

Abstract
Objectives
Hyperglycemia induces oxidative stress and increases inducible nitric oxide synthase (iNOS) expression. We hypothesized that oxidative stress is responsible for hyperglycemia-induced iNOS expression.
Study Design
iNOS-luciferase activities, nitrosylated protein, lipidperoxidation markers 4-HNE and MDA were determined in PYS-2 cells exposed to 5 mM glucose or high glucose (25 mM) with or without SOD1 (copper zinc superoxide dismutase 1) treatment. Levels of iNOS protein and mRNA, nitrosylated protein, and cleaved caspase-3 and -8 were assessed in wild-type embryos and SOD1 overexpressing embryos from non-diabetic and diabetic dams.
Results
SOD1 treatment diminished high glucose-induced oxidative stress, as evidenced by 4-HNE and MDA reductions, and it blocked high glucose-increased iNOS expression, iNOS-luciferase activities, and nitrosylated protein. in vivo SOD1 overexpression suppressed hyperglycemia-increased iNOS expression and nitrosylated protein, and it blocked caspase-3 and -8 cleavage.
Conclusions
We conclude that oxidative stress induces iNOS expression, nitrosative stress, and apoptosis in diabetic embryopathy.
Keywords: SOD1 transgenic mice, oxidative stress, iNOS, nitrosative stress, diabetic embryopathy
INTRODUCTION
Pregestational diabetes significantly increases the risk of a range of congenital malformations, including neural tube defects (NTDs) and cardiovascular defects 1–3. Both in vivo and in vitro studies have demonstrated that hyperglycemia by itself, and not other aberrant changes associated with diabetes, mediate the teratogenicity of diabetes 4–7. It has been shown that hyperglycemia enhances ROS production and decreases endogenous antioxidant enzyme expression, resulting in oxidative stress 7–21.
Suppressing oxidative stress via SOD1 overexpression in SOD1-Tg mice significantly ameliorates maternal diabetes-induced NTDs 22. In addition, both in vivo and in vitro antioxidant treatments have been shown to prevent hyperglycemia-induced malformations 12, 19–21. There is ample evidence, therefore, that hyperglycemia-induced oxidative stress is responsible for the induction of embryonic malformations in diabetic embryopathy 15, 23, 24. However, there is still an urgent need to determine the downstream events of oxidative stress in diabetic embryopathy. This knowledge is a key to understanding the full-range of mechanisms underlying maternal diabetes-induced malformations and their potential prevention.
Aberrant gene expression has been shown to be associated with diabetic embryopathy 18, 25. Studies from our laboratory 13 and others 26 have demonstrated that iNOS (but not eNOS) gene expression is significantly increased in embryos exposed to hyperglycemia. It also has been shown that iNOS largely mediates the teratogenicity of diabetes, because targeted deletion of the inos gene significantly reduces maternal diabetes-induced NTDs 26. Our group has demonstrated that the activation of the c-Jun N-terminal kinases 1 and 2 (JNK1/2) leads to an increase in iNOS gene expression 13. Because JNK1/2 is activated by oxidative stress in diabetic embryopathy 15, 16, 27, we have proposed that hyperglycemia-induced oxidative stress causes increased iNOS gene expression in diabetic embryopathy.
Increased iNOS gene expression has at least two adverse effects on the developing embryo. First, iNOS induction generates very high concentrations of nitric oxide (NO), which, in turn, induces the generation of reactive nitrogen species that leads to nitrosative stress. Abnormal levels of nitric oxide (NO) are associated with adverse pregnancy outcomes in diabetic pregnancies 28. Furthermore, levels of nitrosylated proteins in embryos exposed to hyperglycemia are significantly higher than in embryos cultured under euglycemic conditions 16. The second adverse consequence of increased iNOS gene expression is that it may induce cell apoptosis. For example, targeted inos gene deletion abolishes maternal diabetes-induced cell apoptosis in embryonic organs that are known to be particularly vulnerable to hyperglycemic insults 26. Extensive evidence also supports the assertion that enhanced apoptosis downstream of oxidative stress is the central pathological mechanism in maternal diabetes-induced NTDs. We have shown that mitigating oxidative stress via antioxidant treatments blocks maternal diabetes-induced apoptosis, thus, reducing the incidence of malformations under in vivo diabetic conditions 12, 19–21. Apoptosis, therefore, may bridge the connection between oxidative stress and enhanced iNOS expression in diabetic embryopathy.
The adverse effects of hyperglycemia on the developing embryo are development-stage dependent. Extensive efforts have been focused on the key organogenesis period, embryonic day 7 (E7) to E11 in the mouse. During this critical time, maternal hyperglycemia adversely impacts embryonic vasculogenesis and neural tube closure resulting in vasculopathy and NTDs 12, 14, 16. The yolk sac and the neural tube are the two most susceptible tissues in the developing embryos to hyperglycemic damage. In diabetic embryopathy, early vasculopathy correlates with late structural malformations, such as NTDs 16. Aberrant changes in NO and iNOS have been implicated in diabetic embryonic vasculopathy 28.
We 15 and others 26, 29 have employed a mouse model of diabetic embryopathy at C57BL/6J background, which exhibits a NTD rate of about 25% in embryos exposed to maternal hyperglycemia. Embryonic vasculopathy at early stages (E7–E9) in this mouse model also has been characterized under hyperglycemic conditions 16. The connection between oxidative stress and increased iNOS-nitrosative stress has not been explored. Because both SOD1 overexpression in vivo 22 and iNOS deficiency in iNOS knockout mice 26 reduce hyperglycemia-induced malformations, we used SOD1-Tg mice to test whether SOD1 overexpression in vivo blocks hyperglycemia-increased iNOS expression, consequent nitrosative stress, and apoptosis.
Material and methods
Animals and reagents
C57BL/6J mice (median body weight 22 g) were purchased from the Jackson Laboratory (Bar Harbor, Maine). Streptozotocin (STZ) from Sigma was dissolved in sterile 0.1 M citrate buffer (pH 4.5). Sustained-release insulin pellets were purchased from Linplant (Linshin, Canada). SOD1-Tg mice in C57BL/6J background were revived from frozen embryos by the Jackson Laboratory (Stock number: 002298). The PYS-2 cell line from parietal yolk sac was purchased from ATCC (Cat# CRL-2745).
Cell culture, transient transfection and luciferase assay
PYS-2 cells were cultured in DMEM (Invitrogen) plus 2% FBS. Cells were plated overnight to reach about 80% confluency and were transfected with 0.8 μg mouse iNOS promoter luciferase constructor (iNOS-luc) using Lipofectamine™ 2000 (Invitrogen). The iNOS-luc that contains the mouse iNOS promoter from −1588 to +165, plus the luciferase coding sequence, was provided by Dr. Sang Geon Kim, Seoul National University, South Korea. After co-transfection with Renilla-luc (normalization control, Promega), PYS-2 cells were incubated for 24h with 5 mM glucose or 25 mM glucose in the presence or absence of 400 or 800 Unit of human SOD1 (Sigma). Luciferase activities were measured by a dual luciferase kit (Promega) according to the manufacturer's instructions.
Mouse models of diabetic embryopathy
The procedures for animal use were approved by the Institutional Animal Care and Use Committee of University of Maryland School of Medicine. Eight-week old Wildtype (WT) mice were intravenously injected daily with 75 mg/kg STZ over two days to induce diabetes. Once a level of hyperglycemia indicative of diabetes (≥250 mg/dl) was achieved, insulin pellets were subcutaneously implanted in these diabetic mice to restore euglycemia prior to mating. The mice were then mated with SOD1-Tg male mice at 3:00 PM to generate WT and SOD1-overexpressing embryos. The morning when a vaginal plug was present was designated as embryonic day 0.5 (E0.5) On E5.5, insulin pellets were removed to permit frank hyperglycemia (>250 mg/dl glucose level), so the developing conceptuses would be exposed to a hyperglycemic conditions from E7 onwards. WT, non-diabetic female mice with vehicle injections and sham operation of insulin pellet implants were served as non-diabetic controls. On E8.75, mice were euthanized, and conceptuses were dissected out of the uteri for analysis. To avoid any redundancy, data of malformation incidences were not collected because these have been published elsewhere 22.
Genotyping of Embryos
Embryos from WT diabetic dams mated with SOD1-Tg male mice were genotyped according to the Jackson Laboratory’s protocol using the yolk sac DNA.
Western Blotting
Embryos at E8.75 from different experimental groups were sonicated in 80 μl ice-cold lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 10 mM NaF, 2 mM Na orthovanadate, 1 mM PMSF and 1% Triton 100) containing a protease inhibitor cocktail (Sigma, St. Louis, MO). Equal amounts of protein were resolved by SDS-PAGE and transferred onto Immobilon-P or Immobilon-PSQ (for cleaved caspase) membranes (MILLIPORE). Membranes were incubated for 18 h at 4 °C with the following primary antibodies at 1:1000 to 1:2000 dilutions in 5% nonfat milk: anti-iNOS and anti-SOD1 (Cell Signaling, Beverly, MA); anti-nitrotyrosine and anti-caspase-3 (CHEMICON International, Billerica, MA); rat anti-caspase-8 (Alexis Biochemicals, San Diego, CA); and anti-β-actin (Abcam, Cambridge, MA). Signals were detected using an Amersham ECL Advance Detection Kit (GE Healthcare, Piscataway, NJ). Chemiluminescence emitted from the bands was directly captured using a UVP Bioimage EC3 system (UVP, Upland, CA). Densitometric analysis of chemiluminescence signals was performed by VisionWorks LS software (UVP, Upland, CA).
Real-time PCR (RT-PCR)
Total RNA was isolated from embryonic tissues of cultured conceptuses or conceptuses retrieved from non-diabetic or diabetic mice using an RNeasy Mini Kit (Qiagen, Valencia, CA). RT-PCR assays for iNOS and β-actin were performed using ABI TaqMan Gene Expression Assays (assay ID: Mm01309897_m1 and Mm00607939_s1, respectively, Applied Biosystems, Foster City, CA). Briefly, RNA was reverse transcribed by using the high-capacity cDNA archive kit (Applied Biosystems). RT-PCR and subsequent calculations were performed by the StepOnePlus Real-time PCR system (Applied Biosystems), which detected the signal emitted from fluorogenic probes during PCR.
Statistical Analysis
Densitometric data were presented as means ± SE (standard error). One way ANOVA was performed using SigmaStat 3.5 software. After using a one-way ANOVA in Figure 1, 2, 3 and 4, Tukey was used for multiple comparison testing to estimate the significance of the results. Statistical significance was accepted at p< 0.05.
Figure 1.
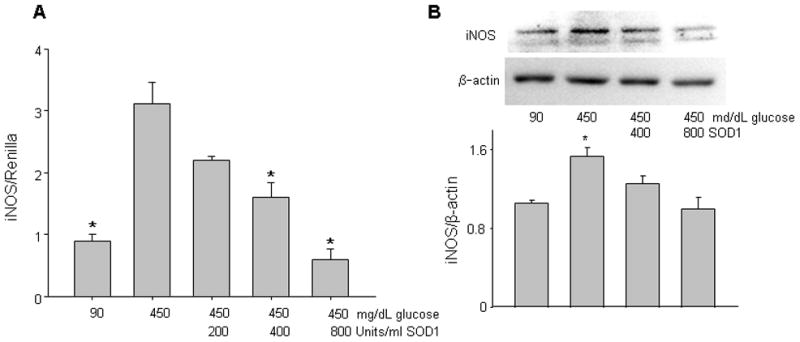
Copper zinc superoxide dismutase 1 (SOD1) treatment blocks high glucose-induced iNOS expression. In A, SOD1 dose-dependently suppressed high glucose-induced iNOS-luciferase activities, which were normalized by Renilla-luciferase activities and expressed as iNOS/Renilla. * indicates significant difference (p < 0.05) when compared to the high glucose (450 mg/dL or 25 mM) alone group. n = 5. In B, SOD1 treatment suppressed high glucose-increased iNOS protein expression. Representative images were shown in the upper panel and the data of densitometric analysis were presented in the bottom graph. * indicates significant difference (p < 0.05) when compared to other groups. n = 3 (three independent experiments).
Figure 2.
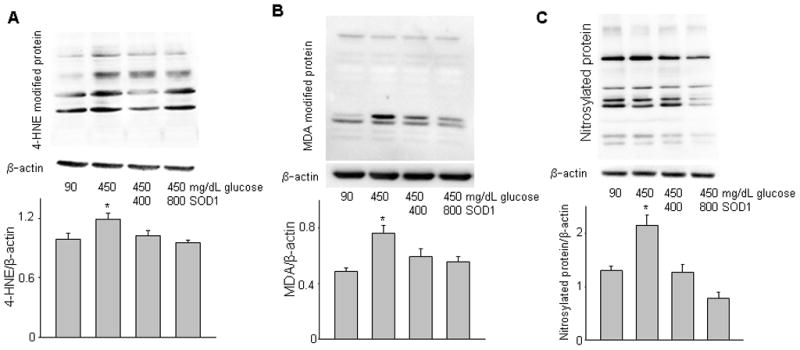
Copper zinc superoxide dismutase 1 (SOD1) treatment suppresses high glucose-induced lipidperoxidation and nitrosative stress. A, levels of 4-hydroxynonenal (4-HNE)-modified protein; B, levels of malondialdehyde (MDA)-modified protein; C, levels of nitrosylated protein, indices of nitrosative stress; Upper panels of A, B and C were representative images of Western blotting. Graphs of lower panels at A, B and C were data of densitometric analysis. * indicates significant difference (p < 0.05) when compared to other groups. n = 3.
Figure 3.
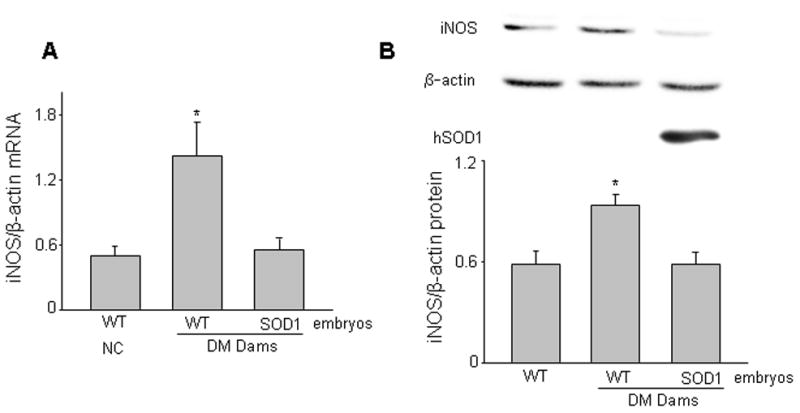
Copper zinc superoxide dismutase 1 (SOD1) overexpression in vivo abrogates maternal hyperglycemia-induced iNOS expression. Levels of iNOS mRNA ( A) and iNOS protein (B) were determined in E8.75 WT embryos from non-diabetic controls (NC), WT embryos and SOD1 overexpressing embryos from diabetic mellitus (DM) WT mice mated with SOD1-Transgenic (Tg) male mice. SOD1 overexpressing embryos harbor the human SOD1 transgene and SOD1 was detected by a human specific antibody. The upper panel of B was representative images of Western blotting. The graph in the lower panel of B was data of densitometric analysis. * indicates significant difference (p < 0.05) when compared to other groups. In A, n = 6, and in B, n = 3. Experiments were repeated three times with embryos from three different mothers in each group.
Figure 4.
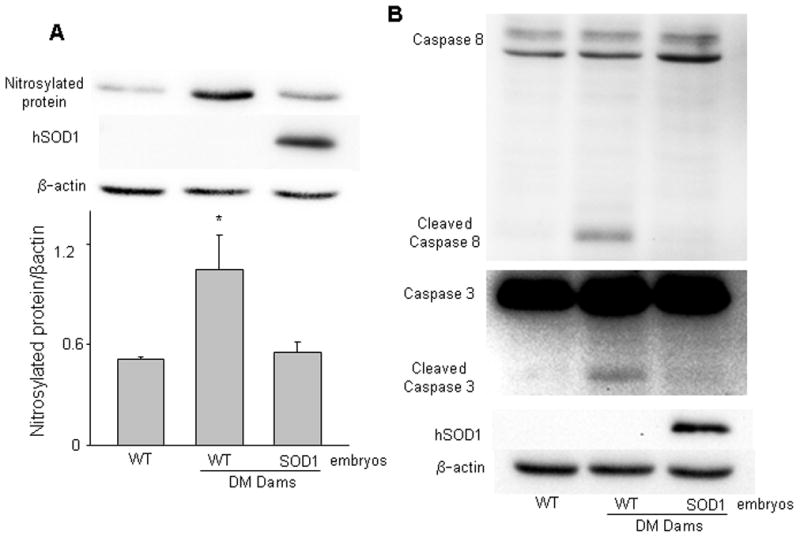
Copper zinc superoxide dismutase 1 (SOD1) overexpression in vivo abolishes maternal hyperglycemia-induced nitrosative stress and caspase activation. Levels of nitrosylated protein (A), and cleaved caspase-8 and caspase-3 (B) were determined in E8.75 WT embryos from non-diabetic controls (NC), WT embryos and SOD1 overexpressing embryos from diabetic mellitus (DM) WT dams mated with SOD1-Transgenic (Tg) males. Representative images of Western blotting were shown in A (upper panel) and B. The graph in the lower panel of A was data of densitometric analysis. * indicates significant difference (p < 0.05) when compared to other groups. n = 3. In B, experiments were repeated three times with embryos from three different mothers in each group, and identical results were obtained. Cleaved products of caspase-8 and -3 were only present in WT embryos of DM dams.
Results
SOD1 treatment suppresses high glucose-increased iNOS transcription and protein expression
We previously have demonstrated that high glucose significantly increases iNOS-luc activities in PYS-2 cells 13. To test whether mitigating oxidative stress using SOD1 treatment blocks high glucose-induced iNOS transcription, cells were cultured under 5 mM or 25 mM glucose with 0, 200, 400, and 800 Unit human SOD1. Consistent with our previous finding 13, 25 mM glucose (high glucose conditions) significantly increased iNOS-luc activities when compared to that of 5 mM glucose group (normoglycemic conditions) (Fig. 1A). SOD1 treatment suppressed high glucose-increased iNOS-luc activities in a dose-dependent manner (Fig. 1A). Both 400 Unit and 800 Unit SOD1 suppressed high glucose-increased iNOS-luc activities to the levels of that found in the normoglycemic group (Fig. 1A). Correlated with the observed increase of iNOS transcription, endogenous iNOS protein expression was up-regulated by high glucose (Fig. 1B). On the other hand, high glucose-increased iNOS protein expression was significantly blunted by SOD1 treatment (Fig. 1B).
SOD1 treatment blocks high glucose-induced lipidperoxidation and nitrosative stress
Previously, we demonstrated that in vivo SOD1 overexpression in SOD1-Tg mice suppressed maternal hyperglycemia-induced lipidperoxidation downstream of PKC activation in diabetic embryopathy 30. The two lipidperoxidation markers, 4-HNE and MDA, are indices of oxidative stress. To test whether SOD1 treatment could effectively mitigate high glucose-induced oxidative stress, we determined 4-HNE- and MDA-modified protein levels and found that high glucose conditions significantly increased levels of 4-HNE- and MDA-modified protein (Fig. 2A, B). Both 400 Unit and 800 Unit SOD1 significantly reduced high glucose-increased 4-HNE- and MDA-modified protein (Fig. 2A, B). Because SOD1 treatment blocked high glucose-induced iNOS expression (Fig. 1), we next analyzed the levels of nitrosylated protein, which are the consequences of increased iNOS expression. As previously reported 13, levels of nitrosylated protein were elevated under high glucose conditions (Fig. 2C), indicating that high glucose induces nitrosative stress. SOD1 significantly blocked high glucose-increased nitrosylated protein in a dose dependent manner (Fig. 2C).
SOD1 overexpression in vivo abrogates maternal hyperglycemia-induced iNOS gene and protein expression
Published data demonstrate that both SOD1 overexpression in SOD1-Tg mice 22 and targeted deletion of the inos gene in iNOS knockout mice 26 significantly ameliorate maternal diabetes-induced malformations. These findings strongly suggest a link between oxidative stress and iNOS in diabetic embryopathy. To test whether increased iNOS expression is downstream of oxidative stress, we assessed the levels of iNOS mRNA and protein in WT embryos from non-diabetic control (NC), WT embryos and SOD1 overexpressing embryos from diabetic WT dams mated with SOD1-Tg males. Consistent with earlier published findings from our group 13 and others 26, maternal hyperglycemia significantly increased the expression of iNOS protein and mRNA in WT embryos (Fig. 3A, B). Maternal hyperglycemia-increased iNOS protein and mRNA expression were diminished in SOD1 overexpressing embryos (Fig. 3A, B). Blood glucose levels in diabetic mice (443.4 ± 14.6 mg/dl) were more than twice as high as those in NC mice (140.9 ± 5.2 mg/dl).
SOD1 overexpression in vivo prevents hyperglycemia-induced nitrosative stress and caspase activation in diabetic embryopathy
Increased iNOS expression leads to nitrosative stress. Because SOD1 overexpression diminished maternal hyperglycemia-induced iNOS expression (Fig. 3), we reasoned that SOD1 overexpression would also abrogate maternal hyperglycemia-induced nitrosative stress. To this end, we determined the levels of nitrosylated protein in WT embryos from non-diabetic control (NC), WT embryos and SOD1 overexpressing embryos from diabetic WT dams mated with SOD1-Tg males. In WT embryos, maternal hyperglycemia significantly increased nitrosylated protein, which manifested one major band in our Westerns (Fig. 4A). In SOD1 overexpressing embryos, maternal hyperglycemia-increased nitrosylated protein was diminished (Fig. 4A). Apoptosis is the central causative event in the induction of embryonic malformations in diabetic embryopathy. We previously characterized the involvement of the initiator caspase (caspase-8) and effector caspase (caspase-3) in maternal hyperglycemia-induced apoptosis in embryonic cells 31. To determine whether mitigating oxidative stress in SOD1-Tg mice could block the activation of the maternal hyperglycemia-induced caspase activation, we measured caspase-3 and -8 cleavage. Maternal hyperglycemia induced robust cleavage of both caspase-3 and -8 (Fig. 4B); whereas, SOD1 overexpression in SOD1 overexpressing embryos abolished maternal hyperglycemia-induced caspase-3 and -8 cleavage (Fig. 4B).
Comment
Studies have demonstrated that oxidative stress 15 and iNOS upregulation 13, 26 downstream of hyperglycemia are two critical events involved in the induction of diabetic embryopathy. In the present study, we establish for the first time a causative relationship between oxidative stress and iNOS upregulation in diabetic embryopathy. More notably, we found that suppressing oxidative stress using both in vitro recombinant human SOD1 treatment and in vivo SOD1 overexpression blocks hyperglycemia-increased iNOS expression and consequent nitrosative stress. Our results, thus, provide strong support that hyperglycemia-induced oxidative stress causes iNOS upregulation and its associated nitrosative stress.
Maternal hyperglycemia induces oxidative stress in embryonic cells by enhancing ROS production and impairing the capability of endogenous antioxidant enzymes 8–11. Previous studies 22, 32 have demonstrated that both in vitro SOD1 treatment and in vivo SOD1 overexpression in SOD1-Tg mice can effectively reduce high glucose- or maternal hyperglycemia-induced oxidative stress. Using the lipidperoxidation markers, 4-HNE and MDA, we have further demonstrated in this study that SOD1 treatments abrogate high glucose-induced oxidative stress in vitro. We previously have reported that SOD1 overexpression in SOD1-Tg mice significantly reduces maternal diabetes-induced embryonic malformations 22.
Two of the most susceptible tissues in the early stages of development, such as E8.75, to hyperglycemic insults are the yolk sac and the neural tube. Our previous studies have demonstrated that exposing PYS-2 cells to high glucose conditions faithfully reflects the changes observed in early stage embryos exposed to maternal hyperglycemia 13. Thus, the PYS-2 cell line, which is an embryonic yolk sac cell line, is an ideal in vitro tool for studying gene transcription in diabetic embryopathy. E8.75 is a critical time of neurulation. At this stage of development, the neural tube consists of the majority of the developing embryo. For these reasons, the E8.75 embryo is an ideal model for studying the causes of NTDs. Both SOD1 overexpression in SOD1-Tg mice 22 and targeted deletion of the inos gene 26 significantly reduce maternal hyperglycemia-induced NTDs. Our present studies, using E8.75 embryos, reveal the causative link between oxidative stress and iNOS upregulation in the context of maternal hyperglycemia-induced NTDs.
We have previously reported that high glucose induces iNOS transcription in a dose dependent manner and the induction of iNOS transcription reaches a plateau at 25 mM (450 mg/dl) glucose 13. Therefore, we used 25 mM glucose, and 200, 400 and 800 Units/ml of SOD1 in our iNOS luciferase studies. Two of these SOD1 doses, 400 and 800 Units/ml SOD1, significantly reduced high glucose-induced iNOS transcription. Thus, our subsequent studies used these two doses. We crossed WT females with SOD1-Tg males to generate SOD1 overexpression embryos. Both WT and SOD1 overexpressing embryos were exposed to the same maternal diabetic conditions. Thus, this experimental design avoids any potential maternal influence due to the SOD1 transgene.
Using genetic approaches, we previously reported strong evidence that JNK1/2 activation is responsible for iNOS upregulation in diabetic embryopathy 13. Moreover, we also have shown that dietary antioxidant supplements 21 can abrogate maternal hyperglycemia-induced JNK1/2. These data suggest that JNK1/2 activation is downstream of oxidative stress in this disease process. Therefore, we propose the oxidative stress-JNK1/2-iNOS pathway, which leads to apoptosis and diabetic embryopathy. Future investigations should aim to determine the roles of transcription factors downstream of JNK1/2 activation in regulation of different nitric oxide synthases, such as iNOS and eNOS, in diabetic embryopathy. iNOS upregulation results in nitrosative stress, which is manifested by increased levels of nitrosylated proteins 13. ROS reacts with nitric oxide generated by excess iNOS leading to nitrosative stress, which is the most detrimental form of oxidative stress. Indeed, under maternal diabetes, oxidative stress and nitrosative stress form a twin cyclone, leading to maximal cellular damage and, ultimately, to embryonic malformations.
Maternal hyperglycemia-induced apoptosis is caspase dependent, and the pro-apoptotic signaling emanating from oxidative stress triggers activation of the initiator caspase, caspase-8 31. Our results link oxidative stress-induced iNOS upregulation and consequent nitrosative stress to caspase-3 and -8 cleavage. This is consistent with the prior observation that targeted deletion of inos gene blocks maternal hyperglycemia-induced caspase-3 activation and apoptosis 26. Enhanced apoptosis is the central mechanism in the induction of diabetic embryopathy. Microarray studies have revealed that an array of apoptotic genes is induced in diabetic embryopathy 18, 25. Further studies are warranted to determine whether iNOS-mediated apoptosis is transcription or non-transcription dependent.
In summary, we have used both in-vitro and in-vivo approaches in this current study to demonstrate that mitigating oxidative stress suppresses high glucose- or maternal hyperglycemia-induced iNOS upregulation and nitrosative stress. The results further strengthen the oxidative stress hypothesis in diabetic embryopathy and reveal that oxidative stress is indeed responsible for iNOS upregulation and nitrosative stress in this disease.
Because oxidative stress causes diabetic embryopathy15, antioxidants have been implicated in therapeutic interventions for this disease. Animal studies have shown that antioxidants are effective in prevention of diabetic embryopathy12, 19–21. However, the effectiveness of antioxidants, particularly vitamins, in human diabetic embryopathy is controversy33, 34. Due to the obesity epidemic35–38, diabetes-associated adverse pregnancy outcomes are urgent public health problems. Glycemic control during pregnancy is difficult to achieve and maintain39, 40. Thus, mechanistic studies will provide the basis for the development of new interventions. Consistent with our previous study13, the present study shows that iNOS and its associated nitrosative stress mediate the adverse effect of oxidative stress downstream of hyperglycemia. Inhibition of iNOS and nitrosative stress may be a new strategy in prevention of human diabetic embryopathy.
Acknowledgments
This study is supported by NIH R01 DK083243 (to Peixin Yang) and R01 DK083770 (to E Albert Reece). The authors are grateful to Ms. Hua Li and Dr. Cheng Xu for their technical support.
Footnotes
Disclosure: None of the authors have a conflict of interest.
Reprint Requests: Not Available
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Becerra JE, Khoury MJ, Cordero JF, Erickson JD. Diabetes mellitus during pregnancy and the risks for specific birth defects: a population-based case-control study. Pediatrics. 1990;85:1–9. [PubMed] [Google Scholar]
- 2.Ramos-Arroyo MA, Rodriguez-Pinilla E, Cordero JF. Maternal diabetes: the risk for specific birth defects. Eur J Epidemiol. 1992;8:503–8. doi: 10.1007/BF00146367. [DOI] [PubMed] [Google Scholar]
- 3.Correa A, Gilboa SM, Besser LM, et al. Diabetes mellitus and birth defects. Am J Obstet Gynecol. 2008;199:237, e1–9. doi: 10.1016/j.ajog.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reece EA, Pinter E, Leranth CZ, et al. Ultrastructural analysis of malformations of the embryonic neural axis induced by in vitro hyperglycemic conditions. Teratology. 1985;32:363–73. doi: 10.1002/tera.1420320306. [DOI] [PubMed] [Google Scholar]
- 5.Pinter E, Reece EA, Leranth CZ, et al. Arachidonic acid prevents hyperglycemia-associated yolk sac damage and embryopathy. Am J Obstet Gynecol. 1986;155:691–702. doi: 10.1016/s0002-9378(86)80001-1. [DOI] [PubMed] [Google Scholar]
- 6.Greene MF, Hare JW, Cloherty JP, Benacerraf BR, Soeldner JS. First- trimester hemoglobin A1 and risk for major malformation and spontaneous abortion in diabetic pregnancy. Teratology. 1989;39:225–31. doi: 10.1002/tera.1420390303. [DOI] [PubMed] [Google Scholar]
- 7.Reece EA, Wiznitzer A, Homko CJ, Hagay Z, Wu YK. Synchronization of the factors critical for diabetic teratogenesis: an in vitro model. Am J Obstet Gynecol. 1996;174:1284–8. doi: 10.1016/s0002-9378(96)70672-5. [DOI] [PubMed] [Google Scholar]
- 8.Yang X, Borg LA, Eriksson UJ. Altered metabolism and superoxide generation in neural tissue of rat embryos exposed to high glucose. Am J Physiol. 1997;272:E173–80. doi: 10.1152/ajpendo.1997.272.1.E173. [DOI] [PubMed] [Google Scholar]
- 9.Yang X, Borg LA, Siman CM, Eriksson UJ. Maternal antioxidant treatments prevent diabetes-induced alterations of mitochondrial morphology in rat embryos. Anat Rec. 1998;251:303–15. doi: 10.1002/(SICI)1097-0185(199807)251:3<303::AID-AR5>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 10.Sakamaki H, Akazawa S, Ishibashi M, et al. Significance of glutathione-dependent antioxidant system in diabetes-induced embryonic malformations. Diabetes. 1999;48:1138–44. doi: 10.2337/diabetes.48.5.1138. [DOI] [PubMed] [Google Scholar]
- 11.Sivan E, Lee YC, Wu YK, Reece EA. Free radical scavenging enzymes in fetal dysmorphogenesis among offspring of diabetic rats. Teratology. 1997;56:343–9. doi: 10.1002/(SICI)1096-9926(199712)56:6<343::AID-TERA1>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 12.Yang P, Li H. Epigallocatechin-3-gallate ameliorates hyperglycemia-induced embryonic vasculopathy and malformation by inhibition of Foxo3a activation. Am J Obstet Gynecol. 2010;203:75, e1–6. doi: 10.1016/j.ajog.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang P, Cao Y, Li H. Hyperglycemia induces inducible nitric oxide synthase gene expression and consequent nitrosative stress via c-Jun N-terminal kinase activation. Am J Obstet Gynecol. 2010;203:185, e5–11. doi: 10.1016/j.ajog.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang P, Reece EA. Role of HIF-1alpha in maternal hyperglycemia-induced embryonic vasculopathy. Am J Obstet Gynecol. 2011;204:332, e1–7. doi: 10.1016/j.ajog.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang P, Zhao Z, Reece EA. Activation of oxidative stress signaling that is implicated in apoptosis with a mouse model of diabetic embryopathy. Am J Obstet Gynecol. 2008;198:130, e1–7. doi: 10.1016/j.ajog.2007.06.070. [DOI] [PubMed] [Google Scholar]
- 16.Yang P, Zhao Z, Reece EA. Blockade of c-Jun N-terminal kinase activation abrogates hyperglycemia-induced yolk sac vasculopathy in vitro. Am J Obstet Gynecol. 2008;198:321, e1–7. doi: 10.1016/j.ajog.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 17.Cao Y, Zhao Z, Eckert RL, Reece EA. Protein kinase Cbeta2 inhibition reduces hyperglycemia-induced neural tube defects through suppression of a caspase 8-triggered apoptotic pathway. Am J Obstet Gynecol. 204:226, e1–5. doi: 10.1016/j.ajog.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reece EA, Ji I, Wu YK, Zhao Z. Characterization of differential gene expression profiles in diabetic embryopathy using DNA microarray analysis. Am J Obstet Gynecol. 2006;195:1075–80. doi: 10.1016/j.ajog.2006.05.054. [DOI] [PubMed] [Google Scholar]
- 19.Reece EA, Wu YK. Prevention of diabetic embryopathy in offspring of diabetic rats with use of a cocktail of deficient substrates and an antioxidant. Am J Obstet Gynecol. 1997;176:790–7. doi: 10.1016/s0002-9378(97)70602-1. discussion 797–8. [DOI] [PubMed] [Google Scholar]
- 20.Reece EA, Wu YK, Wiznitzer A, et al. Dietary polyunsaturated fatty acid prevents malformations in offspring of diabetic rats. Am J Obstet Gynecol. 1996;175:818–23. doi: 10.1016/s0002-9378(96)80005-6. [DOI] [PubMed] [Google Scholar]
- 21.Reece EA, Wu YK, Zhao Z, Dhanasekaran D. Dietary vitamin and lipid therapy rescues aberrant signaling and apoptosis and prevents hyperglycemia-induced diabetic embryopathy in rats. Am J Obstet Gynecol. 2006;194:580–5. doi: 10.1016/j.ajog.2005.08.052. [DOI] [PubMed] [Google Scholar]
- 22.Hagay ZJ, Weiss Y, Zusman I, et al. Prevention of diabetes-associated embryopathy by overexpression of the free radical scavenger copper zinc superoxide dismutase in transgenic mouse embryos. Am J Obstet Gynecol. 1995;173:1036–41. doi: 10.1016/0002-9378(95)91323-8. [DOI] [PubMed] [Google Scholar]
- 23.Horal M, Zhang Z, Stanton R, Virkamaki A, Loeken MR. Activation of the hexosamine pathway causes oxidative stress and abnormal embryo gene expression: involvement in diabetic teratogenesis. Birth Defects Res A Clin Mol Teratol. 2004;70:519–27. doi: 10.1002/bdra.20056. [DOI] [PubMed] [Google Scholar]
- 24.Li R, Chase M, Jung SK, Smith PJ, Loeken MR. Hypoxic stress in diabetic pregnancy contributes to impaired embryo gene expression and defective development by inducing oxidative stress. Am J Physiol Endocrinol Metab. 2005;289:E591–9. doi: 10.1152/ajpendo.00441.2004. [DOI] [PubMed] [Google Scholar]
- 25.Salbaum JM, Kappen C. Neural tube defect genes and maternal diabetes during pregnancy. Birth Defects Res A Clin Mol Teratol. 2010;88:601–11. doi: 10.1002/bdra.20680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sugimura Y, Murase T, Oyama K, et al. Prevention of neural tube defects by loss of function of inducible nitric oxide synthase in fetuses of a mouse model of streptozotocin-induced diabetes. Diabetologia. 2009;52:962–71. doi: 10.1007/s00125-009-1312-0. [DOI] [PubMed] [Google Scholar]
- 27.Yang P, Zhao Z, Reece EA. Involvement of c-Jun N-terminal kinases activation in diabetic embryopathy. Biochem Biophys Res Commun. 2007;357:749–54. doi: 10.1016/j.bbrc.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 28.Nath AK, Enciso J, Kuniyasu M, Hao XY, Madri JA, Pinter E. Nitric oxide modulates murine yolk sac vasculogenesis and rescues glucose induced vasculopathy. Development. 2004;131:2485–96. doi: 10.1242/dev.01131. [DOI] [PubMed] [Google Scholar]
- 29.Kamimoto Y, Sugiyama T, Kihira T, et al. Transgenic mice overproducing human thioredoxin-1, an antioxidative and anti-apoptotic protein, prevents diabetic embryopathy. Diabetologia. 2010;53:2046–55. doi: 10.1007/s00125-010-1784-y. [DOI] [PubMed] [Google Scholar]
- 30.Li X, Weng H, Reece EA, Yang P. SOD1 overexpression in vivo blocks hyperglycemia-induced specific PKC isoforms: substrate activation and consequent lipid peroxidation in diabetic embryopathy. Am J Obstet Gynecol. 205:84, e1–6. doi: 10.1016/j.ajog.2011.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Z, Yang P, Eckert RL, Reece EA. Caspase-8: a key role in the pathogenesis of diabetic embryopathy. Birth Defects Res B Dev Reprod Toxicol. 2009;86:72–7. doi: 10.1002/bdrb.20185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wentzel P, Thunberg L, Eriksson UJ. Teratogenic effect of diabetic serum is prevented by supplementation of superoxide dismutase and N-acetylcysteine in rat embryo culture. Diabetologia. 1997;40:7–14. doi: 10.1007/s001250050636. [DOI] [PubMed] [Google Scholar]
- 33.Conde-Agudelo A, Romero R, Kusanovic JP, Hassan SS. Supplementation with vitamins C and E during pregnancy for the prevention of preeclampsia and other adverse maternal and perinatal outcomes: a systematic review and metaanalysis. Am J Obstet Gynecol. 204:503, e1–12. doi: 10.1016/j.ajog.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Correa A, Gilboa SM, Botto LD, et al. Lack of periconceptional vitamins or supplements that contain folic acid and diabetes mellitus-associated birth defects. Am J Obstet Gynecol. doi: 10.1016/j.ajog.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gunatilake RP, Perlow JH. Obesity and pregnancy: clinical management of the obese gravida. Am J Obstet Gynecol. 204:106–19. doi: 10.1016/j.ajog.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 36.Catalano PM, Hauguel-De Mouzon S. Is it time to revisit the Pedersen hypothesis in the face of the obesity epidemic? Am J Obstet Gynecol. 204:479–87. doi: 10.1016/j.ajog.2010.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hedderson MM, Williams MA, Holt VL, Weiss NS, Ferrara A. Body mass index and weight gain prior to pregnancy and risk of gestational diabetes mellitus. Am J Obstet Gynecol. 2008;198:409, e1–7. doi: 10.1016/j.ajog.2007.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herring SJ, Oken E, Rifas-Shiman SL, et al. Weight gain in pregnancy and risk of maternal hyperglycemia. Am J Obstet Gynecol. 2009;201:61, e1–7. doi: 10.1016/j.ajog.2009.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hod M, Damm P, Kaaja R, et al. Fetal and perinatal outcomes in type 1 diabetes pregnancy: a randomized study comparing insulin aspart with human insulin in 322 subjects. Am J Obstet Gynecol. 2008;198:186, e1–7. doi: 10.1016/j.ajog.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 40.Dunlop AL, Jack BW, Bottalico JN, et al. The clinical content of preconception care: women with chronic medical conditions. Am J Obstet Gynecol. 2008;199:S310–27. doi: 10.1016/j.ajog.2008.08.031. [DOI] [PubMed] [Google Scholar]