

Abstract
Systemic inflammatory response syndrome (SIRS), a serious clinical condition characterized by whole body inflammation, is particularly threatening for elderly patients who suffer much higher mortality rates than the young. A major pathological consequence of SIRS is acute lung injury caused by neutrophil-mediated oxidative damage. Previously, we reported an increase in protein tyrosine nitration (a marker of oxidative/nitrosative damage), and a decrease in antioxidant enzyme, extra-cellular superoxide dismutase (EC-SOD), in the lungs of young mice during endotoxemia-induced SIRS. Here we demonstrate that during endotoxemia, down-regulation of EC-SOD is significantly more profound and prolonged, while up-regulation of iNOS is augmented in aged compared to young mice. Aged mice also showed 2.5-fold higher protein nitration levels, compared to young mice, with particularly strong nitration in the pulmonary vascular endothelium during SIRS. Additionally, by 2-dimensional gel electrophoresis, Western blotting and mass spectrometry, we identified proteins which show increased tyrosine nitration in age- and SIRS-dependent manners; these proteins (profilin-1, transgelin-2, LASP 1, tropomyosin and myosin) include components of the actin cytoskeleton responsible for maintaining pulmonary vascular permeability. Reduced EC-SOD in combination with increased oxidative/nitrosative damage and altered cytoskeletal protein function due to tyrosine nitration may contribute to augmented lung injury in the aged with SIRS.
Keywords: aging, EC-SOD, lung injury, oxidative damage, nitrotyrosine
Introduction
Aging is associated with reduced stress tolerance. Vulnerability to various physiological stresses such as infection, inflammation, and oxidative damage increases with age and is causally related to clinical problems in the elderly. Sepsis is an infection-initiated clinical condition characterized by systemic inflammation [1-3]. The progression of sepsis occurs by a loss of homeostasis, characterized by uncontrolled inflammation, oxidative damage to the vascular endothelium and intravascular coagulation. These conditions lead to multiple organ failure and death for a large number of patients [4]. Sepsis is a particularly serious problem in the geriatric population, as the elderly exhibit significantly elevated mortality. Earlier studies noted 30-40% mortality among the elderly with sepsis, compared to 4-5% for younger patients [5, 6]. More recent analyses estimate that more than 700,000 patients present with sepsis yearly in the United States resulting in 215,000 deaths of which the majority are elderly people [7, 8]. Although age-related dysregulation of the inflammatory response and thrombosis appear to be involved [9-11], the precise mechanisms for the age-dependent vulnerability to sepsis remain unclear.
One of the major pathological consequences of the systemic inflammatory response syndrome (SIRS) is acute lung injury. Oxidative damage due to the production of superoxide during SIRS is highly associated with acute lung injury [12] although a causal relationship still has to be definitively established. 3-nitrotyrosine is a known biomarker of nitric oxide (·NO)-dependent oxidative/nitrosative damage [13-15]. Recently we demonstrated that protein tyrosine nitration is increased in the lungs of young mice during periods of lipopolysaccharide (LPS)-induced systemic inflammation [16]. During the inflammatory response, neutrophils undergo a respiratory burst and produce superoxide (·O2−). The superoxide anion reacts with ·NO (generated by iNOS during the immune response) and produces peroxynitrite (ONOO−), a toxic oxidant and nitrating agent. Breakdown of peroxynitrite produces highly toxic reactive nitrogen and oxygen species (RNS/ROS) such as·NO2 and ·OH and CO·3-. These radicals promote the post-translational modification of tyrosine to 3-nitrotyrosine which alters the structure and conformation of target proteins and can compromise their function [17]. The presence of nitrated proteins has been implicated in many disease states; however, the identification of these proteins has only begun [18].
Although ROS have physiologically essential roles in host defense, overproduction of ROS during SIRS is highly toxic to host tissues, resulting in severe pathophysiological consequences such as vascular endothelial damage [19-21]. In a protective mechanism, superoxide is converted by anti-oxidant enzymes, superoxide dismutases (SOD), to form hydrogen peroxide (H2O2), which is further converted to H2O and O2 by another anti-oxidant enzyme, catalase [22]. There are three different mammalian SODs: intracellular copper-zinc (Cu/Zn-SOD or SOD1), mitochondrial manganese SOD (Mn-SOD or SOD2), and extracellular SOD (EC-SOD or SOD3). EC-SOD is the predominant anti-oxidant enzyme that is localized to the extracellular space [23-28]. It is strongly expressed in vascular smooth muscle cells, alveolar type II cells and lung macrophages among other cell types [29] and localized throughout the blood vessel wall; the major source of the EC-SOD protein is thought to be vascular muscle and not endothelium [30]. An in situ hybridization study in mice demonstrated no mRNA expression of EC-SOD on the capillary wall suggesting that EC-SOD accumulates on the endothelial cells in after injury [31]. In our previous study we demonstrated that EC-SOD levels, but not Cu/Zn- or Mn-SOD, are decreased in the lungs during systemic inflammation. Additionally using EC-SOD transgenic mice, we determined that this enzyme plays a protective role against inflammation mediated oxidative/nitrosative damage and mortality [16].
In the present study, we show that the levels of EC-SOD, but not other SODs, decrease in the lungs of both young and aged mice during systemic inflammation and that the decrease is significantly more profound in the aged. Furthermore, we show that protein tyrosine nitration is significantly elevated in the lungs of aged mice compared to young mice and we identified several of the nitrated proteins whose loss of function may contribute to increased lung injury and vascular permeability in the aged during sepsis.
Materials and Methods
Animals
Young (4-6 month old) and aged (24-26 month old) male C57BL/6 mice were obtained from colonies of the National Institute on Aging (Bethesda, MD). Mice were acclimated for at least 14 days in a 12:12hr light-dark cycle with free access to water and regular chow diet (LabDiet, Brentwood, MO) before experiments began and throughout each experiment. All animal care and surgical procedures were approved by the Institutional Animal Care and Use Committees at the University of Texas Medical Branch and the University of Kentucky.
LPS model for endotoxemia
Acute systemic inflammation was induced by intraperitoneal (ip) injection with bacterial endotoxin lipopolysaccharide (LPS, 2.5 mg/kg) derived from Pseudomonas aeruginosa (Sigma Chemical, St. Louis, MO). For tissue protein or RNA analyses, mice were anesthetized with isoflurane inhalation, the inferior vena cava was cut, and the entire vasculature was perfused with physiological saline through the cardiac ventricles as previously described [16]. Lungs were harvested 0, 6 and 12 hr after LPS injection and flash frozen in liquid nitrogen. Major organs from dead mice were examined, and animals with any evident signs of tumor were excluded from the study.
Histological analysis and Immunohistochemistry
Endotoxic and control mice were anesthetized with isoflurane inhalation, the lungs filled with formalin from the trachea (to maintain the organ structure), harvested and fixed in 10% neutral buffered formalin. The harvested tissues were embedded in paraffin and sections (5 μm) were cut from paraffin blocks, and stained with hematoxylin and eosin (H&E). Immunohistochemistry was performed using Dako Cytomation En Vision+ System-HRP (Dako, Denmark). Sections were stained with 1:1000 diluted anti-nitrotyrosine antibody (Millipore, Billerica, MA) at 4°C overnight and counterstained with hematoxylin. Photomicrograph was taken using Leica DFC295 digital camera and Nikon Eclipse E200 microscope with Leica Application Suite version 3.5.0 (100× magnification).
Protein Extraction, 1D Gel Electrophoresis and Western Blot analysis
Frozen lung tissues were homogenized in an SDS-based lysis buffer; protein concentrations were determined by Bradford Protein Assay (Bio-Rad, Hercules, CA) and equal amounts of proteins (40μg) were resolved on Nu-PAGE Bis-Tris gels (Invitrogen, Carlsbad, CA) and electrophoretically transferred to PVDF membranes [16]. The membranes were incubated with anti-EC-SOD [16], anti-Cu/Zn-SOD, and anti-Mn-SOD (Stressgen, Victoria, BC, Canada) antibodies as previously described [16]. For nitrotyrosine detection, proteins were transferred to nitrocellulose membranes and incubated with a horseradish peroxidase (HRP)-conjugated antibody against nitrotyrosine as previously described [16]. The membranes were washed and the immunoreaction visualized by ECL (Amersham, Arlington Heights, IL). All membranes were reprobed with anti-β-actin antibody (Sigma Chemical, St. Louis, MO) to confirm equal loading of the protein samples. Intensity of each autoradiograph was determined by densitometric analysis (Kodak 1D software, Carestream Health, Rochester, NY) and the level of each protein was normalized for β-actin.
RNA analysis
Total RNA was isolated from tissues using guanidine/phenol solution as previously [32]. The RNA samples (20 μg each) were subjected to Northern blot analysis as recently described [16]. To prepare probe DNA, an EC-SOD cDNA fragment (264-bp) was amplified by reverse transcriptase-polymerase chain reaction (RT-PCR) from mouse lung total RNA [16], and a HincII-EcoRI fragment (817-bp) of mouse iNOS cDNA was obtained by a restriction digestion of a plasmid piNOSL3 which was kindly provided by Dr. Regino Perez-Polo at the University of Texas Medical Branch.
Protein Extraction, 2D Gel Electrophoresis and Western Blot analysis
Frozen lung tissues were homogenized in a urea based-lysis buffer (Bio-Rad, Hercules, CA) and the protein concentrations determined by RC DC Protein Assay (Bio-Rad, Hercules, CA). Two-hundred μg protein samples were prepared by pooling lung homogenates from 4 mice. Isoelectric focusing was performed in the first dimension using 11cm, pH 3-10 immobilized pH gradient strips. In the second dimension, IPG strips were equilibrated with TCEP and iodoacetamide. The strips were then imbedded in agarose above 8-16% TrisHCl gels and run at 150v for 2.5 h. Proteins were electrophoretically transferred to nitrocellulose membranes and incubated with a HRP-conjugated antibody against nitrotyrosine. The membranes were washed and the immunoreaction visualized by enhanced chemiluminescence (ECL). Exposed film was imaged on a ProSCAN ProXPRESS 2D Proteomic Imaging System (Perkin-Elmer, Waltham, MA) for analysis.
Gel Analyses and Spot Excision
Gels were fixed in 10% methanol, 7% acetic acid, stained with SYPRO Ruby (Invitrogen, Carlsbad, CA), destained in 10% ethanol and imaged on the ProSCAN ProXPRESS 2D Proteomic Imaging System. A total of eight 2D gel images and eight western blots (2 from each treatment group) were analyzed using Progenesis Discovery software (Nonlinear Dynamics, U.K.). Western analysis for anti-nitrotyrosine was conducted and the blot images superimposed over the gel images to determine the location of anti-nitrotyrosine immunopositive proteins on the gel. The intensity of each nitrotyrosine positive protein spot was normalized based on the total volume of the protein spot on each gel. Protein gel spots of interest were excised and cut into 1mm size pieces for digestion by trypsin (Promega, Madison, WI). After digestion, 1 μL of sample solution was spotted directly onto a Matrix-Assisted Laser Desorption Ionization (MALDI) target plate and allowed to dry. Alpha-cyano-4-hydroxycinnamic acid matrix solution (Aldrich Chemical, Allentown, PA) was then applied on the sample spot and dried. The dried MALDI spot was blown with compressed air before inserting into the mass spectrometer.
Mass Spectrometry
Matrix-Assisted Laser Desorption Ionization Time-of-Flight Mass Spectrometry (MALDI TOF-MS) was used to analyze the samples and determine protein identification. Data were acquired with a 4800 MALDI TOF/TOF Proteomics Analyzer (Applied Biosystems, Carlsbad, CA). The instrument was operated in positive ion reflectron mode, mass range was 850 – 3000 Da, and the focus mass was set at 1700 Da. For MS data, 2000-4000 laser shots were acquired and averaged from each sample spot. Following MALDI MS analysis, MALDI MS/MS was performed on several (5-10) abundant ions from each sample spot. For MS/MS data, 2000 laser shots were acquired and averaged from each sample spot. Applied Biosystems GPS Explorer™ (v. 3.6) software was used in conjunction with MASCOT to search the protein database using both MS and MS/MS spectral data for protein identification. Protein match probabilities were determined using expectation values and/or MASCOT protein scores. MS peak filtering included the following parameters: mass range 800 Da to 4000 Da, minimum S/N filter = 10, mass exclusion list tolerance = 0.5 Da. For MS/MS peak filtering, the minimum S/N filter = 10. For protein identification, the mouse taxonomy was searched in the NCBI database. The significance of a protein match, based on both the peptide mass fingerprint (PMF) in the first MS and the MS/MS data from several precursor ions, is based on expectation values. The expectation value is the number of matches with equal or better scores that are expected to occur by chance alone. The default significance threshold is p<0.05, so an expectation value of 0.05 is considered to be on this threshold. We used a more stringent threshold of 10-3 for protein identification; the lower the expectation value, the more significant the score.
Results
Exaggerated pulmonary inflammation and edema in aged mouse lung tissue after LPS-induced systemic inflammation
Upon LPS injection, both young and aged mice suffered hypothermia characteristic of LPS-mediated systemic inflammation [9] and a temporal appetite loss. Histology of H&E-stained lung tissues were compared among young (6-mo) and aged (26-mo) mice 24h after LPS-induced systemic inflammation. Non-injected mice were used as controls. Figure 1A shows the histologic characteristics of normal, healthy young lung tissue consisting of thin alveolar walls embedded with capillaries, alveolar cells and occasional alveolar macrophages. Figure 1B shows lung histology associated with normal aging. Upon LPS injection, lung tissue from young mice (Fig. 1C) exhibited slight alveolar wall swelling due to dilated capillaries, and proteinaceous debris was present in the alveoli. Severe edema and inflammation was apparent in the lung tissue from aged mice during systemic inflammation (Fig. 1D). These data demonstrate that aging is associated with increased severity of acute lung injury during systemic inflammation.
Figure 1. Histological analysis demonstrating age-associated severity of acute lung injury during systemic inflammation.
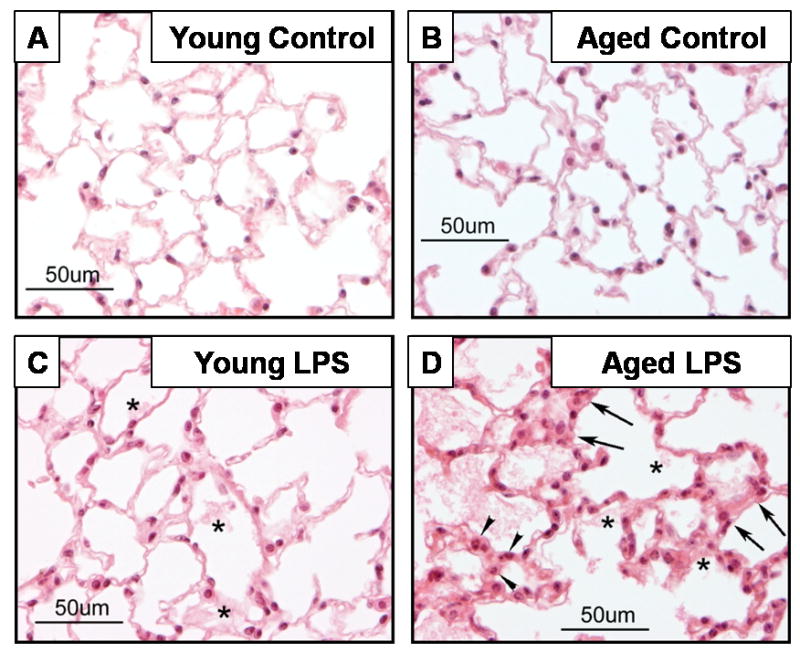
Mice at 6 and 26 months of age were injected with LPS (2.5 mg/kg, ip) and sacrificed 24 h later. Non-injected mice were used as controls. Sections from formalin-fixed, paraffin embedded lung samples were stained with H&E. Arrow heads indicate granular leukocytes, arrows indicate edematous thickened alveolar walls, and stars indicate deposit of proteinaceous debris. Inflammation and edema are distinct in 26-month-old mice with LPS.
Age-associated decrease in antioxidant enzyme, EC-SOD, during LPS-induced systemic inflammation
Previously, we showed that protein and mRNA levels of the antioxidant enzyme EC-SOD are down-regulated during systemic inflammation in young mice and suggested its causal relationship to mortality and increased lung injury due to oxidative damage [16]. Since we found that acute lung injury is more severe in aged mice during systemic inflammation (Fig. 1), we hypothesized that the protective function of EC-SOD may be further impaired by aging. Young (4 months) and aged (24 months) mice were injected with LPS and sacrificed 6 and 12 h later. Whole lungs were obtained and processed for protein analysis by Western blotting. As shown in Figs. 2A and 2B basal levels of EC-SOD (0h time point) are unaltered by aging; however, upon LPS-injection EC-SOD protein level decreased in an age-dependent manner. The level of EC-SOD in young mice decreased from 0 to 6h after LPS injection but recovered to near normal levels by 12h. In aged mice, EC-SOD also decreased by 6h but did not recover, and further decreased by 12h after LPS injection. The protein levels of Cu/Zn- and Mn-SOD were not altered by aging or LPS injection. As most aged mice die after 12h of LPS (2.5mg/kg) injection [10], protein analysis later than this time point was not performed.
Figure 2. Western blot analysis demonstrating that LPS-mediated decrease in pulmonary EC-SOD is more profound in aged mice than young mice.
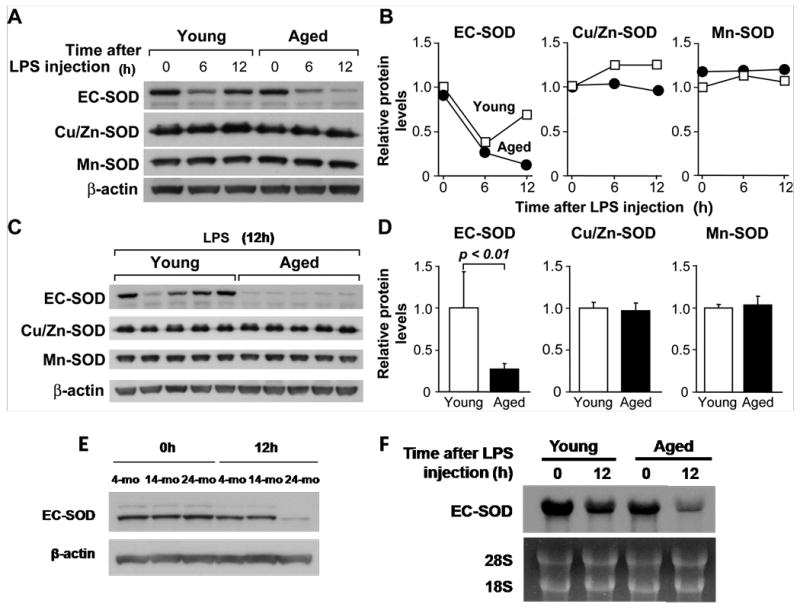
(A) Time course of lung EC-SOD levels in young (4 months) and aged (24 months) mice after LPS injection (2.5 mg/kg, ip). Each lane represents a pooled protein sample derived equally from 5 mice. (B) Densitometric analysis of A. (C) Lung EC-SOD levels in young and aged mice 12 h after LPS injection. Each lane represents a protein sample from an individual mouse (n=5 per age group). (D) Densitometric analysis of C. (E). Lung EC-SOD levels in young (4 months), middle aged (14 months), and aged (24 months) mice 12 h after LPS injection (2.5mg/kg). Each lane represents a pooled protein sample derived equally from 3 mice. (F) Northern hybridization analysis demonstrating that down-regulation of lung EC-SOD mRNA expression after LPS injection was more profound in aged mice than in young mice. Young (4 months) and aged (24 months) mice were injected with LPS (2.5 mg/kg, ip) and total lung RNA was isolated and used for Northern blot analysis. Each lane represents a pooled protein sample derived equally from 5 mice.
To further analyze the age-associated difference of EC-SOD protein expression, western blot analysis was performed on total lung protein from individual young and aged mice sacrificed 12h after LPS injections (which were pooled in Fig. 2A). Figure 2C and 2D depict a clear age-associated difference of EC-SOD down-regulation. The level of EC-SOD was significantly lower in the aged mouse lung compared to the young mouse lung 12h after LPS injection (p<0.001).
We also examined whether age-dependent EC-SOD down-regulation was truly an age-associated phenomenon. Young (4 months), middle-aged (14 months) and aged (24 months) mice were injected with LPS and sacrificed 12h later. As shown in Fig. 2E, EC-SOD levels in young and middle-aged mice recovered to near normal levels by 12h after LPS injection, while only aged mice exhibited a continued down-regulation. This data indicates that profound down-regulation of EC-SOD is a late-life phenomenon.
Age-associated down-regulation of EC-SOD is transcriptionally regulated
To determine whether the age-associated decrease in pulmonary EC-SOD levels is due to transcriptional regulation or a post-translational modification, we examined the mRNA level of EC-SOD in the lung after LPS injection by Northern blot analysis. As shown in Fig. 2F, EC-SOD mRNA was down-regulated by 12h in the lung from young mice but more profoundly down-regulated at 12h in the lung from aged mice. This data closely matches that of EC-SOD protein down-regulation depicted in Fig. 2A and indicates that the down-regulation of EC-SOD during systemic inflammation is regulated at least in part by a transcriptional mechanism.
Age-associated increase in oxidative damage to lung proteins during LPS-induced systemic inflammation
Accumulation of nitrotyrosine is a well known marker of nitric oxide-dependent oxidative stress. Previously, we showed that oxidative damage, measured by accumulation of nitrotyrosine, increases over time during LPS-induced systemic inflammation in young mice [16]. Here, we show that tyrosine nitration is further augmented by aging. Young (4 month) and aged (24 month) mice were injected with LPS and sacrificed 6 and 12h later. High levels of nitrotyrosine were detected throughout the lungs of aged control and aged LPS-injected mice but intense staining of the vascular endothelium was observed only in the aged mouse lung after LPS injection (Fig 3A). Young control and young LPS-injected mice showed much less nitrotyrosine staining than aged mice. Additionally, oxidative/nitrosative damage in the lungs was assessed by western blot analysis for nitrotyrosine. As shown in Fig. 3B, multiple bands ranging in size from 14 – 97 kDa were detected. Overall, an increase in the intensity of the immunoreaction was observed in the lungs from aged mice injected with LPS (lanes 6-10) compared to young mice (lanes 1-5). Densitometric analysis (Fig. 3B) of each lane confirmed that the total intensity of nitrotyrosine positive proteins was greater than 2-fold increased in the lungs from aged mice compared to young mice (p<0.001). We also examined the mRNA levels of inducible nitric oxide synthase (iNOS) in the lungs of young and aged mice after LPS injection. While iNOS expression was slightly elevated 6h after LPS injection in the young, the expression of iNOS in the aged lung was greatly enhanced. In both young and aged mice, iNOS expression in the lung was reduced at 12h compared to 6h with the level of expression still elevated in the aged (Fig. 3C). These results demonstrate that acute lung injury, induced by systemic inflammation, results in an age-associated increase in oxidative/nitrosative damage to lung proteins.
Figure 3. Age-associated increase in protein tyrosine nitration in the lungs during systemic inflammation.
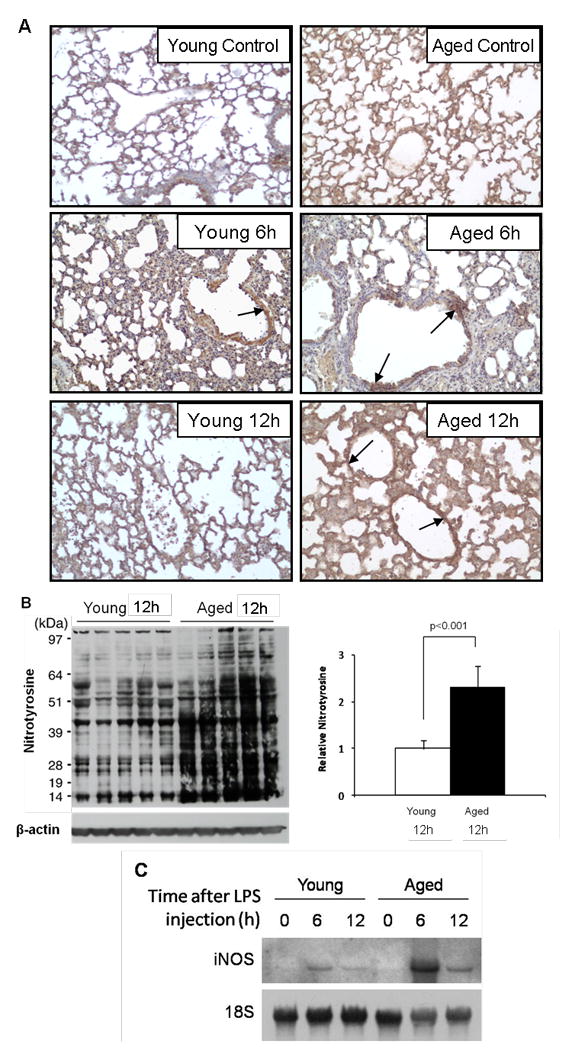
Systemic inflammation was induced in young (4 months) and aged (24 months) mice by injection with LPS (2.5 mg/kg, ip); mice were sacrificed 6 and 12 h later and the lung tissues harvested for analysis. Age and sex-matched non-injected mice were used as controls. (A) Immunohistochemical localization of nitrotyrosine to the vasculature of aged mice after LPS injection. Arrows indicate strongly stained vascular endothelium. (B) Western blot analyses for nitrotyrosine on whole-lung proteins and densitometric analysis of each lane. Total intensity throughout each lane was normalized by β-actin. (C) Northern hybridization analysis demonstrating that induction of lung iNOS mRNA after LPS injection was augmented in aged mice compared to young mice. Young (4 months) and aged (24 months) mice were injected with LPS (2.5 mg/kg, ip) and total lung RNA was isolated and used for Northern blot analysis. Each lane represents a pooled protein sample derived equally from 5 mice.
Identification of lung proteins which are nitrated during LPS-induced oxidative damage
Although protein tyrosine nitration is a well-known marker of oxidative damage, little is known about which proteins are nitrated and how the modification affects their function, particularly with aging. In Fig. 3, we showed that pulmonary protein tyrosine nitration is augmented in aged mice during systemic inflammation. In order to identify which proteins are subject to tyrosine nitration, we performed 2D polyacrylamide gel electrophoresis and western blotting against nitrotyrosine. Young (4-mo) and aged (24-mo) mice were injected with LPS and sacrificed 12h later. Age- and sex-matched non-injected mice were used as controls. Whole lung protein extracts from 4 mice in each group were equally pooled and resolved on 2D gels (not shown). Figure 4 shows images of the resulting western blot membranes that were probed with anti-nitrotyrosine. Protein spots on the membrane were correlated with spots on the gels and the density adjusted for total protein level. The gel spots corresponding to nitrotyrosine positive proteins exhibiting statistically significant age-associated changes were then excised and identified by mass spectrometry. Numbers on the membrane correspond to the identified proteins described in Table 1. Several of the proteins identified, including profilin 1, transgelin 2, voltage dependent anion channel, LIM and SH3 protein 1 and tropomyosin (Spot #'s 1, 2, 4, 5 and 9), are associated with the actin cytoskeleton. The cluster of nitrotyrosine positive spots left of the center of the membrane, between 40-50kDa, were identified as actins. Spots numbered 3, 6, 7 and 8 (carbonyl reductase 2, serine proteinase inhibitor B1a, peroxiredoxin 6, and polymerase I and transcript release factor, respectively) vary in function from oxidative metabolism to transcriptional regulation. The significance of the tyrosine nitration modification to these proteins is described in the Discussion section below.
Figure 4. 2D western blot analyses and mass spectrometry identification of pulmonary proteins that exhibit an age-associated increase in tyrosine nitration during systemic inflammation.
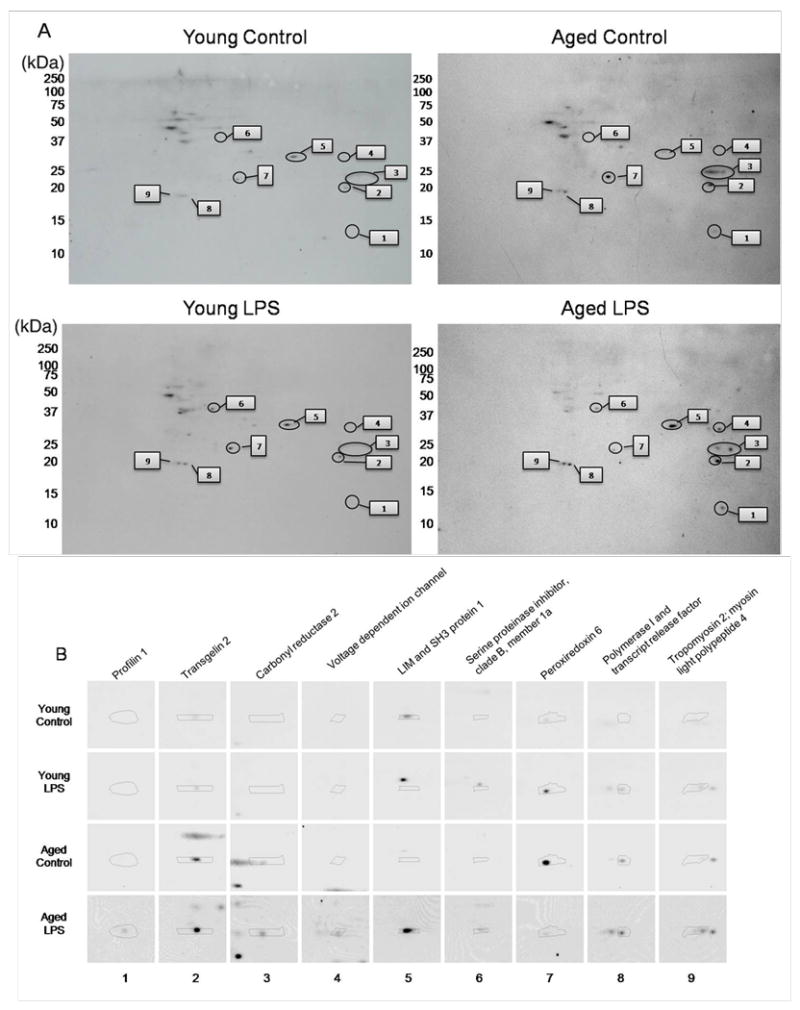
Systemic inflammation was induced in young and aged mice by injection with LPS (2.5 mg/kg, ip); mice were sacrificed 12 h later and the lung tissues harvested for analysis. Age and sex-matched non-injected mice were used as controls. (A) Western blot analysis of 2 dimensional gels detecting tyrosine nitration of lung proteins from young control mice (top left panel); young LPS injected mice (bottom left panel); aged control mice (top right panel); and aged LPS injected mice (bottom right panel). Each gel contained total lung protein derived equally from 4 mice. Numbers indicate proteins identified by mass spectrometry in Table 1. (B) Enlarged view of individual nitrotyrosine positive protein spots. Numbers on bottom correspond to spots in (A).
Table 1. Nitrated proteins identified by MALDI TOF-MS from 2DGE and Western blot analysis of lung proteins.
| SpotA # | Protein | Peptide B Count | Expectation C Value | Function |
|---|---|---|---|---|
| 1 | Profilin 1 | 5 | 7.92E-35 | Actin binding, Rho GTPase binding |
| 2 | Transgelin 2 | 10 | 1.26E-53 | Actin binding, muscle development |
| 3 | Carbonyl reducatse 2 | 5 | 3.15E-17 | Oxidative metabolism |
| 4 | Voltage dependent anion channel | 2 | 9.98E-11 | Mitochondrial ion channel, cell volume regulation, apoptosis |
| 5 | LIM and SH3 protein 1 | 9 | 7.92E-38 | Ion transport, actin binding |
| 6 | Serine proteinase inhibitor, clade B, member 1a | 12 | 6.29E-35 | Lung defense enzyme |
| 7 | Peroxiredoxin 6 | 12 | 6.29E-77 | Protects against oxidative injury |
| 8 | Polymerase I and transcript release factor | 7 | 2.51E-77 | Transcription regulation and termination |
| 9 | Tropomyosin 2 | 14 | 1.26E-08 | Muscle contraction |
| Myosin light polypeptide 4 | 7 | 3.97E-28 | Actin binding, Calcium binding | |
Spot # corresponds to protein spot labeled in Figure 4.
Peptide Count indicates the number of peptides that matched the protein.
Expectation Value is the number of matches with equal or better scores that are expected to occur by chance, score of 5.00E-02 is significant.
Discussion
Acute lung injury, a common consequence of oxidative damage during SIRS and sepsis, is a complex inflammatory syndrome characterized by hypoxemia, non-cardiogenic pulmonary edema, and widespread capillary leakage resulting from endothelial barrier dysfunction [33]. We showed characteristic pathologic changes such as thickened alveolar walls, capillary dilation, and fluid filled alveolar spaces that were more pronounced in the lung from aged mice during systemic inflammation indicating an age-dependent increase in severity of acute lung injury. Gomez et al. previously reported an age-associated increase of neutrophil infiltration and increased myeloperoxidase in the lungs of endotoxic mice; however, they did not observe any lung injury [34]. This difference is likely because we used a higher dose of LPS (2.5 mg/kg, ip) than Gomez et al. (1.5 mg/kg, ip); our dose of LPS is significantly more lethal resulting in 80% mortality in aged mice [10], while the dose used by Gomez et al. resulted in 23% mortality.
The primary function of EC-SOD, the major SOD in the extracellular space [28] is to scavenge superoxide preventing its interaction with nitric oxide and the production of peroxynitrite. Loss of EC-SOD results in elevated superoxide [35], elevated nitrotyrosine-mediated oxidative damage [16], sensitivity to hyperoxia [36] and endothelial cell dysfunction [37]. Since we previously showed that pulmonary EC-SOD is lost during systemic inflammation and this loss coincides with elevated oxidative/nitrosative damage, we speculated that age-associated augmented acute lung injury is due in part to EC-SOD down-regulation. In this study, we confirmed our previous result that pulmonary EC-SOD is down-regulated during LPS-induced systemic inflammation and further discovered that pulmonary EC-SOD down-regulation is more profound in aged mice. Although our previous study used a higher dose of LPS (20 mg/kg) in young mice only, the trend of down-regulation and recovery is the same albeit on a different time-scale. A lower dose of LPS (2.5 mg/kg) was used in the present study because aged mice would not survive for more than a few hours after a dose of 20 mg/kg. To our knowledge, this is the first report to examine an age-associated loss of pulmonary EC-SOD during systemic inflammation. Importantly, while pulmonary EC-SOD levels in young mice recovered by 12h after LPS-injection, EC-SOD down-regulation was sustained in the lungs of aged mice. This age-associated sustained down-regulation of EC-SOD in the late-phase of systemic inflammation could partly explain the different outcome between young and aged with respect to acute lung injury and eventual respiratory organ failure.
The age-associated sustained decline in EC-SOD is at least partly due to transcriptional regulation. Studies by Zelko and Folz identified the promoter region of murine and human EC-SOD genes and have shown that EC-SOD transcription is critically dependent on a single Sp1/Sp3 binding site located within its proximal promoter [38, 39]. More recent findings by the same group indicate that pulmonary cell-specific expression of EC-SOD can be attributed to chromatin remodeling and differential methylation of its promoter [40]. We recently reported that thrombomodulin (TM), an endothelial cell membrane receptor involved in the protein C anti-coagulant pathway, is strongly expressed in the lung and is down-regulated in an age-dependent manner during LPS-induced endotoxemia [10]. This pattern of expression is strikingly similar to the down-regulation of EC-SOD depicted in this study and led us to hypothesize that a common mechanism exists for the down-regulation of these two genes. As both proteins exist in a stable membrane-bound form and a less stable soluble form, it seems that the rapid down-regulation of EC-SOD and TM protein levels in the lung are likely due to post-translational cleavage and elimination from the tissue in addition to a rapid transcriptional decrease as shown previously [16].
In the present study we also show that oxidative/nitrosative damage via protein tyrosine nitration is increased in the lungs of aged mice compared to young during LPS-induced systemic inflammation. Nitrotyrosine in the kidney of young rats was previously localized to the renal cortex with intense staining in the proximal and distal convoluted tubules, initial collecting tubules, glomeruli, and interlobular vascular endothelium [41]. We show broad nitrotyrosine staining throughout the lungs of control and LPS treated young mice with intense nitrotyrosine staining at the vascular endothelial surface of lungs from LPS treated aged mice. Additionally, we show a strong age-associated increase in iNOS mRNA production in the lung after LPS injection suggesting that ·NO is also increased in the aged during systemic inflammation. Increased ·NO, in combination with decreased EC-SOD, in the aged likely results in enhanced production of peroxynitrite. Since intense nitrotyrosine staining was seen at the vascular endothelium but not as extracellular debris, peroxynitrite appears to penetrate the cells and cause tyrosine nitration to intracellular proteins of the lung. Since protein tyrosine nitration is known to affect protein function, we hypothesized that the nitrated proteins may experience a loss of function which ameliorates protective roles and contributes to age-associated acute lung injury. Anti-nitrotyrosine immunodetection via traditional 1D western blot analysis revealed multiple bands, indicating numerous proteins of varying sizes to be nitrated during LPS-induced systemic inflammation. Enhanced nitration was particularly evident in lung proteins from aged mice. By 2D gel electrophoresis and western blot analysis we were able to isolate and excise individual protein spots for identification by mass spectrometry. Half (5 of 10) of the proteins we identified are involved in actin cytoskeleton mechanics; loss of function of such proteins has potential implications in endothelial cell barrier disruption, vascular permeability and leakage [42, 43]. Among these nitrated actin binding proteins were profilin 1, transgelin 2, tropomyosin 2, myosin light polypeptide 4, and LIM and SH3 protein 1.
Profilin 1 is known to play a crucial role in early development and restructuring of the actin cytoskeleton [44]; however its role in disease remains largely unstudied. Plasma fibronectin deficiency magnifies lung vascular permeability [45], thus, as endothelial cell adhesion to fibronectin is dependent on profilin expression [46] absence of functional profilin may be partly responsible for vascular permeability due to inefficient barrier integrity. Transgelin binds actin and facilitates the formation of cytoskeletal structures [47-49]. It was recently proposed that disruption of transgelin expression in stressed vascular smooth muscle cells results in actin cytoskeleton and microtubule remodeling contributing to loss of cytoskeletal integrity and arterial inflammation [50]. LIM 1 and SH3 protein 1 (LASP-1) is a ubiquitously expressed actin binding protein involved in cell migration [51]. A recent study also indicated a crucial role for LASP-1 in platelet cytoskeleton rearrangement [52]. Tropomyosin and myosin are classical actin binding proteins. Tropomyosin binds to actin microfilaments to inhibit myosin binding which keeps the cell at rest. When intracellular Ca2+ increases a conformational change occurs that exposes the myosin binding site on actin allowing myosin to bind and resulting in contraction and endothelial cell gap formation. Protein nitration could interfere with this important mechanism. Interestingly, actin nitration was also observed; however, the pattern of nitration in our study is not clear and seemed to increase by aging but decrease after LPS injection. Actin nitration in vitro was previously observed in rat lung microvascular endothelial cells by treatment with peroxynitrite [53].
A previous study by Aulak et al, reported tyrosine nitration of several proteins including mitochondrial aconitase, fibrinogen, desmin, mitochondrial creatine kinase, malate dehydrogenase, crystalline and voltage dependent anion channel (VDAC) in the lungs of LPS injected rats [18]. With the exception of VDAC, we were not able to identify these same proteins. VDAC a protein which allows for mitochondrial diffusion of small hydrophilic molecules such as superoxide was also recently found to be nitrated in a murine model of ischemia/reperfusion [54]. Some variations in our studies such as type of animal or protein isolation method may explain this difference. For example, we used 4 month and 24 month old C57BL/6 mice, while Aulak's study used Sprague-Dawley rats (age not mentioned but presumably young). Additionally, their data reflects protein tyrosine nitration occurring 18h after LPS (E.coli, 10mg/kg) injection, while our samples were collected 12h after LPS (P. aeruginosa, 2.5 mg/kg) injection.
Other proteins identified to be nitrated during systemic inflammation include carbonyl reducatase 2, Serpinb1, peroxiredoxin 6, and polymerase 1 and transcript release factor. Carbonyl reductase 2 is a lung specific protein thought to function in the metabolism of endogenous carbonyl compounds [55]. Peroxiredoxin 6 is an antioxidant enzyme enriched in the lung which protects alveolar type II epithelial cells against H2O2 mediated oxidative damage [56-58]. In our experiment there was a clear increase in nitration of peroxiredoxin 6 in young mice after LPS injection; however we did not detect high levels of peroxiredoxin 6 nitration in the aged LPS-injected group. Neutrophil serine proteases released by neutrophils during infection are important for bacterial clearance, however during states of severe inflammation the serine proteases can be detrimental to the host. Serine protease inhibitor B1 (Serpinb1) is responsible for inhibiting these serine proteases. Upon infection, Serpinb1 (-/-) mice have increased mortality and decreased bacterial clearance compared to wild-type mice [59]. Additionally, aerosol treatment with Serpinb1 enhances bacterial clearance and protects rat lungs from infection [60].
One limitation of the current study is that we did not establish a direct causal relationship between loss of EC-SOD, increased tyrosine nitration, and acute lung injury with aging. A study using aged EC-SOD transgenic (over-expressing human EC-SOD) mice would be required to provide evidence for such a relationship. We previously reported that such transgenic mice at a young age are more resistant to endotoxemia than age-matched wild-type mice, demonstrating that EC-SOD plays an important protective role during systemic inflammation [16]. Thus, the profound age-associated down-regulation of EC-SOD during systemic inflammation is likely to be causally linked to increased lung protein nitration and tissue damage in old age. Another limitation of our study is that tyrosine nitration is not the sole mechanism leading to oxidative/nitrosative modification of proteins. Oxidative modification to proteins in the lung has been demonstrated to arise from orthotyrosine, metatyrosine, and chlorotyrosine in addition to nitrotyrosine [61]. Modifications caused by chlorotyrosine and nitrotyrosine are more well correlated with neutrophil-mediated oxidative damage [61] and thus these two modifications likely contribute to oxidative damage in the lung during SIRS.
In summary, we have shown that the severity of acute lung injury characterized by pulmonary edema and inflammation is more profound in aged mice during LPS-induced systemic inflammation. We have demonstrated that pulmonary EC-SOD, an anti-oxidant enzyme, is down-regulated during systemic inflammation and this down-regulation is significantly more profound and prolonged in aged mice compared to young mice. LPS-mediated oxidative damage, measured by increased protein tyrosine nitration, was prominent in the vascular endothelium of lungs from aged but not young mice. Identification of the nitrated proteins revealed significant modification to the vascular endothelial cytoskeleton which potentially contributes to barrier dysfunction, increased vascular permeability and pulmonary edema. The results presented here can partly explain the age-associated increase in susceptibility to systemic inflammation, acute lung injury, and respiratory failure.
Acknowledgments
The first and second authors contributed equally to this work. The authors thank Dr. John E. Wiktorowicz, Mrs. Susan J. Stafford and Dr. Zheng Wu of the Biomolecular Resource Facility at the University of Texas Medical Branch for technical advice and assistance with the 2D gel experiments and analysis. The authors also thank Dr. Michael B. Reid of the Department of Physiology at the University of Kentucky for his thoughtful advice and critique of the experimental data. This work was supported by the National Institutes of Health Grant RO1-AG025908. Additional support came from the UTMB, Claude D. Pepper, Older Americans Independence Center, NIH P30A6024832.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cavaillon JM, Adrie C. Sepsis and non-infectious systemic inflammation : from biology to critical care. Weinheim: Wiley-VCH; 2009. Chichester : John Wiley[distributor] [Google Scholar]
- 2.Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250–1256. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- 3.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 4.Astiz ME, Rackow EC. Septic shock. Lancet. 1998;351:1501–1505. doi: 10.1016/S0140-6736(98)01134-9. [DOI] [PubMed] [Google Scholar]
- 5.Stengle J, Dries D. Sepsis in the elderly. Crit Care Nurs Clin North Am. 1994;6:421–427. [PubMed] [Google Scholar]
- 6.Stanley M. Sepsis in the elderly. Crit Care Nurs Clin North Am. 1996;8:1–6. [PubMed] [Google Scholar]
- 7.Martin GS, Mannino DM, Moss M. The effect of age on the development and outcome of adult sepsis. Crit Care Med. 2006;34:15–21. doi: 10.1097/01.ccm.0000194535.82812.ba. [DOI] [PubMed] [Google Scholar]
- 8.Gorina Y, Hoyert D, Lentzner H, Goulding M. Trends in causes of death among older persons in the United States. Aging Trends. 2005:1–12. [PubMed] [Google Scholar]
- 9.Saito H, Sherwood ER, Varma TK, Evers BM. Effects of aging on mortality, hypothermia, and cytokine induction in mice with endotoxemia or sepsis. Mech Ageing Dev. 2003;124:1047–1058. doi: 10.1016/j.mad.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Starr ME, Ueda J, Takahashi H, Weiler H, Esmon CT, Evers BM, Saito H. Age-dependent vulnerability to endotoxemia is associated with reduction of anti-coagulant factors activated protein C and thrombomodulin. Blood. 2010;115:4886–4893. doi: 10.1182/blood-2009-10-246678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamamoto K, Shimokawa T, Yi H, Isobe K, Kojima T, Loskutoff DJ, Saito H. Aging accelerates endotoxin-induced thrombosis : increased responses of plasminogen activator inhibitor-1 and lipopolysaccharide signaling with aging. Am J Pathol. 2002;161:1805–1814. doi: 10.1016/s0002-9440(10)64457-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roth E, Manhart N, Wessner B. Assessing the antioxidative status in critically ill patients. Curr Opin Clin Nutr Metab Care. 2004;7:161–168. doi: 10.1097/00075197-200403000-00010. [DOI] [PubMed] [Google Scholar]
- 13.Kristof AS, Goldberg P, Laubach V, Hussain SN. Role of inducible nitric oxide synthase in endotoxin-induced acute lung injury. Am J Respir Crit Care Med. 1998;158:1883–1889. doi: 10.1164/ajrccm.158.6.9802100. [DOI] [PubMed] [Google Scholar]
- 14.Gorg B, Wettstein M, Metzger S, Schliess F, Haussinger D. Lipopolysaccharide-induced tyrosine nitration and inactivation of hepatic glutamine synthetase in the rat. Hepatology. 2005;41:1065–1073. doi: 10.1002/hep.20662. [DOI] [PubMed] [Google Scholar]
- 15.Ischiropoulos H. Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Arch Biochem Biophys. 1998;356:1–11. doi: 10.1006/abbi.1998.0755. [DOI] [PubMed] [Google Scholar]
- 16.Ueda J, Starr ME, Takahashi H, Du J, Chang LY, Crapo JD, Evers BM, Saito H. Decreased pulmonary extracellular superoxide dismutase during systemic inflammation. Free Radic Biol Med. 2008;45:897–904. doi: 10.1016/j.freeradbiomed.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Radi R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc Natl Acad Sci U S A. 2004;101:4003–4008. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aulak KS, Miyagi M, Yan L, West KA, Massillon D, Crabb JW, Stuehr DJ. Proteomic method identifies proteins nitrated in vivo during inflammatory challenge. Proc Natl Acad Sci U S A. 2001;98:12056–12061. doi: 10.1073/pnas.221269198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bhattacharyya J, Biswas S, Datta AG. Mode of action of endotoxin: role of free radicals and antioxidants. Curr Med Chem. 2004;11:359–368. doi: 10.2174/0929867043456098. [DOI] [PubMed] [Google Scholar]
- 20.Fattman CL, Schaefer LM, Oury TD. Extracellular superoxide dismutase in biology and medicine. Free Radic Biol Med. 2003;35:236–256. doi: 10.1016/s0891-5849(03)00275-2. [DOI] [PubMed] [Google Scholar]
- 21.Macdonald J, Galley HF, Webster NR. Oxidative stress and gene expression in sepsis. Br J Anaesth. 2003;90:221–232. doi: 10.1093/bja/aeg034. [DOI] [PubMed] [Google Scholar]
- 22.Kinnula VL, Crapo JD. Superoxide dismutases in the lung and human lung diseases. Am J Respir Crit Care Med. 2003;167:1600–1619. doi: 10.1164/rccm.200212-1479SO. [DOI] [PubMed] [Google Scholar]
- 23.Marklund SL. Human copper-containing superoxide dismutase of high molecular weight. Proc Natl Acad Sci U S A. 1982;79:7634–7638. doi: 10.1073/pnas.79.24.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marklund SL, Holme E, Hellner L. Superoxide dismutase in extracellular fluids. Clin Chim Acta. 1982;126:41–51. doi: 10.1016/0009-8981(82)90360-6. [DOI] [PubMed] [Google Scholar]
- 25.Sandstrom J, Karlsson K, Edlund T, Marklund SL. Heparin-affinity patterns and composition of extracellular superoxide dismutase in human plasma and tissues. Biochem J. 1993;294(Pt 3):853–857. doi: 10.1042/bj2940853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Folz RJ, Crapo JD. Extracellular superoxide dismutase (SOD3): tissue-specific expression, genomic characterization, and computer-assisted sequence analysis of the human EC SOD gene. Genomics. 1994;22:162–171. doi: 10.1006/geno.1994.1357. [DOI] [PubMed] [Google Scholar]
- 27.Folz RJ, Guan J, Seldin MF, Oury TD, Enghild JJ, Crapo JD. Mouse extracellular superoxide dismutase: primary structure, tissue-specific gene expression, chromosomal localization, and lung in situ hybridization. Am J Respir Cell Mol Biol. 1997;17:393–403. doi: 10.1165/ajrcmb.17.4.2826. [DOI] [PubMed] [Google Scholar]
- 28.Fukai T. Extracellular SOD and aged blood vessels. Am J Physiol Heart Circ Physiol. 2009;297:H10–12. doi: 10.1152/ajpheart.00425.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nozik-Grayck E, Suliman HB, Piantadosi CA. Extracellular superoxide dismutase. Int J Biochem Cell Biol. 2005;37:2466–2471. doi: 10.1016/j.biocel.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 30.Faraci FM, Didion SP. Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol. 2004;24:1367–1373. doi: 10.1161/01.ATV.0000133604.20182.cf. [DOI] [PubMed] [Google Scholar]
- 31.Fukui S, Ookawara T, Nawashiro H, Suzuki K, Shima K. Post-ischemic transcriptional and translational responses of EC-SOD in mouse brain and serum. Free Radic Biol Med. 2002;32:289–298. doi: 10.1016/s0891-5849(01)00804-8. [DOI] [PubMed] [Google Scholar]
- 32.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 33.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 34.Gomez CR, Hirano S, Cutro BT, Birjandi S, Baila H, Nomellini V, Kovacs EJ. Advanced age exacerbates the pulmonary inflammatory response after lipopolysaccharide exposure. Crit Care Med. 2007;35:246–251. doi: 10.1097/01.CCM.0000251639.05135.E0. [DOI] [PubMed] [Google Scholar]
- 35.Jung O, Marklund SL, Geiger H, Pedrazzini T, Busse R, Brandes RP. Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability: in vivo and ex vivo evidence from ecSOD-deficient mice. Circ Res. 2003;93:622–629. doi: 10.1161/01.RES.0000092140.81594.A8. [DOI] [PubMed] [Google Scholar]
- 36.Carlsson LM, Jonsson J, Edlund T, Marklund SL. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc Natl Acad Sci U S A. 1995;92:6264–6268. doi: 10.1073/pnas.92.14.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lund DD, Chu Y, Miller JD, Heistad DD. Protective effect of extracellular superoxide dismutase on endothelial function during aging. Am J Physiol Heart Circ Physiol. 2009;296:H1920–1925. doi: 10.1152/ajpheart.01342.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zelko IN, Folz RJ. Sp1 and Sp3 transcription factors mediate trichostatin A-induced and basal expression of extracellular superoxide dismutase. Free Radic Biol Med. 2004;37:1256–1271. doi: 10.1016/j.freeradbiomed.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 39.Zelko IN, Mueller MR, Folz RJ. Transcription factors sp1 and sp3 regulate expression of human extracellular superoxide dismutase in lung fibroblasts. Am J Respir Cell Mol Biol. 2008;39:243–251. doi: 10.1165/rcmb.2007-0378OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zelko IN, Mueller MR, Folz RJ. CpG methylation attenuates Sp1 and Sp3 binding to the human extracellular superoxide dismutase promoter and regulates its cell-specific expression. Free Radic Biol Med. 2010;48:895–904. doi: 10.1016/j.freeradbiomed.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bian K, Davis K, Kuret J, Binder L, Murad F. Nitrotyrosine formation with endotoxin-induced kidney injury detected by immunohistochemistry. Am J Physiol. 1999;277:F33–40. doi: 10.1152/ajprenal.1999.277.1.F33. [DOI] [PubMed] [Google Scholar]
- 42.Finigan JH. The coagulation system and pulmonary endothelial function in acute lung injury. Microvasc Res. 2009;77:35–38. doi: 10.1016/j.mvr.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 43.Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–1500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- 44.Witke W, Sutherland JD, Sharpe A, Arai M, Kwiatkowski DJ. Profilin I is essential for cell survival and cell division in early mouse development. Proc Natl Acad Sci U S A. 2001;98:3832–3836. doi: 10.1073/pnas.051515498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wheatley EM, Vincent PA, McKeown-Longo PJ, Saba TM. Effect of fibronectin on permeability of normal and TNF-treated lung endothelial cell monolayers. Am J Physiol. 1993;264:R90–96. doi: 10.1152/ajpregu.1993.264.1.R90. [DOI] [PubMed] [Google Scholar]
- 46.Moldovan NI, Milliken EE, Irani K, Chen J, Sohn RH, Finkel T, Goldschmidt-Clermont PJ. Regulation of endothelial cell adhesion by profilin. Curr Biol. 1997;7:24–30. doi: 10.1016/s0960-9822(06)00024-8. [DOI] [PubMed] [Google Scholar]
- 47.Fu Y, Liu HW, Forsythe SM, Kogut P, McConville JF, Halayko AJ, Camoretti-Mercado B, Solway J. Mutagenesis analysis of human SM22: characterization of actin binding. J Appl Physiol. 2000;89:1985–1990. doi: 10.1152/jappl.2000.89.5.1985. [DOI] [PubMed] [Google Scholar]
- 48.Lees-Miller JP, Heeley DH, Smillie LB. An abundant and novel protein of 22 kDa (SM22) is widely distributed in smooth muscles. Purification from bovine aorta. Biochem J. 1987;244:705–709. doi: 10.1042/bj2440705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lees-Miller JP, Heeley DH, Smillie LB, Kay CM. Isolation and characterization of an abundant and novel 22-kDa protein (SM22) from chicken gizzard smooth muscle. J Biol Chem. 1987;262:2988–2993. [PubMed] [Google Scholar]
- 50.Shen J, Yang M, Ju D, Jiang H, Zheng JP, Xu Z, Li L. Disruption of SM22 promotes inflammation after artery injury via nuclear factor kappaB activation. Circ Res. 2010;106:1351–1362. doi: 10.1161/CIRCRESAHA.109.213900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Raman D, Sai J, Neel NF, Chew CS, Richmond A. LIM and SH3 protein-1 modulates CXCR2-mediated cell migration. PLoS One. 2010;5:e10050. doi: 10.1371/journal.pone.0010050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Traenka J, Hauck CR, Lewandrowski U, Sickmann A, Gambaryan S, Thalheimer P, Butt E. Integrin-dependent translocation of LASP-1 to the cytoskeleton of activated platelets correlates with LASP-1 phosphorylation at tyrosine 171 by Src-kinase. Thromb Haemost. 2009;102:520–528. doi: 10.1160/TH09-03-0143. [DOI] [PubMed] [Google Scholar]
- 53.Neumann P, Gertzberg N, Vaughan E, Weisbrot J, Woodburn R, Lambert W, Johnson A. Peroxynitrite mediates TNF-alpha-induced endothelial barrier dysfunction and nitration of actin. Am J Physiol Lung Cell Mol Physiol. 2006;290:L674–L684. doi: 10.1152/ajplung.00391.2005. [DOI] [PubMed] [Google Scholar]
- 54.Liu B, Tewari AK, Zhang L, Green-Church KB, Zweier JL, Chen YR, He G. Proteomic analysis of protein tyrosine nitration after ischemia reperfusion injury: mitochondria as the major target. Biochim Biophys Acta. 2009;1794:476–485. doi: 10.1016/j.bbapap.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakanishi M, Deyashiki Y, Ohshima K, Hara A. Cloning, expression and tissue distribution of mouse tetrameric carbonyl reductase. Identity with an adipocyte 27-kDa protein. Eur J Biochem. 1995;228:381–387. [PubMed] [Google Scholar]
- 56.Kim TS, Dodia C, Chen X, Hennigan BB, Jain M, Feinstein SI, Fisher AB. Cloning and expression of rat lung acidic Ca(2+)-independent PLA2 and its organ distribution. Am J Physiol. 1998;274:L750–761. doi: 10.1152/ajplung.1998.274.5.L750. [DOI] [PubMed] [Google Scholar]
- 57.Mo Y, Feinstein SI, Manevich Y, Zhang Q, Lu L, Ho YS, Fisher AB. 1-Cys peroxiredoxin knock-out mice express mRNA but not protein for a highly related intronless gene. FEBS Lett. 2003;555:192–198. doi: 10.1016/s0014-5793(03)01199-2. [DOI] [PubMed] [Google Scholar]
- 58.Wang Y, Feinstein SI, Fisher AB. Peroxiredoxin 6 as an antioxidant enzyme: protection of lung alveolar epithelial type II cells from H2O2-induced oxidative stress. J Cell Biochem. 2008;104:1274–1285. doi: 10.1002/jcb.21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Benarafa C, Priebe GP, Remold-O'Donnell E. The neutrophil serine protease inhibitor serpinb1 preserves lung defense functions in Pseudomonas aeruginosa infection. J Exp Med. 2007;204:1901–1909. doi: 10.1084/jem.20070494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Woods DE, Cantin A, Cooley J, Kenney DM, Remold-O'Donnell E. Aerosol treatment with MNEI suppresses bacterial proliferation in a model of chronic Pseudomonas aeruginosa lung infection. Pediatr Pulmonol. 2005;39:141–149. doi: 10.1002/ppul.20167. [DOI] [PubMed] [Google Scholar]
- 61.Lamb NJ, Gutteridge JM, Baker C, Evans TW, Quinlan GJ. Oxidative damage to proteins of bronchoalveolar lavage fluid in patients with acute respiratory distress syndrome: evidence for neutrophil-mediated hydroxylation, nitration, and chlorination. Crit Care Med. 1999;27:1738–1744. doi: 10.1097/00003246-199909000-00007. [DOI] [PubMed] [Google Scholar]