three loci, FOXP1, GRIN2B, and CHD8, are recognized for duplication of the locus as the predominant mutational mechanism, yet all three loci are implicated by heterozygous inactivation from BCA disruption in this study. Two of these genes, FOXP1 and GRIN2B are also supported by de novo mutation in ASD families (O’Roak et al., 2011). CHD8 is a completely novel candidate not previously implicated in ASD or any human abnormality. These data suggest annotation of the human genome is insufficient to reliably predict the impact of various mutational mechanisms of specific loci involved in neurodevelopment.
Disrupted genes do not converge on a single pathway in neurodevelopment
The genomics approach in this study enabled direct interpretation of locus specificity for further downstream analyses. We evaluated the networks in which these genes may participate and whether any biological pathways emerged as significantly enriched for genes disrupted by these BCAs. A qualitative network analysis based on interactions from PubMed abstracts using a natural language processing algorithm identified a network of 429 interacting genes (Fig. 5 and Fig. S4). Fourteen of the original genes, many of which are involved in transcriptional regulation, were found to interact indirectly in a large, interconnected network, and TCF4, SNRPN, CHD8, and GTF2F1 were confirmed as interacting partners of the RNA Polymerase II complex. Analysis of GO terms found enrichment of transcription factors, phosphoproteins, and protein heterodimerization activity (p < 0.005). A quantitative network assessment did not find statistically significant first or second-order interactions compared to chance expectations (given the size and composition of the given networks). The nominally significant networks (statistical enrichment of pathways at p < 0.05) included shared interactions between SNTG2, UTRN, GNA14, and CDKRAP2 (p = 0.01) as well as KCND2 and GRIN2B (p = 0.03) (Fig. S4). The SNURF-SNRPN complex was nominally significant in both analyses and in an assessment of physical interactions (SI). No results were significant after correction for multiple testing. There was also no convergence on one or a few pathways involved in neurodevelopment, likely a reflection of the modest number of genes studied and the unknown function of many of the novel genes. These results could also signify that interaction of a diverse range of functional networks at many levels is critical to normal human neurodevelopment and that there is ample opportunity for genetic lesions to disrupt different functional pathways while still leading to similar neurodevelopmental outcomes.
Figure 5. Network analysis of genes implicated in autism or neurodevelopment in this study.

A large network of genes disrupted by BCAs in this study are connected by first, second, or higher order interactions. No networks were significantly enriched for genes disrupted by BCAs after correction for multiple comparisons, though a number of loci have limited functional annotation available or remain of unknown function.
DISCUSSION
Direct sequencing of BCA breakpoints, followed by targeted assessment of molecular diagnostic CNV findings in independent subjects, proved to be an efficient strategy for individual gene discovery in abnormal neurodevelopment. As this approach begins with sequence resolution of individual BCAs, it is not subject to notable limitations of other de novo mutation studies that rely purely on CNVs or on exome sequencing (i.e., primarily identification of large multi-gene regions in the former and failure to assess most non-coding sequence in the latter) and therefore provides an effective complement to these approaches. The yield of this study is considerable, with 22 novel loci being disrupted by BCAs and supported by secondary analyses either by a statistical enrichment of CNV burden or disruption in multiple cases and absence of dosage alterations in controls, with several additional genes and sequences whose potential for contribution merits further examination. These findings not only extend specific knowledge of neurodevelopmental genes implicated by microdeletion syndromes, GWA, CNV, and exome sequencing studies, but also reveal entirely new, unsuspected genes in ASD and NDD. This work also presents direct evidence for a complex genetic architecture that connects neurodevelopmental and adult onset psychiatric disorders by implicating robustly associated schizophrenia loci as contributors to neurodevelopmental abnormalities.
Our analyses were based upon the premise that alone, a de novo BCA disruption in a single case, CNVs spanning a given region, or the presence of a de novo mutation within the coding region of an interesting biological candidate does not by itself represent compelling evidence that a gene contributes to neurodevelopment. Instead, we sought convergent genomic evidence combining disruption by a de novo BCA (or in two subjects a BCA that co-segregated with the phenotype) with association by CNV or mutation in independent and similarly affected cases, followed by comparison with risk loci from GWAS studies. The strength of evidence varied between loci, as shown in Table 1 and Table S2. In some instances, genes were statistically supported by secondary CNV analyses, in others, the statistical support was inconclusive, and in several the CNV data do not nominate the locus as an NDD candidate (e.g. ZBTB20; see Table 1, Table S2, and Table S3). Additionally, like any genetic study, we cannot discount a potential contribution from altered expression of other genes at a distance from the site of the disruption. While such an effect could result from the transposition of functional elements, the comprehensive testing of this possibility is problematic, since it could involve dysregulation that is tissue-specific, that occurs due to the potentially altered nuclear organization of the rearranged chromosomes, or that is a secondary physiological consequence of the primary gene disruption. Nonetheless, secondary CNV analyses from independent NDD cases indicate a profound collective contribution of the disrupted genes on neurodevelopment. Simulations and subset analyses to evaluate empirical significance established that the increased CNV burden of the loci disrupted by BCAs was robust, unusually specific to neurodevelopmental disorders compared to other phenotypic presentations referred for molecular diagnostic testing, and was not driven by any single gene, individual disruption category, cluster of symptoms, or discrete diagnostic category. Rather, it was an accumulation of risk factors from all four categories that collectively contributed to the significant burden observed (see SI). For some genes, developmental abnormalities were predominantly associated with dosage alteration in only one direction while for others the increased CNV burden involved both deletions and duplications. These data illustrate the immature state of annotation of the human genome with respect to dosage sensitivity and to prediction of phenotypic outcomes from genetic lesions, a limitation that may be alleviated at least in part by the growing capacity to sequence BCAs in a relatively high-throughput manner.
A surprising number of genes previously associated with adolescent or adult onset psychiatric disorders were disrupted in children with autism and NDD in this study. The concept of schizophrenia as a neurodevelopmental disorder has long been proposed (Murray and Lewis, 1987; Weinberger, 1986), and a growing consensus in the recent literature suggests that there are shared risk factors across what are viewed clinically as distinct phenotypic classifications, although few of these have been described at the individual gene level (Owen et al., 2011). Our study supports a shared genetic etiology for at least a portion of the phenotypic spectrum of schizophrenia, autism, and the neurodevelopmental abnormalities studied here. We find unambiguous gene disruption by BCAs in ASD and NDD subjects, coupled with an increased CNV burden and a substantial over-representation of polygenic risk compared to null expectations from schizophrenia GWAS. Individual genes may thus show a differential risk depending on the nature of the genetic lesion, with heterozygous inactivation from BCA, CNV, or deactivating point mutation being a relatively penetrant contributor to ASD or NDD while subtle effects from common variants contribute to later onset disorders. However, contrary to this simple hypothesis, we were surprised to find persistent enrichment of these genes also among common variant associations from autism GWAS studies, suggesting that even subtle perturbation of genes important in normal human development can contribute to abnormal outcomes across the lifespan, presumably in interaction with other genetic and environmental influences.
This initial sequence-based delineation of a large collection of subjects harboring chromosomal aberrations in autism and related neurodevelopmental disorders establishes an approach that can be exploited for efficient discovery of individual genetic factors contributing to otherwise complex disorders. Each individual gene revealed provides a new, specific hypothesis concerning the disease to be tested with further genetic and biological study. If supported, each then represents a foundation for investigations into the role that the biochemical activity and regulation of its product play in pathogenesis and into the potential for treatment through their manipulation. Ultimately, such data will also provide invaluable annotation of the human genome and profoundly impact the clinical interpretation of genomic events in subjects referred to diagnostic laboratories for autism and other developmental abnormalities.
EXPERIMENTAL PROCEDURES
Patients
Subjects were obtained from the Developmental Genome Anatomy Project (DGAP) (Higgins et al., 2008), the Autism Consortium of Boston, the Center for Human Genetic Research (CHGR) Neurodevelopmental Repository, and the Autism Genome Resource Exchange (AGRE). These studies were approved by the Institutional Review Board of Partners HealthCare System. Clinical information was obtained by direct questionnaires, medical records, or structured clinical interviews (see SI).
Sequencing
Sequencing was performed on the Illumina platform (Illumina Inc). Libraries were created by four different methods optimized for delineating BCAs, including 1) Illumina standard insert paired-end sequencing, 2) Illumina mate-pair sequencing (long 2000–4000 bp inserts), 3) our customized jumping libraries (long 3000–4500 bp inserts), and 4) our capture of breakpoints method (CapBP) for rearrangements previously localized (see SI with complete protocols in (Talkowski et al., 2011a)). Interestingly, some rearrangements proved to be far more complex than suspected by karyotyping, and in at least two subjects this complexity represented balanced germline chromothripsis similar to but distinct from previously described events in cancer cells (DGAP127 and DGAP203; see Table S4 and (Chiang et al., 2012)). In complex rearrangements where more than two genes were disrupted by BCA breakpoints, we conservatively excluded these cases from interpretation in our secondary CNV/GWAS analyses.
Bioinformatics
Sequencing reads were aligned using publicly available alignment programs and custom scripts, followed by processing of BAM files using a C++ program, Bamstat, to tabulate mapping statistics and output lists of anomalous read-pairs (defined as having ends that map to two different chromosomes, having an abnormal insert size, and/or unexpected strand orientations) (Talkowski et al., 2011a). Anomalous pairs were clustered by their mapped mates with a program that performs a single-linkage clustering of paired reads if corresponding ends map within a specified distance. All junction fragments predicted from paired-end sequencing were PCR amplified from genomic DNA, independent of the libraries and sequencing reads, and all breakpoints presented in this study were confirmed by capillary sequencing (Table S1 and also Table S5).
Evaluation of CNVs in independent subjects
Complete details on the CNV data, statistical analyses, and simulations to determine empirical significance are fully described in SI. We compiled CNV data from 33,573 cases from molecular diagnostic laboratories of Signature Genomic Laboratories (SG) and Children’s Hospital Boston (CHB) analyzed by oligonucleotide aCGH. A subset of the SG data was included in a recent CNV study (Cooper et al., 2011). We fully annotated both datasets and collapsed all subjects with an indication for study (but without additional clinical information to verify a specific diagnosis) of autism, ASD, or a related neurodevelopmental abnormality into a combined NDD cohort (19,556 cases) and those for which the clinical indication did not indicate an NDD (14,017 cases). Control data were obtained across multiple published resources as described in the SI (n = 13,991 independent controls). Only 6,239 controls were analyzed for CNVs on the X chromosome, and this reduced comparison group was used for analyses of CDKL5. The resolution between the clinical aCGH and higher resolution control SNP microarrays varied extensively. To overcome this disparity but retain the native resolution of each individual array platform, we analyzed all CNVs that disrupted any documented coding or non-coding exon from all transcripts available from multiple database sources (Table S1). Notably, post-hoc analyses reveal that a size filter of 100 kb, or the resolution of the most sparse control arrays, would have resulted in an almost identical CNV burden test to our exon disruption model (p = 1.18×10−47), but would have omitted high confidence CNV calls in controls that could point towards reduced penetrance or non-significant loci in this study such as ZBTB20. Empirical significance was established by simulations performed in MATLAB (Mathworks Inc.) using custom scripts with all methods and findings described in the SI. In brief, one million gene-sets of 33 loci were randomly selected from the genome, and case-control CNV burden tests were performed for each random gene-set. These same analyses were performed for NDD cases compared to all other cases in the molecular diagnostic cohort. Neither simulation detected a random gene-set exceeding the significance of the experimental gene-set. Another set of experiments performed subset analyses, randomly selecting up to 1,000 gene-sets of each possible number of k genes from the experimental gene-set of 33 loci, ranging from k = 5 to k = 33, finding a minimum of 90.5% of all subsets at a level as low as k = 5 exceeded multiple testing correction and 100% of the 1,000 gene-sets to be significant at k > 13 (see SI).
Genome-wide association studies
We performed gene-set enrichment analysis from GWAS data using published methods (Rossin et al., 2011) on a recent schizophrenia meta-analysis (Ripke et al., 2011) and two autism genome-wide association studies (Weiss et al., 2009) (Wang et al., 2009). Briefly, linkage disequilibrium blocks across the genome are scored with the maximum Z-score achieved in the block. That score is corrected for the number of tests across the block using linear regression and the residuals are then used as the new, corrected score for each block. Genes nominate scores based upon the unique set of blocks they overlap, and the nominated scores are compared to background scores from all genes in the genome using a one-tailed rank-sum test.
Network analysis
A qualitative assessment of interacting genes and complexes for each of the genes in which functional information was available (little data existed for some of the novel genes) was initially performed using Natural Language Processing (NLP) from published abstracts (see SI). Secondary analyses of GO and KEGG enrichments were then performed followed by quantitative network building of first- and second-order interactions of the set of proteins coded by the genes disrupted by BCAs. The significance of these networks was determined by permutation testing (SI).
Supplementary Material
Highlights.
Balanced chromosomal abnormality breakpoints harbor individual strong effect genes
Disrupted genes implicate epigenetic and transcriptional regulation in autism
Dosage sensitive loci confer risk to autism from a spectrum of mutational mechanisms
Genes disrupted are associated with diverse childhood and adult clinical outcomes
Acknowledgments
We are grateful to all participating subjects and families, and to the many healthcare professionals who have contributed to this study, including Mary-Alice Abbott, Darius J. Adams, Kwame Anyane-Yeboa, Stephen G. Bamforth, Tina Bartell, David P. Bick, Joann N. Bodurtha, Carol Clericuzio, Stephanie Cohen, Kristin Dalton, Maria Descartes, Joanne Milisa Drautz, Dawn L. Earl, Luis F. Escobar, Shannon Gerner, Edwin Guzman, Kenneth Handelman, Tim Heshka, Robert J. Hopkin, Micheil Innes, Debby Lambert, Emmanuelle Lemyre, Cynthia Lim, Livija Medne, Graciela Moya, Katie Rutledge, Wendy Smith, Mark Stephan, Darci Sternen, Katie Stoll, Paulien van Galen, Nancy J. Van Vranken, Erica Wahl, Susan E. Wiley, Amy L White, Anne Woods, and Elaine H. Zackai. The invaluable control data for this study were provided by Pamela Sklar, Shaun Purcell, the International Schizophrenia Consortium, the Wellcome Trust Case Control Consortium, Evan Eichler, Bradley Coe, and Greg Cooper. We thank Tammy Gillis, Mary Anne Anderson, Jayla Ruliera, and Thon de Boer for technical assistance. We also thank Dennis Gurgul, Nilay Roy, and Brent Richter of Partners Research Computing at Massachusetts General Hospital, and contributing staff from Signature Genomic Laboratories and Children’s Hospital Boston. This work was funded by grants GM061354 and HD065286 from the NIH, the Simons Foundation Autism Research Initiative, and Autism Speaks. This research was also supported by the Division of Intramural Research, National Human Genome Research Institute (NHGRI), National Institutes of Health and Human Services, United States of America. M. Talkowski was supported by an NIMH National Research Service Award (MH087123) and an MGH ECOR Fund for Medical Discovery Award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amiel J, Rio M, de Pontual L, Redon R, Malan V, Boddaert N, Plouin P, Carter NP, Lyonnet S, Munnich A, et al. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. Am J Hum Genet. 2007;80:988–993. doi: 10.1086/515582. Epub 2007 Mar 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkaloglu B, O’Roak BJ, Louvi A, Gupta AR, Abelson JF, Morgan TM, Chawarska K, Klin A, Ercan-Sencicek AG, Stillman AA, et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet. 2008;82:165–173. doi: 10.1016/j.ajhg.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batsukh T, Pieper L, Koszucka AM, von Velsen N, Hoyer-Fender S, Elbracht M, Bergman JE, Hoefsloot LH, Pauli S. CHD8 interacts with CHD7, a protein which is mutated in CHARGE syndrome. Human molecular genetics. 2010;19:2858–2866. doi: 10.1093/hmg/ddq189. [DOI] [PubMed] [Google Scholar]
- Blake DJ, Forrest M, Chapman RM, Tinsley CL, O’Donovan MC, Owen MJ. TCF4, schizophrenia, and Pitt-Hopkins Syndrome. Schizophr. 2010;36:443–447. doi: 10.1093/schbul/sbq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Jacobsen JC, Ernst C, Hanscom C, Heilbut A, Blumenthal I, Mills RE, Kirby A, Lindgren AM, Rudiger SR, et al. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nature Genetics. 2012 doi: 10.1038/ng.2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, et al. Genome-wide Translocation Sequencing Reveals Mechanisms of Chromosome Breaks and Rearrangements in B Cells. Cell. 2011;147:107–119. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, et al. A copy number variation morbidity map of developmental delay. Nature genetics. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endele S, Rosenberger G, Geider K, Popp B, Tamer C, Stefanova I, Milh M, Kortum F, Fritsch A, Pientka FK, et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat Genet. 2010;42:1021–1026. doi: 10.1038/ng.677. [DOI] [PubMed] [Google Scholar]
- Ferreira MA, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, Fan J, Kirov G, Perlis RH, Green EK, et al. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nature genetics. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nature Genetics. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins AW, Alkuraya FS, Bosco AF, Brown KK, Bruns GA, Donovan DJ, Eisenman R, Fan Y, Farra CG, Ferguson HL, et al. Characterization of apparently balanced chromosomal rearrangements from the developmental genome anatomy project. Am J Hum Genet. 2008;82:712–722. doi: 10.1016/j.ajhg.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehler J. PDE10A Inhibitors: Novel Therapeutic Drugs for Schizophrenia. Curr Pharm Des. 2011;17:137–150. doi: 10.2174/138161211795049624. [DOI] [PubMed] [Google Scholar]
- Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, Moran J, Chambert K, Toncheva D, Georgieva L, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Molecular psychiatry. 2012;17:142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra T, Brunner HG, Amiel J, Oudakker AR, Nillesen WM, Magee A, Genevieve D, Cormier-Daire V, van Esch H, Fryns JP, et al. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am J Hum Genet. 2006;79:370–377. doi: 10.1086/505693. Epub 2006 Jun 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147:95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakics V, Karran EH, Boess FG. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology. 2010;59:367–374. doi: 10.1016/j.neuropharm.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meerabux JM, Ohba H, Iwayama Y, Maekawa M, Detera-Wadleigh SD, DeLisi LE, Yoshikawa T. Analysis of a t(18;21)(p11.1;p11.1) translocation in a family with schizophrenia. J Hum Genet. 2009;54:386–391. doi: 10.1038/jhg.2009.47. [DOI] [PubMed] [Google Scholar]
- Morikawa Y, Komori T, Hisaoka T, Ueno H, Kitamura T, Senba E. Expression of mKirre in the developing sensory pathways: its close apposition to nephrin-expressing cells. Neuroscience. 2007;150:880–886. doi: 10.1016/j.neuroscience.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Murray RM, Lewis SW. Is schizophrenia a neurodevelopmental disorder? Br Med J (Clin Res Ed) 1987;295:681–682. doi: 10.1136/bmj.295.6600.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama M, Oshikawa K, Tsukada Y, Nakagawa T, Iemura S, Natsume T, Fan Y, Kikuchi A, Skoultchi AI, Nakayama KI. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nature cell biology. 2009;11:172–182. doi: 10.1038/ncb1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, Nikolov I, Hamshere M, Carroll L, Georgieva L, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nature genetics. 2008;40:1053–1055. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nature genetics. 2011;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen MJ, O’Donovan MC, Thapar A, Craddock N. Neurodevelopmental hypothesis of schizophrenia. Br J Psychiatry. 2011;198:173–175. doi: 10.1192/bjp.bp.110.084384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravel C, Berthaut I, Bresson JL, Siffroi JP. Prevalence of chromosomal abnormalities in phenotypically normal and fertile adult males: large-scale survey of over 10,000 sperm donor karyotypes. Hum Reprod. 2006;21:1484–1489. doi: 10.1093/humrep/del024. [DOI] [PubMed] [Google Scholar]
- Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, Lin DY, Duan J, Ophoff RA, Andreassen OA, et al. Genome-wide association study identifies five new schizophrenia loci. Nature genetics. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Paredes M, Ceballos-Chavez M, Esteller M, Garcia-Dominguez M, Reyes JC. The chromatin remodeling factor CHD8 interacts with elongating RNA polymerase II and controls expression of the cyclin E2 gene. Nucleic Acids Res. 2009;37:2449–2460. doi: 10.1093/nar/gkp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Ballif BC, Lucas A, Spence EJ, Powell C, Aylsworth AS, Torchia BA, Shaffer LG. Small deletions of SATB2 cause some of the clinical features of the 2q33.1 microdeletion syndrome. PLoS One. 2009a;4:e6568. doi: 10.1371/journal.pone.0006568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld JA, Ballif BC, Torchia BS, Sahoo T, Ravnan JB, Schultz R, Lamb A, Bejjani BA, Shaffer LG. Copy number variations associated with autism spectrum disorders contribute to a spectrum of neurodevelopmental disorders. Genetics in medicine : official journal of the American College of Medical Genetics. 2010;12:694–702. doi: 10.1097/GIM.0b013e3181f0c5f3. [DOI] [PubMed] [Google Scholar]
- Rosenfeld JA, Leppig K, Ballif BC, Thiese H, Erdie-Lalena C, Bawle E, Sastry S, Spence JE, Bandholz A, Surti U, et al. Genotype-phenotype analysis of TCF4 mutations causing Pitt-Hopkins syndrome shows increased seizure activity with missense mutations. Genet Med. 2009b;11:797–805. doi: 10.1097/GIM.0b013e3181bd38a9. [DOI] [PubMed] [Google Scholar]
- Rossin EJ, Lage K, Raychaudhuri S, Xavier RJ, Tatar D, Benita Y, Cotsapas C, Daly MJ. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet. 2011;7:e1001273. doi: 10.1371/journal.pgen.1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serizawa S, Miyamichi K, Takeuchi H, Yamagishi Y, Suzuki M, Sakano H. A neuronal identity code for the odorant receptor-specific and activity-dependent axon sorting. Cell. 2006;127:1057–1069. doi: 10.1016/j.cell.2006.10.031. [DOI] [PubMed] [Google Scholar]
- Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R, Yu CE, Yu J, Munson J, Chen D, Hua W, Estes A, Cortes F, de la Barra F, Yu D, et al. Identification of a novel gene on chromosome 7q11.2 interrupted by a translocation breakpoint in a pair of autistic twins. Genomics. 2002;80:129–134. doi: 10.1006/geno.2002.6810. [DOI] [PubMed] [Google Scholar]
- Talkowski ME, Ernst C, Heilbut A, Chiang C, Hanscom C, Lindgren A, Kirby A, Liu S, Muddukrishna B, Ohsumi TK, et al. Next-generation sequencing strategies enable routine detection of balanced chromosome rearrangements for clinical diagnostics and genetic research. Am J Hum Genet. 2011a;88:469–481. doi: 10.1016/j.ajhg.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, Mullegama SV, Rosenfeld JA, van Bon BW, Shen Y, Repnikova EA, Gastier-Foster J, Thrush DL, Kathiresan S, Ruderfer DM, et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. American Journal of Human Genetics. 2011b;89:551–563. doi: 10.1016/j.ajhg.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura S, Morikawa Y, Hisaoka T, Ueno H, Kitamura T, Senba E. Expression of mKirre, a mammalian homolog of Drosophila kirre, in the developing and adult mouse brain. Neuroscience. 2005;133:615–624. doi: 10.1016/j.neuroscience.2005.03.030. [DOI] [PubMed] [Google Scholar]
- Thompson BA, Tremblay V, Lin G, Bochar DA. CHD8 is an ATP-dependent chromatin remodeling factor that regulates beta-catenin target genes. Molecular and cellular biology. 2008;28:3894–3904. doi: 10.1128/MCB.00322-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters JT, Corvin A, Owen MJ, Williams H, Dragovic M, Quinn EM, Judge R, Smith DJ, Norton N, Giegling I, et al. Psychosis susceptibility gene ZNF804A and cognitive performance in schizophrenia. Archives of general psychiatry. 2010;67:692–700. doi: 10.1001/archgenpsychiatry.2010.81. [DOI] [PubMed] [Google Scholar]
- Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PM, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger D. The pathogenesis of schizophrenia: a neurodevelopmental theory. In: Weinberger RND, editor. The Neurology of Schizophrenia. Elsevier; 1986. pp. 387–405. [Google Scholar]
- Weiss LA, Arking DE, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–808. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, et al. Association between microdeletion and microduplication at 16p11.2 and autism. The New England journal of medicine. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Williams HJ, Craddock N, Russo G, Hamshere ML, Moskvina V, Dwyer S, Smith RL, Green E, Grozeva D, Holmans P, et al. Most genome-wide significant susceptibility loci for schizophrenia and bipolar disorder reported to date cross-traditional diagnostic boundaries. Hum Mol Genet. 2011;20:387–391. doi: 10.1093/hmg/ddq471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.