

Abstract
The neuronal doctrine, developed a century ago regards neuronal networks as the sole substrate of higher brain function. Recent advances in glial physiology have promoted an alternative hypothesis, which places information processing in the brain into integrated neuronal-glial networks utilizing both binary (neuronal action potentials) and analogue (diffusional propagation of second messengers/metabolites through gap junctions or transmitters through the interstitial space) signal encoding. It has been proposed that the feed-forward and feed-back communication between these two types of neural cells, which underlies information transfer and processing, is accomplished by the release of neurotransmitters from neuronal terminals as well as from astroglial processes. Understanding of this subject, however, remains incomplete and important questions and controversies require resolution. Here we propose that the primary function of peri-synaptic glial processes is to create an “astroglial cradle” that shields the synapse from a multitude of extrasynaptic signaling events and provides for multifaceted support and long-term plasticity of synaptic contacts through variety of mechanisms, which may not necessarily involve the release of “glio”transmitters.
“What is the function of glial cells in neural centers? The answer is still not known, and the problem is even more serious because it may remain unsolved for many years to come until physiologists find direct methods to attack it”
It has been more than 20 years since the first evidence challenging the role of glia as passive bystanders was published. In 1990 Ann Cornell-Bell, Stephen Smith and others reported that glutamate triggers oscillatory and propagating Ca2+ waves in cultured astrocytes (Cornell-Bell et al. 1990). At first glance, this discovery may not seem particularly striking, because functional expression of glutamate receptors in neuroglia was already established (Bowman and Kimelberg 1984; Kettenmann et al. 1984). Yet the fact that astrocytes respond to a neurotransmitter with propagating intra- and intercellular signals suggested that astroglia are capable of sensing and possibly integrating synaptic transmission, and therefore can be involved in information processing in the brain. The notion that neurotransmitters can evoke signaling in non-excitable cells in the brain was a radical departure from the established view of how the brain functions. At the same time, this initial observation highlighted the fundamental differences between neuronal and glial signaling that occur in different temporal domains: the glial Ca2+ waves propagate at a speed of 4 - 20 □m/s, whereas the velocity of action potential-mediated conductance between neurons approaches 10 - 100 m/s (Hartline and Colman 2007).
Several years later, two studies added additional support to the emerging understanding of astrocytes’ role in higher brain processes. These reports demonstrated that in co-cultures, astrocytes can trigger calcium increases in adjacent neurons (Nedergaard 1994; Parpura et al. 1994). This was a fundamental finding because it indicated that the ‘housekeeper cells’ not only receive information from neurons and communicate it to their own kind, but they can also feed signals back to neuronal networks. These findings led naturally to the question of whether astrocytes are capable of modulating synaptic transmission. In 1998, four independent groups showed (a) that Schwann cells modulate synaptic efficacy in the neuromuscular junction (Robitaille 1998); (b) that glial cells in co-cultures can increase the frequency of miniature postsynaptic currents (Araque et al. 1998); (c) that glial cells in the retina modulate the activity of ganglion cells (Newman and Zahs 1998); and (d) that hippocampal astrocytes can increase the strength of inhibitory transmission through a Ca2+-dependent pathway (Kang et al. 1998). These four studies, in combination with the finding that astrocytes release glutamate in response to increases in cytosolic Ca2+ triggered by metabotropic receptor activation (Bezzi et al. 1998), was the basis for a novel model of synaptic function in which astrocytes modify synaptic activity by the Ca2+-dependent release of gliotransmitters. This idea caught on, and over the past twelve years, more than a hundred publications have reported that astrocytes indeed influence synaptic communication in many areas the of CNS. In 1996, these findings were synthesized into the concept of “synaptic triate” (Kettenmann et al. 1996), which later developed into ‘the tripartite synapse’, a model that suggests that signal integration and transduction by each individual synapse should be considered in terms of the pre- and post-synaptic terminals and adjacent perisynaptic astrocytic processes (Araque et al. 1999). The tripartite synapse concept assumes (Fig. 1) that activation of astroglia by neurotransmitters released from the presynaptic terminal triggers additional release of transmitters (usually glutamate and/or ATP) from astroglial compartment, which can directly participate in the synaptic event.
Figure 1. The tripartite synapse.

The model of the tripartite synapse is based on the observation that glutamate or GABA trigger Ca2+ increases in peri-synaptic glial processes in an mGlu1/5 receptor-dependent pathway (or by GABAB receptors in inhibitory synapses). In turn, the increase in astrocytic Ca2+ triggers vesicular gliotransmitters release, which modulates synaptic strength by binding to pre- and postsynaptic receptors. Six recent diagrams of the tripartite synapse illustrating that the tripartite synapse is viewed as the epicenter for neuron-glia signaling. A (Eroglu and Barres, 2010), B (Gordon and Bains 2006), C (Koizumi et al. 2005), D (Nadkarni et al. 2008) E (Allen and Barres 2009), F (Sotero and Martinez-Cancino 2010).
At present, the glial field is no longer avant-garde, and most textbooks provide a section on neuronal-glial signaling. Recently, however, several studies (Agulhon et al. 2010; Petravicz et al. 2008) have questioned the paradigm of astroglial transmitter release and modulation of synaptic transmission and plasticity, providing evidence to the contrary. Although this contrary evidence is open to counter-interpretations and leaves many unanswered questions, it adds to a growing number of voices who are concerned that non-physiological nature of many experiments investigating astrocytic function at synapses might have led to an inaccurate understanding of the way how the astrocytes operate. This topic has been reviewed recently in several excellent publications, some which are in favor of astrocytic transmitter release (Araque et al. 1999; Halassa and Haydon 2010; Volterra and Meldolesi 2005), or provide evidence against it (Agulhon et al. 2008), or are skeptical (Hamilton and Attwell, 2010). In this article, we suggest that the inability to reconcile recent observations in the field may result from misinterpretations arising from designing experiments based on the model of the tripartite synapse. The model does not take into account that structural and functional barrier erected by astroglial membranes around individual synapses as well as highly localized homeostatic interactions (e.g. providing metabolic support and transmitter clearance) are essential for ascertaining a high spatial specificity and local plasticity of synaptic transmission. We suggest that the role of astrocytes in information processing lies in global and tonic modulation of neural networks. Simultaneously astroglia keeps the spatial precision of synaptic transmission and provide local homeostatic support for individual synapses. We also contemplate a novel model of gliotransmitter-independent interactions in glial-neuronal networks that may stimulate further discussion and future studies.
Lack of selective experimental tools in glia research
The contradictory observations on the nature, mechanism and physiological relevance of gliotransmission might be partially explained by the use of neuronal function recordings (neuronal ‘readout’, such as an EPSC) as an indication of astrocytic function, combined with the difficulty in selectively perturbing astrocytes without simultaneously influencing the synapse. Neural readouts therefore provide an indirect measure of astrocytic actions and hence experimental findings are difficult to interpret conclusively. As an example of ambiguous experimental design, removal of Mg2+ from the bath solution does not only relieve the voltage-dependent block on NMDA receptors, but has multiple effects on astrocytes including the initiation of spontaneous Ca2+ signaling (Stout and Charles 2003) and inhibition of glutamine synthetase (Deuel and Stadtman 1970; Dowton and Kennedy 1985). Similarly, endothelin-1, PAR-1, P2Y1, and bradykinin receptors presumed to be specifically expressed on the perisynaptic astrocytic terminals, may also appear in other cell types, including subpopulations of neurons, endothelial cells and microglia. Another popular experimental paradigm uses fluoacetate to inhibit glycolysis in astrocytes. Although fluoroacetate primarily, albeit not exclusively, targets astrocytes, lack of metabolic support from astrocytes will affect synaptic transmission. Therefore fluoroacetate cannot be used as a direct tool to investigate astroglial-mediated transmission, since metabolically compromised neurons are less likely to respond to stimulation. Moreover, fluoacetate effectively blocks the glutamine-glutamate shuttle as conversion of glutamate to glutamine is an energy requiring process. Lack of glutamate will not only suppress glutamate accumulation in excitatory neurons, but also affect the inhibitory activity since GABA production is similarly glutamine-dependent (Ortinski et al., 2010). Likewise, dialyzing compounds such as BAPTA into the astrocytes not only occludes Ca2+ signaling, but also alters a multitude of intracellular Ca2+-dependent signaling/metabolic pathways that may affect synaptic transmission indirectly (see (Agulhon et al. 2008; Hamilton and Attwell, 2010) for a detailed update on experimental modulation of gliotransmission). In fact, simply touching an astrocyte with a patch pipette without breaking the plasma membrane is sufficient to evoke intracellular signaling events and consequent vasodilatation (Zonta et al. 2003). Several studies have employed depolarization pulses to stimulate gliotransmitter release (Bekar et al. 2008; Jourdain et al. 2007; Kang et al. 1998). However, the astroglial plasma membrane contains hemichannels, which open in response to depolarization thus resulting in a nonspecific leakage of cytosolic content, including glutamate and ATP from astrocytes; the latter two possibly triggering variety of indirect effects.
Currently, photolysis of caged Ca2+ or InsP3 loaded into astrocytes is the most specific method for selective stimulation of astroglia, but probably increases cytosolic Ca2+ to pathological levels and bypasses potentially crucial intracellular signaling pathways (i.e. G-protein related processes) that are activated following physiological receptor stimulation (Fiacco and McCarthy 2004; Liu et al. 2004). It is also important to consider the limitations of acute slice preparations for studying astroglia, as astrocytes are engineered to maintain an optimal environment at the expense of their own health. Astrocytes in acute slice preparation exhibit signs of reactive changes and loss of glycogen granula (Fiala et al. 2003). Astrocytic modulation of neural activity should necessarily always include adult animals, as it is well-known that glial cells during development initiate the first electrical activity by release of transmitters, including ATP and glutamate. For example, immature glial cells initiate the first electrical activity by release of ATP in the developing auditory system (Tritsch et al. 2007), and glutamate in the subventricular zone (Platel et al. 2010), but this activity ceases as mature synapses are established. All in all, it is not sufficient to base studies on neuroglia signaling on slices prepared from yoing pups only. in vivo confirmation in adult animals should be obtained whenever possible. Luckily, much improved tools for selective stimulation of astrocytes in the adult brain are underway. For example, optogenetic tools that stimulate relevant astrocytic signaling pathways such as opto-adrenoceptors composed of chimeras of rhodopsin and either α1 or β2 adrenoceptors (Airan et al. 2009). Chemogenetic tools such as, for example, Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) (Armbruster et al. 2007)) have the potential to transform the field. The selectivity of action of these experimental manipulations and the potential to activate large number of astrocytes in selected brain region bear great promise. One of the major advantages of these novel approaches is that behavioral assays can be included in the analysis. We predict that studies based on opto- and chemogenetics approaches may radically transform our view on astrocytic function and their role in neural circuit within just a few years.
When, where, and why do astrocytes release gliotransmitters?
The concept of the tripartite synapse originated from observations that astroglial Ca2+ signals produced in response to neural activity, activate the release of ‘gliotransmitters’ such as D-serine, ATP and glutamate that modulate synaptic transmission (Agulhon et al. 2008; Allen and Barres 2009; Araque et al. 1999; Hamilton and Attwell, 2010; Volterra and Meldolesi 2005) (Fig. 1). According to this model, neurotransmitters released from the neuronal terminal activate receptors on adjacent glial processes, which triggers a Ca2+ increase in astrocytes, and gliotransmitter release that in turn can modulate both pre- and post-synaptic neuronal compartments. The Ca2+-dependent release of glutamate from astroglial cells in vitro is well established, with several groups directly detecting glutamate release from cultured astrocytes in response to receptor stimulation, as reviewed in (Volterra and Meldolesi 2005). However, two important questions concerning the physiological significance of these observations remain: is gliotransmitter release regulated and spatially confined (as opposed to non-specific leakage of cytosolic compounds) and does it occur in vivo? One potential problem is that glutamate is a small compound (180 Da) that can permeate most anion channels. Any disturbances of membrane permeability or simply the opening of anion channels can result in efflux of cytosolic glutamate that acts as a transmitter once released. A key to demonstrating the specificity of Ca2+-mediated glutamate release is to provide evidence that glutamate is released in a controlled fashion in response to physiological stimulation. To our knowledge, this has not been tested directly, except in a single study reporting that receptor-mediated glutamate release was accompanied by the release of other amino acids, including aspartate, taurine, glycine, and D-serine from cultured astrocytes. The relative ratio of amino acids released matched that of the cytosolic compartment, suggesting that this event reflected non-specific leakage (Nedergaard et al. 2002; Takano et al. 2005)(Fig. 2).
Figure 2. Ca2+ signaling in cultured astrocytes trigger release of multiple amino acids, not only glutamate.

(A) Amino acids in the cytosol of cultured rat astrocytes. (B) Amino acid release in response to exposure to a hypotonic solution (214 mOsm). (C) Amino acid release in response to the purinergic receptor agonist ATP (100 μM). Glutamate is not released in isolation, but rather together with other amino acids present in the cytosol of astrocytes. Modified from (Takano et al. 2005).
What then is the evidence for regulated glutamate release from astrocytes in situ? In slices, glutamate release from astrocytes has been detected by monitoring neuronal slow inward currents (SICs) sensitive to ifenprodil, an antagonist of NR2B subunit-containing extrasynaptic NMDA receptors (Halassa and Haydon, 2010). Concurrent Ca2+-imaging has documented that the SIC is accompanied by an increase in cytosolic Ca2+ in groups of neurons consistent with the occurrence of a substantial release of glutamate (Fellin et al. 2004; Tian et al. 2005). However, it has been difficult to definitively link Ca2+ signaling in astrocytes to glutamate release, because Ca2+ increases in astrocytes evoked SICs inconsistently and with a delay of 1-20 sec. Most troublesome, however, has been the absence of a definitive demonstration that the glutamate that triggers SICs is released from astroglia. Although it is true that SICs are largely TTX-insensitive, this does not unequivocally identify astrocytes as the only source of glutamate, as other cellular elements can provide for non-synaptic glutamate release. For example, depolarization or stimulation of climbing fibers evokes somatodendritic release of glutamate from cerebellar Purkinje cells (Duguid et al. 2007; Shin et al. 2008a). Activation of excitatory autoreceptors by dendritic release of glutamate has also been demonstrated in mitral cells of the rat olfactory bulb (Salin et al. 2001), and TTX-resistant SICs in hippocampal slices treated with high concentrations of 4-AP and TEA have been ascribed to glutamate release from axons of CA3 pyramidal neurons (Strowbridge 1999).. The similarity between the SICs in these studies suggests that astrocytes are not the only source of non-synaptic glutamate release. In fact, due to the expression of glutamine synthetase, the cytosolic concentration of glutamate is lower in astrocytes (~2 mM) than in neurons (~10 mM) (Nedergaard et al. 2002).
Pharmacological tools have provided indirect evidence for astrocytic glutamate release. In these studies, glutamate release from astrocytes has been detected by comparing the effect of astrocytic stimulation on synaptic function in the absence and in the presence of Group I mGlu receptor antagonists (Fiacco and McCarthy 2004; Navarrete and Araque, 2010). Group I mGluRs agonists, however, are known to potentiate excitatory transmission by increasing neurotransmitter release, enhancing AMPA/NMDA receptor responses, and modulating several types of voltage-gated channels (Anwyl 1999). Conversely, group 1 mGluR inhibitors reduce baseline neuronal responsiveness to stimulation (Anwyl 1999). Therefore, evidence for astrocytic glutamate release based on comparing astrocytic-mediated modulation in presence and absence of mGluR antagonists is not definitive, since these agents broadly suppress excitatory transmission. In other words, the lack of changes in synaptic transmission in response to astrocytic activation in the presence of mGluR antagonist may reflect that the synapse is less responsive to any input, rather than astroglia-specific modulation of synaptic activity by release of glutamate. Overall, neither of these studies provided definitive proof for astrocytic glutamate release.
Conversely, two types of experiments have recently claimed to provide direct evidence against the notion of astroglia-dependent modulation of synaptic transmission. First, it was demonstrated that receptor-mediated increases in astrocytic Ca2+ do not affect synaptic input to hippocampal CA1 neurons. This study employed a transgenic mouse in which the expression of a Gq-coupled receptor (MrgA1) was driven by an inducible promoter (Fiacco et al. 2007). Native MrgA1 receptors are not expressed in the brain and is activation induced robust increases in cytosolic Ca2+ in transgenic astrocytes, which was designed to mimic the physiological activation of astrocytes by neuronally released glutamate. The key observation was that Ca2+ signaling in astrocytes did not affect the frequency or amplitude of neuronal EPSPs. A second set of studies used InsP3R2 knock out mice, in which astrocytes were unable to mobilize Ca2+ from intracellular stores (InsP3R2 are the only InsP3 receptor expressed by astrocytes) (Petravicz et al. 2008). Despite the lack of astroglial Ca2+ signaling, both short- and long-term synaptic plasticity appeared normal, suggesting that astrocytes do not play a significant role in these processes (Agulhon et al. 2010).
This finding was unexpected and led to a debate on how essential astrocytes are for synaptic transmission and plasticity. The strength of McCarthy and co-workers analysis was that these studies employed selective agonist-stimulated Ca2+ signaling in astrocytes to show that receptor activated Ca2+ signaling in astrocytes is not sufficient to induce gliotransmitter release. The observation naturally led to a re-evaluation of past studies and called attention to the non-physiological nature of many of the protocols. However, as with previous studies, these more recent findings also have limitations. Specifically, Ca2+ increases driven by MrgA1 receptors may not engage the intracellular pathways involved in gliotransmitter release or initiate other mechanism(s) by which astrocytes may modulate neuronal activity. The non-polarized expression of MrgA1 receptors in combination with a lack of downstream effectors may prevent their effective coupling to appropriate molecular pathways and physiological responses. Likewise, mobilization of cytosolic Ca2+ stores may not be the only pathway by which astrocytes increase cytosolic Ca2+ and compensation during development may lessen the consequences of InsP3 receptor deletion; a developmental adaptation cannot be ruled out until conditional knockouts are produced. Nevertheless, these results cast doubt on the importance of gliotransmitter modulation of synaptic function, or at least suggest that a certain care is required when interpreting data on astrocytic modulation of synaptic transmission via gliotransmitter release.
However, a serious problem is that the experimental design of these and other studies is based on the model of tripartite synapse for interactions between astrocytes and neurons. The concept of the tripartite synapse assumes that perisynaptic glia processes dynamically feedback to modulate ongoing synaptic transmission and aim at detecting rapid changes in the efficacy of transfer of signal across a single synapse. This approach ignores that the primary role of astrocytes is to isolate the individual synapse and that their modulatory roles likely is more diffuse and directed at multisynaptic neuronal networks (see later). In fact, multiple lines of evidence suggest that withdrawal of peri-synaptic astrocytes processes lead to increased glutamatergic transmission, and that the primary function of astrocytes thereby is to control synaptic transmission by clearing extracellular glutamate rather than to modulate synaptic transmission through glutamate release.
Astrocytic shielding of the synapse
If the glial part of the tripartite synapse is not at the core of synaptic transmission and plasticity, what about the other reported roles of astrocytes such as neurotransmitter clearance and control of ambient glutamate? What are the implications for the tripartite synapse hypothesis and the roles for astrocytes in general?
The traditional view of astrocytic processes is that they provide a structure that shields the synapse from neurotransmitter interference from nearby synapses. We believe this important role needs to be incorporated into a new unifying concept that can accommodate all available data.
An important issue to reconcile is the apparent paradox that astrocytes appear to be able to both release glutamate and also participate in its rapid removal from the synaptic cleft. Indeed, multiple studies indicate that withdrawal of perisynaptic glia processes does not result in a decrease in glutamatergic signaling as would be expected if astrocytes released glutamate. On the contrary, reduction in synaptic glial coverage prolongs EPSPs in the cerebellum (Iino et al. 2001), increases activation of mGlu receptors in the supraoptic nucleus (Oliet et al. 2001; Piet et al. 2004), and results in synaptic spillover of glutamate during simulation of a typical CA1 hippocampal synapse (Rusakov 2001). Thus, in the context of the core process of pre-to-post synaptic transmission, the primary function of perisynaptic glial processes appears to be uptake, rather than release of glutamate.
It is also clear that glutamate released from astrocytes or other extrasynaptic sources do not have access to the postsynaptic density and does not trigger action potentials or even AMPA receptor-mediated EPSPs. Slow inward currents trigger a prolonged depolarization of neurons (0.3 -1.0 sec) that in many aspects resembles the membrane depolarization observed in response to the raising extracellular K+ concentration. Several studies of cortical and hippocampal excitatory synapses have documented that slow inward currents activate extrasynaptic NR2b-containing NMDA receptors (Jourdain et al. 2007; Kato et al. 2006; Volterra and Meldolesi 2005). This observation is important, because it implies that glutamate released by astrocytes is not targeted to specific synapses, but dissipates and induces non-specific activation of nearby cells. Most strikingly, despite the intense interest in the concept of gliotransmission for more than a decade, we are not aware that any data have been collected in adult intact animals that support a physiological role of glutamate release from astrocytes. A transgenic mouse was developed 7 years ago to study the role of astrocytic glutamate release in synaptic plasticity. In this mouse expression of a dominant-negative mutation of synaptobrevin 2 (dnSNARE) interferes with the exocytosis in GFAP expressing astrocytes (Pascual et al. 2005). The dnSNARE mice were generated based on the in vitro observations that formation of SNARE complexes was required for astrocytic glutamate release (Zhang et al. 2004). Despite being available for years, the dnSNARE mice have so far not been utilized for studies on exocytotic glutamate release from astrocytes. We conclude that astrocytic glutamate release only definitively has been demonstrated in cultures, and thus far has only be proven to play a role in synaptic plasticity using non-physiological stimulation paradigms or under pathological conditions.
Astrocytic regulation of trans-synaptic communication
Based on the discussion above, we propose that the tripartite synapse is not a centre for neuroglial signaling, but rather that perisynaptic glial processes provide a structure that shields the synapse from interference from nearby synapses and other extrasynaptic signaling events, thus creating an astroglial cradle (Fig. 4). The idea of neuroglia as an insulator in the nervous system is very old and was already in vogue at the end of 19th century (Santiago Ramon-y-Cajal credited this idea to his brother Pedro - Ramon-y-Cajal, 1909/1911) (Cajal 1909; Cajal and Ramon 1995). Astrocytic sheaths provide a degree of physical separation between synapses and the insulator function seems to represent a generic function of neuroglia at large including myelinating oligodendrocytes. Perhaps more importantly, however, astroglial perisynaptic membranes additionally act as a dynamic functional barrier by employing active uptake of transmitters. This astroglial cradle may reduce both “spill-in” of transmitters released during extrasynaptic signaling events and “spill-out” of transmitters from the cleft, thus effectively isolating the single synapse from the rest of CNS. This allows the co-existence of spatially and temporally precise synaptic transmission with a multitude of slower and less specific extrasynaptic signaling events. This latter point reflects an important critique of the tripartite synapse model related to the general assumption that the synapse is the center for all signaling in CNS. While synaptic transmission certainly is responsible for information transfer in neuronal networks, a multitude of signaling occurs outside the synapses among the many other cell types comprising the CNS, utilizing transmitters including ATP, PGE2, and BDNF that can all potentially interfere with synaptic transmission (Coull et al. 2005). Thus, a key function of peri-synaptic astrocytic processes is to prevent “spill-in” of transmitters release by non-neuronal cells, as for example microglial cells (Noda et al. 1999) that directly may interfere with the precision of synaptic transmission.
Figure 4. Morphological plasticity of astrocytes.
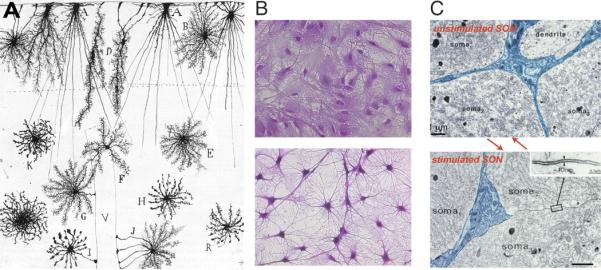
(A) Golgi-stained astrocytes from a 2 month-old human infant in cortical layers I-III. A, B, C, and D are cells in layer I, whereas E,F,G and H are astrocytes in layer II and II. I and J are cells with endfeet contacting blood vessels. V, blood vessel. (Ramón y Cajal, 1913). (B) Cultured rat astrocytes before (top panel) and after addition of dBcAMP (1 μM, lower panel), cresyl violet. Courtesy of Drs. Fujita and Abe, Hoshi University. (C) Electron microscopy of the rat SON during parturition (top panel) and lactation (lower panel). Astrocytes withdraw their processes in lactating animals. From (Theodosis et al. 2008).
More importantly, Ca2+-regulated gliotransmitter release, may not necessarily represent the main pathway for astroglia-dependent regulation of synaptic transmission. Astrocytes are in a possession of ionotropic receptors (glutamate and P2X receptors) that mediate rapid influx of Na+ and Ca2+ (Lalo et al. 2006; Lalo et al. 2008). The functional importance of these glial receptors is necessarily distinct from those of neurons, because the astrocytic plasma membrane is electrically non-excitable (Verkhratsky and Kettenmann, 2010). In all probability, the primary function of astroglial ionotropic receptors is to generate local increases in [Na+]i and [Ca2+]i in perisynaptic processes, which in multiply ways enable astrocyte to both control and support synaptic transmission. For example, an increase in [Na+]i contributes to fast Ca2+ signals in perisynaptic astrocytic processes though reverse activation of the Na+/Ca2+ exchanger (NCX) (Figure 3) (Kirischuk et al. 1997). The decrease in the transmembrane Na+ gradient is also expected to reduce the efficacy of astroglia glutamate transport and thereby prolong the duration of EPSP (Lalo et al. 2010). Perhaps, the most important consequence of an increase in astrocytic [Na+]i is stimulation of the Na+/K+ ATPase. [Na+]i is the rate-limiting step for the Na+/K+ ATPase in many cell types, including astrocytes (Lupfert et al. 2001; Pellerin and Magistretti 1997). Thus, ionotropic receptor-mediated Na+ influx (similarly to Na+ influx generated by glutamate transporter) in astrocytes activates the Na+/K+ ATPase activity. Since the Na+/K+ ATPase exchanges 3 intracellular Na+ ions for 2 extracellular K+ ions, the net result is decline in extracellular K+. Novel data suggest that astrocytic Ca2+ signaling may also directly potentiate the activity of the Na+/K+ ATPase, in addition to the increase triggered by channel-mediated Na+ influx. Since extracellular K+ is a key determinant of neuronal membrane potential, even minor decreases in extracellular K+ concentration may suppress the excitability by increasing the gap between resting membrane potential and the threshold for activation of voltage-gated Na+ channels. Thus, the ability of astrocytes to lower extracellular K+ provides an elegantly simple mechanism for rapid astrocytic modulation of synaptic transmission. Another important consequence of the increase in the Na+/K+ ATPase pump activity is that astrocytes will generate lactate to fuel active synapses (Pellerin and Magistretti 1997). In this regard it is important to note that the astrocytic Na+/K+ ATPase is exceedingly sensitive to minor increases in extracellular K+ and that K+ increases associated with synaptic activity leads to an obligatory rise in lactate (Ransom et al. 2000; Rose and Ransom 1996). Further discussion of this important topic is beyond the scope of this review. However, it is important to acknowledge that multiple potential routes of gliotransmitter-independent neuroglia signaling exist and that activation of the astrocytic Na+/K+ ATPase will provide metabolic support synaptic transmission by release of lactate (Adachi et al. 1995). In summary, the astroglial cradle may not only isolate synapses, but also control synaptic strength by dynamic control of extracellular K+, and at the same time assist with local delivery of energy substrate to active synapses.
Figure 3. The astroglial cradle.
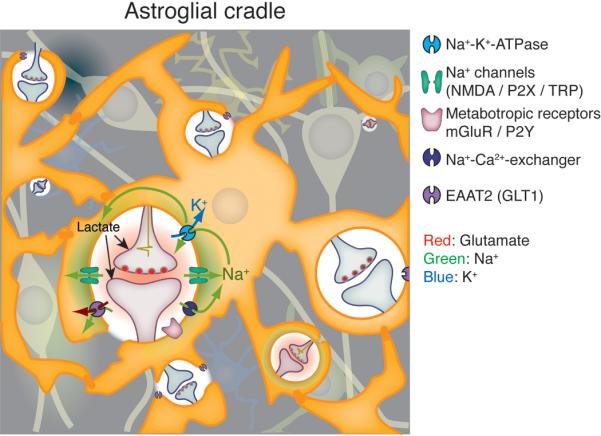
In the “astroglial cradle” model, the main function of peri-synaptic glial processes is to isolate and support the function of individual synapses, thus ensuring the spatial specificity of synaptic transmissions. The physical isolation of synapses first noted by Ramon y Cajal is complemented by transporters (GLT1 and other), which together provide a functional barrier isolating the synapse from signaling events in the extrasynaptic space. The astroglial membranes contain several types of ionotropic receptors, including NMDA and P2X receptors located close to the sites of neurotransmitter release from presynaptic terminals (Lalo et al. 2006; Lalo et al. 2008). Although these receptors are functionally similar to those expressed by neurons, ionotropic receptors assume a different function in the electrically non-excitable membranes of astrocytes (indeed, the amplitudes of synaptically induced ionotropic receptor-mediated currents measured from astrocytes rarely exceed 50 pA, whereas typical input resistance of astrocytes in situ is ~ 50 mOhm (Lalo et al. 2006); the resulting depolarization cannot, therefore, exceed 2 - 3 mV). Activation of astrocytic NMDA and P2X receptors generate inward cationic (mainly carried by Na+) currents, which rapidly increase cytosolic Na+ concentration in perisynaptic astrocytic processes. The transient increase of [Na+]I, has several functional consequences: (1) The increase in [Na+]i forces the reverse mode of Na+/Ca2+ exchanger (NCX; the latter is strategically co-localized with glutamate transporters in perisynaptic astroglial processes) (Minelli et al. 2007), which produces rapid local Ca2+ increases in the perisynaptic processes of active synapses. (2) The decrease in transmembrane Na+ gradient transiently reduces the efficacy of Na+-dependent glutamate transport, thereby slowing down elimination of synaptically released glutamate. (Kirischuk et al. 1997). (3) The excess of [Na+]i activates Na+/K+ ATPase, which in turn decreases extracellular K+ and enhances lactate production. The latter provides focal metabolic support to active synapses (Pellerin et al. 2007). In addition, elevation in [Ca2+]i (due to activation of metabotropic receptros with subsequent Ca2+ mobilization from the stores or due to plasmalemmal Ca2+ entry) directly enhances the activity of the Na+/K+ ATPase providing a longer lasting decrease in extracellular K+ and further maintenance of lactate shuttle. Combined activity of Na+/K+ pump and NCX eventually lower [Na+]i thus restoring glutamate accumulation.
In summary, activation of ionotropic receptors ion transporters and exchngers in astroglia membranes initiates fast neuronal-glial-neuronal interactions (msec) at the level of the individual synapse, whereas Ca2+ signaling provides for longer lasting modulation (seconds). Combined, the signaling events evoked by activation of astrocytic ionotropic and metabotropic receptors enable astrocytes to modulate synaptic transmission on multiple levels, including control of the peak amplitude and duration of glutamate increase, extracellular K+ concentration, and energy substrate delivery (lactate) employing pathways that do not involve gliotransmitter release.
Towards a new understanding of what astrocytes do - and what they do not do
Although all considerations discussed above on gliotransmitter release constrain our interpretations of studies probing the mechanisms and significance of gliotransmission, it is encouraging that in vivo experiments demonstrate that astrocytes participate in essentially all CNS functions studied thus far. For example, astrocytes display complex patterns of Ca2+ signals in mice during motor activity (Dombeck et al. 2007; Hoogland et al. 2009; Nimmerjahn et al. 2009), and cortical astrocytes are activated during sensory stimulation (Schummers et al. 2008; Wang et al. 2006). In order to understand why our current concepts of neuroglial signaling lead to such contradictory data, it is worth considering the basic evolutionary relationship between astrocytes and neurons. Neurons and neuroglia share common phylogenetic ancestry, both being derived from epithelial cells endowed with secretory apparatus, plasmalemmal ion channels and intercellular gap junctions (Reichenbach and Pannicke 2008). The diffuse nervous system that appeared in primitive multicellular organisms favored electrical signaling, which offered rapid reaction times with obvious evolutionary benefits. At later evolutionary stages, the appearance of neuronal conglomerates prompted the development of supportive cells, which provided for metabolic support and acted as cellular isolators. These proto-astrocytes assisted neurons in their functional activity (Bacaj et al. 2008). Subsequent evolution of the nervous system resulted in the specialization of these cellular elements: neurons perfected electrical excitability and fast synaptic transmission whereas astroglia assumed responsibility for nervous tissue homeostasis, morphological organization, metabolic support and defense. Conceptually astrocytes are engineered to analyze their surroundings and correct for any changes that may affect brain homeostasis and neurotransmission, typically utilizing several alternative mechanistic strategies to accomplish these tasks. Anyone who has worked with astrocytes in culture knows how these cells can rapidly alter their shape, size and functional phenotype. This phenomenon was initially described as a response to an increase in intracellular cAMP, which triggered rapid stellation and shrinkage of astrocytes in cultures (Chiu and Goldman 1985; Goldman and Abramson 1990) (Fig. 4). In the hypothalamus, astrocytes withdraw their processes in response to lactation, dehydration or oxytocin, significantly reducing the astroglial coverage of nearby neurons (Theodosis et al. 2008). In cerebellum, removal of the GluR2 subunit from Bergmann glial cells leads to retraction of their perisynaptic processes (Iino et al. 2001). In the hippocampus, LTP is accompanied by astroglial remodeling involving rapid extension and retraction of filopodia and lamellopodia-like protrusions and processes (Haber et al. 2006; Hirrlinger et al. 2004). The astroglial capacity to rapidly change shape and adjust their volume is provided by volume regulated channels that respond to changes in the environment by releasing osmolytes, including glutamate, aspartate and taurine (Kimelberg et al. 1990). Changes in astroglial cell volume are also triggered by receptor stimulation and are, at least in part, responsible for glutamate release from cultured astrocytes subjected to appropriate agonists (Lee et al. 2007; Takano et al. 2005) (Fig. 1). In addition, astrocytes demonstrate a remarkable functional plasticity. For example astroglial K+ uptake is mediated by at least 3 competing mechanisms, including KIR channels, a variety of K+/Na+/Cl- and K+/Cl- co-transporters, and the Na+/K+ ATPase. At least two of these pathways must be blocked to reduce astrocytic K+ uptake, demonstrating that astrocytes are capable of fulfilling their task in maintaining ion homeostasis using interchangeable mechanisms (D'Ambrosio et al. 2002; Walz 2000). Astrocytes are also metabolically opportunistic cells endowed with multiple enzymatic pathways allowing them to adjust energy metabolism to substrate availability. Astrocytes produce glutamate for synaptic transmission, but can also use glutamate as a metabolic substrate when needed. Likewise, astrocytes have the ability to generate large quantities of lactate, which can be oxidized as an energy source (McKenna et al. 2006). Considering the structural and functional plasticity of astrocytes, it is not surprising that contradictory data have accumulated over the years. Astrocytes are so sensitive to minor differences in experimental approaches that their functional responses likely varies from lab to lab. The most important practical consequence of the shared heritage of neurons and astrocytes is that no experimental manipulation can be assumed to solely affect one cell type without impinging upon the function of the other.
As far as glial signaling is concerned, we assume that Ca2+ waves are important may be only because we are able to visualize them. At the same time, it is clear that the astroglial syncytia may facilitate alternative and perhaps more sophisticated signaling pathways than currently recognized. Second, and probably even more important, the vast majority of experimental approaches currently in use involve the recording of neuronal electrical signals as readout for a glial function. The evidence collected so far seems to indicate that astroglia do not directly participate in synaptic transmission or in the rapid modulation of synaptic function. On the contrary, the most important roles of astrocytes likely relate to longer-lasting and more generalized integrative processes, which are simply too slow, too diffuse, and too subtle to be measured with conventional electrophysiological techniques. All in all, the paradigm that identifies Ca2+-induced gliotransmitter release as an ultimate mechanism for rapid neuronal-glial communications appears oversimplified, since existing evidence shows that astrocytic Ca2+ signaling can cause either inhibition, potentiation or have no effect on both excitatory and inhibitory transmission (Agulhon et al. 2010; Araque et al. 1999; Kang et al. 1998; Navarrete and Araque ; Pascual et al. 2005). The fact that experimental work of recent decades has failed to identify a common pathway by which astroglial Ca2+ signaling affects synaptic transmission suggests that more complex mechanisms are involved. The neuronal-glial network likely utilizes multiple intercellular signaling pathways, of which Ca2+-regulated gliotransmission may prove to be neither the sole nor even the most important mechanism.
Thus, in our efforts to seek a fundamental understanding of glial function, it is critical to abandon the neurocentric experimental designs and define new approaches that generate glial rather than neuronal readouts. Sadly, this remains the major limitation of the field. After 20 years of studying gliotransmission, we have not yet defined measures of astrocytic function, except for simple assays based on glutamate and K+ uptake, or gliotransmitter release in vitro. This lack of knowledge regarding the basic tasks and signaling capacities of astrocytes prevents us from meaningfully defining their roles in complex brain functions. It is also important to acknowledge that a given input will not necessarily result in the same output in every time, since astrocytes are plastic supportive cells that dynamically change their intracellular concentration of glutamate and their degree of synaptic coverage. The role of astrocytes in higher brain function is likely not executed by local exocytotic release of gliotransmitters that modulate the function of a few synapses within a short time scale. Rather, the role of astrocytes is to isolate synapses locally, while on a more global scale exert tonic and long-lasting modulation of neural network. Experiments based on the concept of the tripartite model as outlined in a recent review (Hamilton and Attwell 2010), which are aimed at a detailed analysis of exocytotic release of gliotransmitters and their role in rapid modulation of fast synaptic events are likely to produce inconsistent data that reflect pathophysiological alterations in astrocytes rather than reveal biologically relevant responses.
Brain function is based on connections and networking; the number and plasticity of connections being crucial for the development of intellect and cognition. Neurons and glia are equally important for casting neural circuitry; their functions are congruent and complementary. Do astrocytes participate in higher brain functions? The answer is certainly yes because without them, executive neuronal networks would not operate. Do astrocytes actively participate in fast information transfer? Most certainly not, because their function lies in a different temporal domain. Are astrocytes involved in information processing? This remains a central question of neuroscience; the astroglial involvement in the generation of thoughts, in memory and learning can be, at present, neither confirmed nor denied. To solve this problem we need to move from the neuronal doctrine to a more inclusive framework of brain function, integrating neuronal-glial circuitry as a whole. Furthermore we must expand this framework to accommodate the many intercellular signaling mechanisms that coexist with and complement neuronal action potentials and chemical synaptic transmission.
Acknowledgements
We thank Takahiro Takano for discussion and graphics and Mary McKenna and Jeffrey Iliff for discussions and comments on the manuscript. Supported by G. Harold and Leila Y. Mathers Charitable Foundation and National Institute of Health
References
- Adachi K, Cruz NF, Sokoloff L, Dienel GA. Labeling of metabolic pools by [6-14C]glucose during K+-induced stimulation of glucose utilization in rat brain. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 1995;15:97–110. doi: 10.1038/jcbfm.1995.11. [DOI] [PubMed] [Google Scholar]
- Agulhon C, Fiacco TA, McCarthy KD. Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science. 2010;327:1250–4. doi: 10.1126/science.1184821. [DOI] [PubMed] [Google Scholar]
- Agulhon C, Petravicz J, McMullen AB, Sweger EJ, Minton SK, Taves SR, Casper KB, Fiacco TA, McCarthy KD. What is the role of astrocyte calcium in neurophysiology? Neuron. 2008;59:932–46. doi: 10.1016/j.neuron.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Airan RD, Thompson KR, Fenno LE, Bernstein H, Deisseroth K. Temporally precise in vivo control of intracellular signalling. Nature. 2009;458:1025–9. doi: 10.1038/nature07926. [DOI] [PubMed] [Google Scholar]
- Allen NJ, Barres BA. Neuroscience: Glia - more than just brain glue. Nature. 2009;457:675–7. doi: 10.1038/457675a. [DOI] [PubMed] [Google Scholar]
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–15. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- Araque A, Sanzgiri RP, Parpura V, Haydon PG. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci. 1998;18:6822–9. doi: 10.1523/JNEUROSCI.18-17-06822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl Acad Sci U S A. 2007;104:5163–8. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacaj T, Tevlin M, Lu Y, Shaham S. Glia are essential for sensory organ function in C. elegans. Science. 2008;322:744–7. doi: 10.1126/science.1163074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekar L, Libionka W, Tian GF, Xu Q, Torres A, Wang X, Lovatt D, Williams E, Takano T, Schnermann J. Adenosine is crucial for deep brain stimulation-mediated attenuation of tremor. Nat Med. 2008;14:75–80. doi: 10.1038/nm1693. others. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–5. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- Bowman CL, Kimelberg HK. Excitatory amino acids directly depolarize rat brain astrocytes in primary culture. Nature. 1984;311:656–9. doi: 10.1038/311656a0. [DOI] [PubMed] [Google Scholar]
- Chiu FC, Goldman JE. Regulation of glial fibrillary acidic protein (GFAP) expression in CNS development and in pathological states. J Neuroimmunol. 1985;8:283–92. doi: 10.1016/s0165-5728(85)80067-9. [DOI] [PubMed] [Google Scholar]
- Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 1990;247:470–3. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–21. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- D'Ambrosio R, Gordon DS, Winn HR. Differential role of KIR channel and Na(+)/K(+)-pump in the regulation of extracellular K+ in rat hippocampus. J Neurophysiol. 2002;87:87–102. doi: 10.1152/jn.00240.2001. [DOI] [PubMed] [Google Scholar]
- Deuel TF, Stadtman ER. Some kinetic properties of Bacillus subtilis glutamine synthetase. J Biol Chem. 1970;245:5206–13. [PubMed] [Google Scholar]
- Dombeck DA, Khabbaz AN, Collman F, Adelman TL, Tank DW. Imaging large-scale neural activity with cellular resolution in awake, mobile mice. Neuron. 2007;56:43–57. doi: 10.1016/j.neuron.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowton M, Kennedy I. Purification and properties of glutamaine synthetase from fleshfly muscle. Insect Biochem. 1985;15:763–770. [Google Scholar]
- Duguid IC, Pankratov Y, Moss GW, Smart TG. Somatodendritic release of glutamate regulates synaptic inhibition in cerebellar Purkinje cells via autocrine mGluR1 activation. J Neurosci. 2007;27:12464–74. doi: 10.1523/JNEUROSCI.0178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–31. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43:729–43. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, Taves SR, Petravicz J, Casper KB, Dong X, Chen J, McCarthy KD. Selective stimulation of astrocyte calcium in situ does not affect neuronal excitatory synaptic activity. Neuron. 2007;54:611–26. doi: 10.1016/j.neuron.2007.04.032. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J Neurosci. 2004;24:722–32. doi: 10.1523/JNEUROSCI.2859-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC, Kirov SA, Feinberg MD, Petrak LJ, George P, Goddard CA, Harris KM. Timing of neuronal and glial ultrastructure disruption during brain slice preparation and recovery in vitro. J Comp Neurol. 2003;465:90–103. doi: 10.1002/cne.10825. [DOI] [PubMed] [Google Scholar]
- Goldman JE, Abramson B. Cyclic AMP-induced shape changes of astrocytes are accompanied by rapid depolymerization of actin. Brain Res. 1990;528:189–96. doi: 10.1016/0006-8993(90)91657-3. [DOI] [PubMed] [Google Scholar]
- Gordon GR, Bains JS. Can homeostatic circuits learn and remember? J Physiol. 2006;576:341–7. doi: 10.1113/jphysiol.2006.110270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber M, Zhou L, Murai KK. Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J Neurosci. 2006;26:8881–91. doi: 10.1523/JNEUROSCI.1302-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol. 2010;72:335–55. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton NB, Attwell D. Do astrocytes really exocytose neurotransmitters? Nat Rev Neurosci. 2010;11:227–38. doi: 10.1038/nrn2803. [DOI] [PubMed] [Google Scholar]
- Hartline DK, Colman DR. Rapid conduction and the evolution of giant axons and myelinated fibers. Curr Biol. 2007;17:R29–35. doi: 10.1016/j.cub.2006.11.042. [DOI] [PubMed] [Google Scholar]
- Hirrlinger J, Hulsmann S, Kirchhoff F. Astroglial processes show spontaneous motility at active synaptic terminals in situ. Eur J Neurosci. 2004;20:2235–9. doi: 10.1111/j.1460-9568.2004.03689.x. [DOI] [PubMed] [Google Scholar]
- Hoogland TM, Kuhn B, Gobel W, Huang W, Nakai J, Helmchen F, Flint J, Wang SS. Radially expanding transglial calcium waves in the intact cerebellum. Proc Natl Acad Sci U S A. 2009;106:3496–501. doi: 10.1073/pnas.0809269106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino M, Goto K, Kakegawa W, Okado H, Sudo M, Ishiuchi S, Miwa A, Takayasu Y, Saito I, Tsuzuki K. Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science. 2001;292:926–9. doi: 10.1126/science.1058827. others. [DOI] [PubMed] [Google Scholar]
- Jourdain P, Bergersen LH, Bhaukaurally K, Bezzi P, Santello M, Domercq M, Matute C, Tonello F, Gundersen V, Volterra A. Glutamate exocytosis from astrocytes controls synaptic strength. Nat Neurosci. 2007;10:331–9. doi: 10.1038/nn1849. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman S, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–92. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Kang N, Xu J, Xu Q, Nedergaard M, Kang J. Astrocytic glutamate release-induced transient depolarization and epileptiform discharges in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2005;94:4121–30. doi: 10.1152/jn.00448.2005. [DOI] [PubMed] [Google Scholar]
- Kato H, Narita M, Miyatake M, Yajima Y, Suzuki T. Role of neuronal NR2B subunit-containing NMDA receptor-mediated Ca2+ influx and astrocytic activation in cultured mouse cortical neurons and astrocytes. Synapse. 2006;59:10–7. doi: 10.1002/syn.20213. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Backus KH, Schachner M. Aspartate, glutamate and gamma-aminobutyric acid depolarize cultured astrocytes. Neurosci Lett. 1984;52:25–9. doi: 10.1016/0304-3940(84)90345-8. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Faissner A, Trotter J. Neuron-Glia Interactions in Homeostasis and Degeneration. In: Greger R, Windhort U, editors. Comprehensive Human Physiology. Springer Verlag; Berlin Heidelberg: 1996. pp. 533–543. [Google Scholar]
- Kimelberg H, Goderie S, Higman S, Pang S, Waniewski R. Swelling-induced release of glutamate, aspartate, and taurine from astrocytic cultures. J Neurosci. 1990;10:1583–1591. doi: 10.1523/JNEUROSCI.10-05-01583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirischuk S, Kettenmann H, Verkhratsky A. Na+/Ca2+ exchanger modulates kainate-triggered Ca2+ signaling in Bergmann glial cells in situ. FASEB J. 1997;11:566–72. doi: 10.1096/fasebj.11.7.9212080. [DOI] [PubMed] [Google Scholar]
- Koizumi S, Fujishita K, Inoue K. Regulation of cell-to-cell communication mediated by astrocytic ATP in the CNS. Purinergic Signal. 2005;1:211–7. doi: 10.1007/s11302-005-6321-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalo U, Pankratov Y, Kirchhoff F, North RA, Verkhratsky A. NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J Neurosci. 2006;26:2673–83. doi: 10.1523/JNEUROSCI.4689-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalo U, Pankratov Y, Wichert SP, Rossner MJ, North RA, Kirchhoff F, Verkhratsky A. P2X1 and P2X5 subunits form the functional P2X receptor in mouse cortical astrocytes. J Neurosci. 2008;28:5473–80. doi: 10.1523/JNEUROSCI.1149-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CJ, Mannaioni G, Yuan H, Woo DH, Gingrich MB, Traynelis SF. Astrocytic control of synaptic NMDA receptors. J Physiol. 2007;581:1057–81. doi: 10.1113/jphysiol.2007.130377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QS, Xu Q, Arcuino G, Kang J, Nedergaard M. Astrocyte-mediated activation of neuronal kainate receptors. Proc Natl Acad Sci U S A. 2004;101:3172–7. doi: 10.1073/pnas.0306731101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupfert C, Grell E, Pintschovius V, Apell HJ, Cornelius F, Clarke RJ. Rate limitation of the Na+,K+-ATPase pump cycle. Biophys J. 2001;81:2069–81. doi: 10.1016/S0006-3495(01)75856-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna M, Gruetter G, Sonnewald U, Waagepetersen H, Schousboe A. Energy Metabolism of the Brain. Basic Neurochemistry: Molecular, Cellular and Medical Aspects Elsevier. 2006:531–558. [Google Scholar]
- Minelli A, Castaldo P, Gobbi P, Salucci S, Magi S, Amoroso S. Cellular and subcellular localization of Na+-Ca2+ exchanger protein isoforms, NCX1, NCX2, and NCX3 in cerebral cortex and hippocampus of adult rat. Cell Calcium. 2007;41:221–34. doi: 10.1016/j.ceca.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Nadkarni S, Jung P, Levine H. Astrocytes optimize the synaptic transmission of information. PLoS computational biology. 2008;4:e1000088. doi: 10.1371/journal.pcbi.1000088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarrete M, Araque A. Endocannabinoids potentiate synaptic transmission through stimulation of astrocytes. Neuron. 2010;68:113–26. doi: 10.1016/j.neuron.2010.08.043. [DOI] [PubMed] [Google Scholar]
- Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994;263:1768–71. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Takano T, Hansen AJ. Beyond the role of glutamate as a neurotransmitter. Nat Rev Neurosci. 2002;3:748–55. doi: 10.1038/nrn916. [DOI] [PubMed] [Google Scholar]
- Newman EA, Zahs KR. Modulation of neuronal activity by glial cells in the retina. J Neurosci. 1998;18:4022–8. doi: 10.1523/JNEUROSCI.18-11-04022.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Mukamel EA, Schnitzer MJ. Motor behavior activates Bergmann glial networks. Neuron. 2009;62:400–12. doi: 10.1016/j.neuron.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M, Nakanishi H, Akaike N. Glutamate release from microglia via glutamate transporter is enhanced by amyloid-beta peptide. Neuroscience. 1999;92:1465–74. doi: 10.1016/s0306-4522(99)00036-6. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Piet R, Poulain DA. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science. 2001;292:923–6. doi: 10.1126/science.1059162. [DOI] [PubMed] [Google Scholar]
- Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, Haydon PG, Coulter DA. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci. 2010;13:584–91. doi: 10.1038/nn.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–7. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–6. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, Magistretti PJ. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55:1251–62. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake stimulates Na+,K+-ATPase activity in astrocytes via activation of a distinct subunit highly sensitive to ouabain. J Neurochem. 1997;69:2132–7. doi: 10.1046/j.1471-4159.1997.69052132.x. [DOI] [PubMed] [Google Scholar]
- Petravicz J, Fiacco TA, McCarthy KD. Loss of IP3 receptor-dependent Ca2+ increases in hippocampal astrocytes does not affect baseline CA1 pyramidal neuron synaptic activity. J Neurosci. 2008;28:4967–73. doi: 10.1523/JNEUROSCI.5572-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piet R, Vargova L, Sykova E, Poulain DA, Oliet SH. Physiological contribution of the astrocytic environment of neurons to intersynaptic crosstalk. Proc Natl Acad Sci U S A. 2004;101:2151–5. doi: 10.1073/pnas.0308408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platel JC, Dave KA, Gordon V, Lacar B, Rubio ME, Bordey A. NMDA receptors activated by subventricular zone astrocytic glutamate are critical for neuroblast survival prior to entering a synaptic network. Neuron. 2010;65:859–72. doi: 10.1016/j.neuron.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramón-y-Cajal S. Histologie du Système Nerveux de l'Homme et des Vertébrés. Maloine; Paris, France: 1909, 1911. (reviewed and updated by the author, translated from Spanish by L. Azoulay) English translation: Ramon-y-Cajal, S. (1995): Histology of the Nervous system System of Man and Vertebrates, translated by N. Swanson & L. Swanson, OUP, New York
- Ransom CB, Ransom BR, Sontheimer H. Activity-dependent extracellular K+ accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. The Journal of physiology. 2000;522(Pt 3):427–42. doi: 10.1111/j.1469-7793.2000.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenbach A, Pannicke T. Neuroscience. A new glance at glia. Science. 2008;322:693–4. doi: 10.1126/science.1166197. [DOI] [PubMed] [Google Scholar]
- Robitaille R. Modulation of synaptic efficacy and synaptic depression by glial cells at the frog neuromuscular junction. Neuron. 1998;21:847–55. doi: 10.1016/s0896-6273(00)80600-5. [DOI] [PubMed] [Google Scholar]
- Rose CR, Ransom BR. Intracellular sodium homeostasis in rat hippocampal astrocytes. The Journal of physiology. 1996;491:291–305. doi: 10.1113/jphysiol.1996.sp021216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusakov DA. The role of perisynaptic glial sheaths in glutamate spillover and extracellular Ca2+ depletion. Biophys J. 2001;81:1947–59. doi: 10.1016/S0006-3495(01)75846-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA, Lledo PM, Vincent JD, Charpak S. Dendritic glutamate autoreceptors modulate signal processing in rat mitral cells. J Neurophysiol. 2001;85:1275–82. doi: 10.1152/jn.2001.85.3.1275. [DOI] [PubMed] [Google Scholar]
- Schummers J, Yu H, Sur M. Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science. 2008;320:1638–43. doi: 10.1126/science.1156120. [DOI] [PubMed] [Google Scholar]
- Shin JH, Kim YS, Linden DJ. Dendritic glutamate release produces autocrine activation of mGluR1 in cerebellar Purkinje cells. Proc Natl Acad Sci U S A. 2008;105:746–50. doi: 10.1073/pnas.0709407105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotero RC, Martinez-Cancino R. Dynamical mean field model of a neural-glial mass. Neural computation. 2010;22:969–97. doi: 10.1162/neco.2009.04-09-1002. [DOI] [PubMed] [Google Scholar]
- Stout C, Charles A. Modulation of intercellular calcium signaling in astrocytes by extracellular calcium and magnesium. Glia. 2003;43:265–73. doi: 10.1002/glia.10257. [DOI] [PubMed] [Google Scholar]
- Strowbridge BW. Glutamate receptors mediate TTX-resistant synchronous activity in the rat hippocampus. J Neurosci. 1999;19:5758–67. doi: 10.1523/JNEUROSCI.19-14-05758.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Kang J, Jaiswal JK, Simon SM, Lin JH, Yu Y, Li Y, Yang J, Dienel G, Zielke HR. Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proc Natl Acad Sci U S A. 2005;102:16466–71. doi: 10.1073/pnas.0506382102. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodosis DT, Poulain DA, Oliet SH. Activity-dependent structural and functional plasticity of astrocyte-neuron interactions. Physiol Rev. 2008;88:983–1008. doi: 10.1152/physrev.00036.2007. [DOI] [PubMed] [Google Scholar]
- Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–81. doi: 10.1038/nm1277. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Yi E, Gale JE, Glowatzki E, Bergles DE. The origin of spontaneous activity in the developing auditory system. Nature. 2007;450:50–5. doi: 10.1038/nature06233. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Kettenmann H. Calcium signalling in glial cells. Trends Neurosci. 1996;19:346–52. doi: 10.1016/0166-2236(96)10048-5. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–40. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. 2000;36:291–300. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- Wang X, Lou N, Xu Q, Tian GF, Peng WG, Han X, Kang J, Takano T, Nedergaard M. Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat Neurosci. 2006;9:816–23. doi: 10.1038/nn1703. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Pangrsic T, Kreft M, Krzan M, Li N, Sul JY, Halassa M, Van Bockstaele E, Zorec R, Haydon PG. Fusion-related release of glutamate from astrocytes. J Biol Chem. 2004;279:12724–33. doi: 10.1074/jbc.M312845200. [DOI] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. doi: 10.1038/nn980. [DOI] [PubMed] [Google Scholar]