

Abstract
Monitoring changes in the fluorescence of metabolic chromophores, reduced nicotinamide adenine dinucleotide and flavin adenine dinucleotide, and the absorption of cytochromes, is useful to study neuronal activation and mitochondrial metabolism in the brain. However, these optical signals evokedby stimulation, seizures and spreading depression in intact brain differ from those observed in vitro. The responses in vivo consist of a persistent oxidized state during neuronal activity followed by mild reduction during recovery. In vitro, however, brief oxidation is followed by prolonged and heightened reduction, even during persistent neuronal activation. In normally perfused, oxygenated and activated brain tissue in vivo, partial pressure of oxygen (PO2) levels often undergo a brief ‘dip’ that is always followed by an overshoot above baseline, due to increased blood flow (neuronal–vascular coupling). By contrast, in the absence of blood circulation, tissue PO2 in vitro decreases more markedly and recovers slowly to baseline without overshooting. Although oxygen is abundant in vivo, it is diffusion-limited in vitro. The disparities in mitochondrial and tissue oxygen availability account for the different redox responses.
Changes in redox level of mitochondrial respiratory chain components can be monitored in live brain tissue by optical imaging
Changes in fluorescence of metabolic cofactors [i.e. reduced nicotinamide adenine dinucleotide (NADH)‡ and flavin adenine dinucleotide (FAD)] involved in energy processes, and in the absorption spectrum of cytochromes, provide a measure of their redox level. Mitochondrial energy metabolism is tightly coupled with neuronal activity, and changes in metabolic activity alter the redox level of these cofactors. Optical changes related to redox state have been used for many years to gauge the metabolic activity in brain and in other organs. Relating redox responses to electrical, mechanical or secretory processes has provided a rich source of data on the coupling of metabolic activity to their function. Such investigations in mammalian brain shed light on the mechanism of seizures, of spreading depression (SD) and of hypoxia and ischemia. Recently, however, a discrepancy has become apparent between the pattern of redox changes in intact, perfused brain ‘in vivo’*, and isolated preparations of central neurons, tissue slices and slice cultures maintained ‘in vitro’† in an organ bath. This essay aims at resolving the apparent discrepancy.
Mitochondrial oxidative metabolism utilizes the substrate pyruvate, derived from glucose, to generate the reduced form of the coenzyme NAD (NADH) from its oxidized form NAD+, in addition to reduced FAD (FADH2) from FAD in the tricarboxylic acid (TCA) cycle (Figure 1). NADH is also produced during glycolysis, and intercompartmental redox gradients can be resolved between the cytosol and mitochondria through the malate–aspartate shuttle. The subsequent oxidation of NADH and FADH2 in the electron transport chain establishes a potential across the inner mitochondrial membrane, which enables the production of ATP [1,2].
Figure 1.
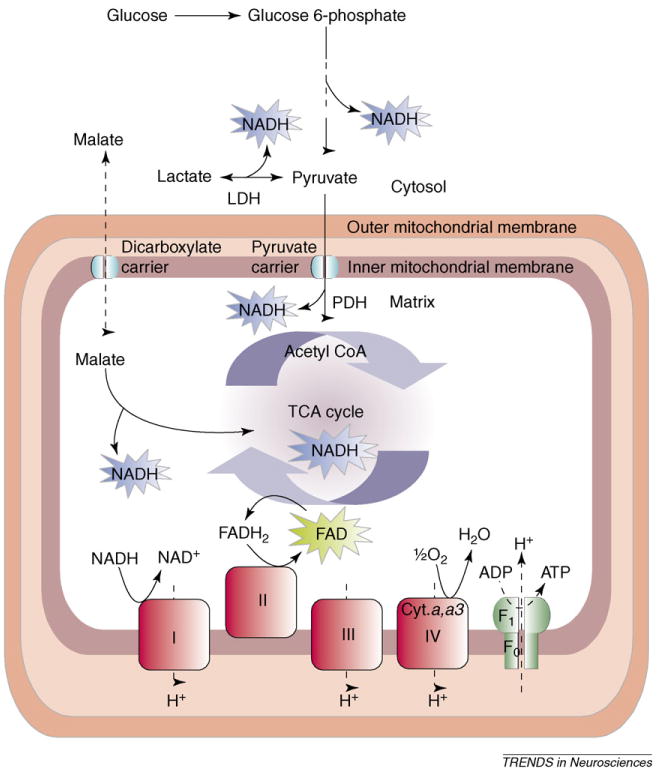
Simplified diagram of sites of reduction and oxidation in energy metabolism. NADH and FADH2 are generated during glycolysis and in the TCA cycle. NADH is oxidized at complex I of the electron transport train. FAD is reduced in the TCA cycle and oxidized by complex II. Molecular oxygen is reduced to H2O by complex IV (cytochrome a,a3). Abbreviation: cyt. a,a3, cytochrome a,a3.
Because NADH is fluorescent when excited with UV light (at 340–370 nm, emission at 450 nm) while NAD+ is not, NADH oxidation leads to a decrease in fluorescence. FAD is fluorescent (excitation at 450 nm and emission above 510 nm), so that oxidation of FADH2 to FAD causes an increase in its fluorescence [3–9]. The fluorescence of NADH cannot be separated from that of NADPH, but the two cofactors are known to act in concert [10]. The fluorescence of FAD cannot be distinguished from other flavoproteins, but presumably the fraction of the signal that varies with metabolic activity is derived from the FADH2–FAD pair [11].
Cytochrome a,a3 acts at the last step in electron transport, the reduction of O2 to water at complex IV, while NADH–NAD+, at the other end of the transport chain in complex I, mediates the entry of reducing equivalents (Figure 1) [2]. Changes in redox level of the respiratory chain pigments cytochrome a,a3 can be detected by spectrophotometry, but a cannot be distinguished from a3. The two subunits are, however, functionally yoked within complex IV (Figure 1). The light absorption maximum for the reduced form of cytochrome a,a3 is at 603–605 nm. Fluctuations in the light absorption at these wavelengths are directly related to the redox state of these respiratory pigments and can be detected as the difference in reflected light intensity between two different spectral bands, one at the specific wavelength for the cytochrome absorbance and the other at a ‘neutral’ (isosbestic) wavelength, which serves as a reference [5].
Redox levels of the metabolic cofactors NADH, FADH2 and cytochromes have been presumed to be related to the activation of, and net flux in, metabolic pathways (glycolysis, TCA cycle, mitochondrial electron transport), and hence to the production of ATP [12–14] (see also Discussion and conclusions). Jöbsis and associates [4,5] attributed the observed NADH fluorescence in intact cortex primarily to the mitochondrial fraction rather than to the cytosolic fraction, as the close interaction between the high concentration of NADH molecules in the matrix and associated dehydrogenase enzymes can enhance the fluorescence compared with that in free solution, such as in cytosol (see also Refs [16,17]). The signals attributed to FAD and cytochromes originate entirely from mitochondria where these cofactors are confined as long as mitochondria are intact. Jöbsis and associates also point out that in mammalian neocortex, 91% of the mitochondria reside in neurons and only a minority are found in glial cells (e.g. Ref. [5]; but for hippocampal neuropil, see Ref. [18]).
Various technical difficulties complicate the imaging of redox levels. Non-metabolic chromophores and tissue light scattering can interfere with the monitoring of NADH, FAD and cytochromes. In intact brain, hemoglobin (Hb) in the microvessels is the most important source of interference because it absorbs excitation light and quenches the emitted fluorescence [4]. Also, NADH in red blood cells can interfere with the measurement of NADH in tissue when blood flow is changing [4]. To correct for the interference by Hb, the light reflected at the isosbestic wavelength of Hb is subtracted from the fluorescence or absorbance signal at the wavelength relevant to the redox state of the compound that is being studied. Effectiveness of the correction was demonstrated by the intra-arterial injection of a bolus of saline to dilute Hb, which brightened the cortical surface yet did not affect the corrected NADH signal [19]. Also, during both hypoxia or ischemia, NADH and cytochrome a,a3 redox levels became reduced, even though blood volume increases in the former but decreases in the latter case [5,20–23].
Except for minimal residual amounts in small vessels in tissue slices, Hb is absent in preparations in vitro but changes in light scattering and reflectance can still interfere with the NADH and FAD fluorescence in tissue slices and cultures [24]. Fluorescence can decrease not only owing to redox level change but also if the total pool (NAD+ + NADH, or FAD + FADH2) is depleted, such as with irreversible neuronal damage manifested by loss of evoked neuronal responses (e.g. Refs [25,26]).
Redox responses in excited brain tissue in vivo
Autofluorescence of NADH in cerebral cortex of anesthetized animals was first recorded decades ago by Jöbsis and collaborators [3,4,27]. Neuronal activation was induced by electrical stimulation, seizure discharges (Figure 2) or spreading depression (SD). SD involves massive depolarization of neurons and glial cells, leading to redistribution of ions and cell swelling [28]. Each of these interventions caused a decrease in corrected NADH fluorescence, indicating NADH oxidation (Figure 2). During the period of recovery following activation, NADH fluorescence returned to a level slightly higher than baseline (increased reduction). The responses were consistently oxidative as long as cerebral blood flow (CBF) and oxygen supply were adequate. By contrast, during moderate hypoxia or impaired CBF, the NADH baseline redox level became reduced and neuron excitation was accompanied by a further transient reduction [20,22,27,29,30].
Figure 2.
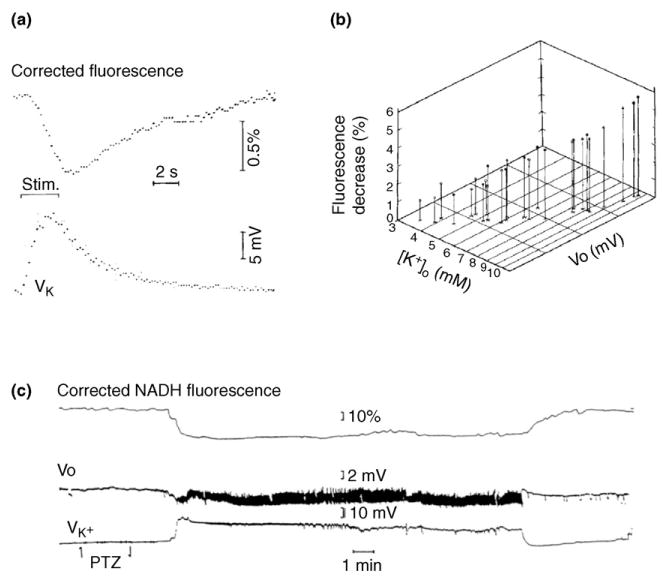
NADH fluorescence recorded in vivo in anesthetized cat neocortex decreases in response to electrical stimulation and during seizure discharge, signaling oxidation. (a) NADH fluorescence corrected for changes in reflectance, and voltage of K+-selective electrode. Each trace is the average of eight responses to 3 s stimulus trains. The fluorescence decrease is delayed relative to the increase in [K+]o. (b) The three-way correlation of the maximal amplitudes of the fluorescence decrease, the peak extracellular potassium concentration ([K+]o) and the negative DC potential shift (Vo). The stimulus intensities and train durations were varied for each point in the graph. Each point represents a single response, all from one cortical site. (c) Corrected NADH fluorescence, Vo shift and K+-selective electrode potential during a seizure induced by i.v. administration of pentylenetetrazol (PTZ). The original experiments shown were performed in the laboratory of our late colleague, Frans Jöbsis, to whose memory this article is dedicated. Modified, with permission, from Ref. [15].
To monitor the redox state of cytochrome a,a3, Jöbsis, Rosenthal and collaborators used variations in its absorbance spectrum (detected as changes in reflected light) [5]. Similarly to NADH, cytochrome a,a3 also became oxidized during electrical stimulation, seizures and SD in normally oxygenated cortex, but was reduced in response to the same stimuli during hypoxia or when tissue perfusion became insufficient [22,20,31]. Jöbsis and associates showed that 20–40% of the ‘labile’ (i.e. utilizable) fraction of cytochrome a,a3 was in the reduced form in the ‘resting’ (unstimulated and anesthetized) cat, rabbit and rat neocortex [5,12,23].
Combining optical with electrical recording, a three-way correlation was detected among the oxidative responses of NADH, the increases in interstitial potassium [K+]o and the sustained [direct current (DC)] negative potential shifts evoked by repetitive electrical stimulation [15,32,33] (Figure 2a,b). Only during seizure discharges was the correlation broken: NADH oxidation exceeded what could have been expected from the increase in [K+]o. It was inferred that the metabolic response was in large part determined by the demands of restoring ion distributions, but that during seizures more than the usual fraction of the energy was expended on processes other than ion transport.
Numerous reports confirmed that neuronal excitation in intact brain was accompanied by sustained oxidation of NADH [19,30,34–45]. A close correlation was also observed between NADH oxidation and oxygen consumption in cat and rat neocortex [46,47]. Following brief (∼5 s) direct electrical stimuli in intact cortex, the initial oxidation was followed by a slight reduction and oxygen consumption was elevated only during the initial oxidation [46].
Of particular importance are the recent studies by Strong et al. [40,41,48], who imaged NADH fluorescence in the region surrounding ischemic or traumatized brain foci, and tracked the course of peri-infarct depolarization (PID) waves. PID waves are similar to SD. NADH fluorescence increased transiently while a PID propagated within the penumbral region (see also Ref. [49]), whereas NADH fluorescence decreased with each wave as it emerged into adjacent healthy, well-perfused cortex [40,48].
Vascular responses and tissue oxygen tension appear to vary among species. In gerbil neocortex, Hashimoto et al., [50] found NADH fluorescence increased during the depolarization phase of SD, and then decreased (oxidation) during the recovery. In this species, however, unlike in cat and rat, during the depolarization, the blood flow did not change, but it increased during the subsequent recovery, coincident with NADH oxidation. Earlier, Mayevsky et al. [51] reported that during SD, the gerbil cortex was more prone to hypoxia than rat cortex. In a recent paper, Takano et al. [52] report that in mouse neocortex, partial pressure of oxygen (PO2) decreased precipitously during the passage of an SD wave in spite of the increased blood flow. The NADH response was recorded at high spatial resolution by two-photon microscopy. The response varied from one micro-region to the next. In the close vicinity of a blood-perfused capillary, NADH became oxidized during the SD-related voltage shift and reduced thereafter, but at some distance from O2 sources NADH was reduced directly after the onset of the SD.
Recently Shibuki et al. [53] recorded flavoprotein (mainly FAD) fluorescence changes from rat sensory cortex in response to direct cortical as well as peripheral sensory stimulation. An increase in fluorescence (oxidation) (uncorrected fluorescence recording) outlasted the stimulation. Other studies have shown biphasic stimulation-evoked responses of flavoprotein fluorescence in rat sensory cortex [54] and in mouse cerebellar cortex [55]. During stimulation, fluorescence briefly increased, indicating oxidation, and then decreased. Weber et al. [54] consider it plausible that the apparent delayed decrease in light intensity was in fact caused by interference by Hb. However, Reinert et al. [55] report evidence that the delayed decrease in FAD fluorescence (reduction) after the stimulation was due neither to changes in blood flow nor to Hb oxygenation.
In summary, many reports agree that NADH, FADH2 and cytochrome a,a3 become oxidized during electrical or ‘physiological’ stimulation as well as seizure discharges and SD in the intact brain, provided that vascular responses and oxygen supply respond normally. Thereafter, during recovery, redox levels shift into a reduced state in some, but not in all, cases. Careful control experiments show that these recordings were not significantly contaminated by artifact.
Redox responses of mitochondrial cofactors in isolated neurons and brain tissue in vitro
Duchen adapted optical detection methods to gauge the redox state of mitochondrial cofactors in isolated cells [6]. Depolarization of dissociated dorsal root ganglion neurons was associated with an initial brief oxidation followed by a more prolonged reduction (increased NADH fluorescence) of the NADH–NAD+ pair. This response was related to mitochondrial depolarization and it appeared to be calcium dependent [56].
Subsequently, several laboratories recorded NADH and/or FAD fluorescence responses in brain tissue slices and in organotypic slice cultures. Typically, investigators have found that electrical stimulation in vitro was accompanied by transient oxidation followed by amore prolonged shift to the reduced state of both the NADH– NAD+ and FADH2–FAD pairs [7–9,13,17,18,25,57–59]. Seizure-like discharges and spreading depression were also accompanied by these biphasic NADH and FAD responses (Figure 3). The initial oxidation rapidly reached maximum and then started to diminish, crossing the baseline to shift to reduction in a few seconds, even if the neuronal activity (or SD) continued. The reduction phase consistently outlasted the electrical activity (Figure 3). Except for very short stimulus trains, the duration of the transient oxidation was independent of the duration of the stimulation. The more intense the neuronal activation, the larger and more prolonged was the reduction phase. Consistent with these findings, Shibuki et al. [53] reported primarily oxidation of flavoprotein in rat cortical slices during very brief (1 s) electrical stimulation, but the recordings were not sufficiently long to detect any later flavoprotein reduction.
Figure 3.
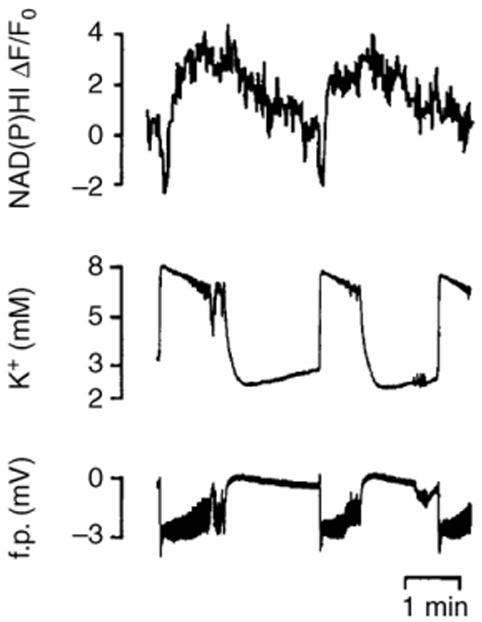
Changes in NAD(P)H fluorescence, extracellular potassium and DC field potential, during low Mg2+-induced epileptiform activity in a hippocampal slice under interface conditions. Note the short NAD(P)H oxidation and prolonged reduction during each burst of seizure-like discharge. Abbreviation: f.p., field potential. Adapted, with permission, from Ref. [13].
Whereas in cat cortex, in vivo NADH oxidation was linearly related to the increase in [K+]o [15] (Figure 2), in rat organotypic slice cultures, the reduction following the oxidation was correlated with [K+]o [7]. When NADH and FAD were recorded simultaneously, the FAD optical signal evoked by stimulation was similar to the NADH signal but it was inverted. Because the two fluorescence signals move in opposite directions, it is unlikely that non-metabolic optical effects produced the biphasic sequence, although light scattering changes might have influenced their magnitude. Kosterin et al. [60] also observed in nerve fiber terminals in isolated mouse neurohypophysis oxidation of NADH and FADH2 during brief (up to 2 s) stimulation that outlasted the stimulus train but that was then followed by a delayed, large persistent reduction. In cultured embryonal mouse Purkinje cells, Hayakawa et al. [61] recorded NAD(P)H oxidation correlated with calcium-induced mitochondrial depolarization.
The origin of the reduction phase of the in vitro NADH– NAD+ response is controversial. Kasischke et al. [18], using high resolution imaging, concluded that the early oxidation originates in neuronal mitochondria, while the later reduction is generated by glycolysis in astrocytic cytoplasm. However, Brennan et al. [17] concluded that both the oxidation and the reduction phase of the fluorescent signal represent mainly neuronal mitochondrial processes (see also Ref. [59]). However, it appears that little oxygen is utilized during the recovery (i.e. reduction) phase that follows stimulation in both in vivo and in vitro preparations [46,59], perhaps suggesting that this phase represents the restoration of the previously depleted reduced fraction of metabolic intermediates.
Thus, reports from numerous laboratories agree that, in isolated neurons or brain slices maintained in an organ bath, stimulation evokes transient oxidation NADH and FADH2 initially, which is followed by reduction even if the excited state persists.
Oxygen availability – resolving the discrepancy between in vivo and in vitro preparations
Similarly to the redox response in vivo, the onset of neuronal stimulation in vitro is accompanied by an immediate oxidation of metabolic cofactors. However, there are two crucial differences: (i) in the intact brain, oxidation continues for as long as the tissue is excited (Figure 2c), whereas in vitro it is limited in duration (Figure 3); (ii) the delayed phase of reduction is weak in vivo and it occurs only during the recovery that follows activation, whereas in vitro the reduction is frequently larger in amplitude than the oxidation and it occurs earlier, regardless of the duration of the excitation. In this article, we suggest that these differences can be explained by the relative oxygen availability in each situation.
Neuronal stimulation in the intact brain frequently causes a brief decrease in tissue PO2 which is referred to as the ‘initial dip’ [62,63]. According to Offenhauser et al. [63], the dip is more pronounced in cerebellar cortex than elsewhere in the central nervous system (CNS), presumably because of different neuronal–vascular coupling. They suggest that the dip occurs while neurons are using up tissue oxygen reserve [63]. The dip is not detected under all conditions or in all regions [64], and is generally followed by overabundant blood flow and an overshoot of PO2. Leão [65] first observed that during SD cerebral veins fill with bright red oxygenated blood, indicating an increase in blood flow in excess of the oxygen demand. An ‘oversupply’ of oxygen has been confirmed for all forms of increased neuronal excitation, evoked by ‘physiological’ stimulation as well as by seizures and SD, except in cases of circulatory or respiratory failure. It can be detected both as an increase in tissue PO2 above its ‘resting’ level and also in so-called blood oxygen level dependent (BOLD) signals (see e.g. Refs [63,64,66–71] and many others). The increase in blood flow and hence of tissue oxygen availability can, however, be suppressed by blocking the production of the vasodilator agent nitric oxide [63]. When neuronal–vascular coupling is so prevented, the stimulus-induced PO2 decrease (‘dip’) is enhanced and prolonged, resembling in vitro conditions [59]. In the case of SD, hyperemia predominates while neurons are depolarized but this is usually followed by prolonged hypo-perfusion [72–78]. The late hypo-perfusion following SD can cause delayed reduction of NADH–NAD+ [44]. In the cortex of anesthetized mice, the response varies among micro-domains, according to the distance from capillaries [52].
In contrast to the intact brain, vascular responses are absent in brain slices, and oxygen availability is diffusion limited. The gas phase of the tissue chamber (for interface slices) usually contains 95% O2, resulting in a PO2 in the bathing fluid that is much higher than physiological conditions in vivo. However, within a 350–450 μm thick tissue slice or 500–600 μm thick neurohypophysis the distance for O2 diffusion is much greater than from capillaries to neurons in gray matter in vivo, depressing the PO2 deep within the tissue [9,59,79,80]. Additionally, in the absence of Hb, the amount of oxygen dissolved in an aqueous solution at equal partial pressure is much lower than blood oxygen content.
A PO2 gradient exists within unstimulated tissue slices, declining from 500 mmHg at the surface to less than 200 mmHg in the center of a slice [59]. The PO2 measured with similar polarographic microelectrodes or electron paramagnetic resonance in cerebral cortex in vivo varied between 15 and 80 mmHg when blood oxygen level was normal [79,81,82]. During excitation in hippocampal tissue slices, rapid uptake of O2 into mitochondria results in a profound decrease in local tissue PO2 (Figure 4) [59]. In relating PO2 microelectrode recordings to NADH autofluorescence one must ask whether the two signals originate from comparable components in the tissue. The depth from which fluorescence of ultraviolet excitation light can be collected has been variously estimated to be effective within 0.4 to 1.2 mm from the surface (refer to Refs [3,13,83]). The signal decreases with distance. For the trials shown in Figure 4, the O2 electrode was inserted to a depth of ∼280 μm. At a more superficial depth of 110 μm, the pre-stimulus baseline PO2 was higher but the stimulus-induced PO2 change was similar to the one recorded deeper [59]. The PO2 signal was therefore well within the layers of origin of the NADH autofluorescence. It should also be remembered that the excitation light for FAD is of a longer wavelength than the ultraviolet light required for NADH excitation, hence it penetrates deeper, yet it shows oxidation–reduction responses comparable to NADH.
Figure 4.
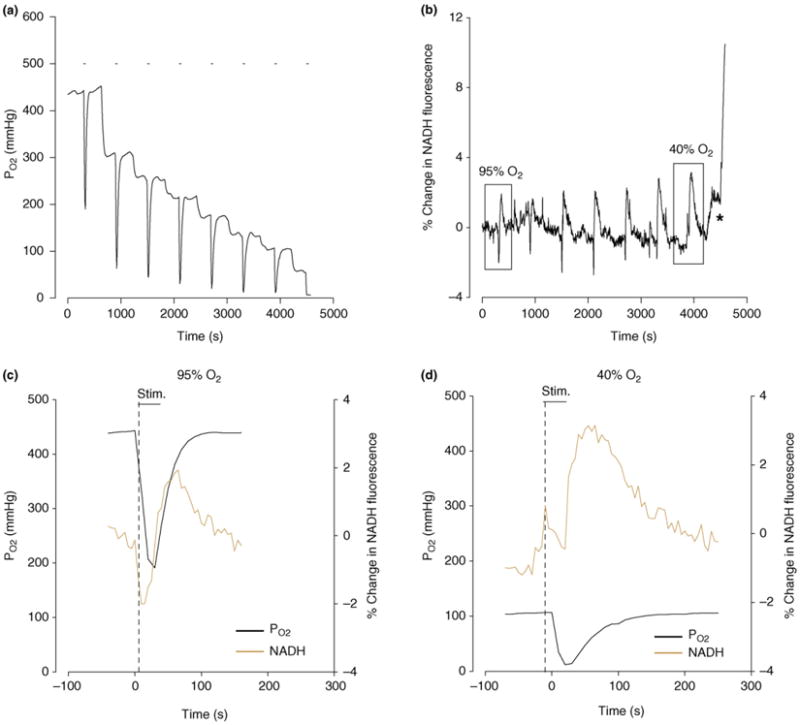
Partial pressure of oxygen (PO2) and change in NADH fluorescence induced by electrical stimulation in a hippocampal tissue slice at different ambient oxygen levels. (a) PO2 recorded by micro-polarography at ∼260 μm depth in stratum radiatum in the CA1 region. Stimulation of Schaffer bundle by eight consecutive 30 s trains is indicated by short bars above traces. Oxygen fraction in gas phase was decreased in steps between stimulations. (b) NADH fluorescence responses in the same region of the same slice as in (a). Initially NADH responses are biphasic indicating oxidation-reduction sequence but, as ambient O2 levels are decreased, the oxidation is suppressed and the reduction enhanced. The asterisk marks the onset of hypoxic spreading depression. (c) and (d) are superimposed, expanded versions of the 1st and 7th responses in (a) and (b) [identified by rectangles in (b)]. Modified, with permission, from Ref. [59].
It therefore appears that local oxygen availability becomes low in vitro during and after stimulation (although not necessarily ‘hypoxic’ – see Discussion), compared with the excess of oxygen supply in vivo. In the intact brain during hypoxia, reduction replaces the normal oxidation. Similarly, lowering the ambient oxygen concentration in tissue slices augmented the reduction phase (Figure 4) [59]. Also, the amplitude of the reduction phase in neurosecretory terminals in vitro was significantly lower in the presence of 95% oxygen than in room air [60] indicating that local oxygen availability can modulate the NADH signal.
During stimulation of blood-perfused brain tissue, the rise in PO2 begins at a time comparable to the onset of local oxygen decrease in isolated brain tissue preparations, regardless of the PO2 level that prevailed before the stimulus (Figure 4). Correspondingly, in intact brain the oxidation of metabolic cofactors persists, whereas in vitro the redox signals convert to a reduction. The differences in local oxygen availability can thus account for the consistently observed differences in the sequence of redox responses.
Discussion and conclusions
During intense neuronal activity, increased energy demand stimulates both the reduction of oxygen to water and the generation of reduced cofactors such as NADH in glycolysis, in addition to NADH and FADH2 in the TCA cycle. But at the same time, as the production of ATP in the electron transport chain accelerates, excess NADH and FADH2 are oxidized to NAD+ and to FAD (Figure 1). Both Ca2+ influx and increase in ADP are important mediators of enhanced ATP production and of TCA cycle activation [2,14,57,60,84].
The autofluorescence of NADH and FAD is related to the (weighted) average redox levels of these compounds in the cell. The rate at which ATP is being produced does not depend on the average redox level of metabolic cofactors, but on the net flux and turnover of reducing equivalents in the electron transport chain. The rates of both oxidation and reduction accelerate the demand for metabolic energy increases, but the rates are, apparently, not exactly balanced. If NADH and FADH2 utilization in the electron chain lead while dehydrogenase and TCA cycle stimulation follow, a temporary imbalance of the redox ratios occurs, resulting in the observed initial oxidation. Fast Ca2+ influx stimulates the mitochondrial electron transport chain [25,56,57,60], which can account for the rapid shift in the direction of oxidation. Preponderance of oxidation can be maintained as long as oxygen supply remains abundant. When O2 availability decreases, more reducing equivalents accumulate at the other end of the reaction chain so that average redox levels become more reduced. Yet the gradient required for electron transport and ATP production can remain adequate even if the O2 level in the extracellular fluid decreases below the ‘resting’ baseline level. It is not the external concentration (or partial pressure) that is critical [21], but the flow of O2 from the extracellular phase to the inner mitochondrial membrane. Pathological hypoxia arises only if the turnover of the metabolic cofactors in energy-generating metabolic processes becomes insufficient.
In summary, the metabolic pathways involved in the turnover of metabolic cofactors during neuronal activation are similar for both in vitro and in vivo preparations. Therefore, findings from both preparations reflect oxidative metabolism and neuronal activity. Advantages of the in vitro preparations include the ability to both monitor and directly manipulate the biochemistry and physiology of the tissue, and the exclusion of the effects of vascular reactivity. However, during synaptic stimulation in vitro, the local tissue PO2 diminishes, advancing the onset and amplitude of the reduction phase. O2 decreases in vitro at the time when the vascular response would normally augment the oxygen availability in the intact brain. Therefore, we propose that differences observed in the changes of NADH and FAD redox levels between intact brain and in vitro preparations arise from the differences in oxygen diffusion and local mitochondrial oxygen availability.
Acknowledgments
We dedicate this essay to the memory of our friend and colleague, the late Frans F. Jöbsis. Supported by grants from NIH (RO1 NS 051586) and VAMC (Merit Review).
Footnotes
The term ‘reduction’ is used to denote shifts in redox state away from oxidation, not a decrease in a variable.
To avoid ambiguity, we use ‘in vivo’ to mean brain tissue in its anatomical location (in situ) and perfused by blood, usually studied in an anesthetized animal with functioning lungs, heart and autonomic reflexes.
‘In vitro’ means isolated cells, tissue slices, cell cultures and organotypic slice cultures maintained in an oxygenated, saline-perfused organ bath.
References
- 1.Duchen MR. Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium. 2000;28:339–348. doi: 10.1054/ceca.2000.0170. [DOI] [PubMed] [Google Scholar]
- 2.Beattie DS. Bioenergetics and oxidative metabolism. In: Devlin TM, editor. Textbook of Biochemistry with Clinical Correlations. Wiley-Liss; 2002. pp. 537–595. [Google Scholar]
- 3.Chance B, et al. Intracellular oxidation-reduction states in vivo. Science. 1962;137:499–508. doi: 10.1126/science.137.3529.499. [DOI] [PubMed] [Google Scholar]
- 4.Jöbsis FF, et al. Intracellular redox changes in functioning cerebral cortex. I. Metabolic effects of epileptiform activity. J Neurophysiol. 1971;34:735–749. doi: 10.1152/jn.1971.34.5.735. [DOI] [PubMed] [Google Scholar]
- 5.Jöbsis FF, et al. Reflectance spectrophotometry of cytochrome aa3 in vivo. J Appl Physiol. 1977;43:858–872. doi: 10.1152/jappl.1977.43.5.858. [DOI] [PubMed] [Google Scholar]
- 6.Duchen MR, Biscoe TJ. Mitochondrial function in Type I cells isolated from rabbit arterial chemoreceptors. J Physiol. 1992;450:13–31. doi: 10.1113/jphysiol.1992.sp019114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kann O, et al. Coupling of neuronal activity and mitochondrial metabolism as revealed by NAD(P)H fluorescence signals in organotypic hippocampal slice cultures. Neuroscience. 2003;119:87–100. doi: 10.1016/s0306-4522(03)00026-5. [DOI] [PubMed] [Google Scholar]
- 8.Shuttleworth CW, et al. NAD(P)H fluorescence imaging of postsynaptic neuronal activation in murine hippocampal slices. J Neurosci. 2003;23:3196–3208. doi: 10.1523/JNEUROSCI.23-08-03196.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foster KA, et al. Optical and pharmacological tools for the study of mitochondrial function and dysfunction in brain tissue. Prog Neurobiol. 2006;79:136–171. doi: 10.1016/j.pneurobio.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.York JL. Enzymes: classification, kinetics and control. In: Devlin TM, editor. Textbook of Biochemistry with Clinical Correlations. Wiley-Liss; 2002. pp. 413–463. [Google Scholar]
- 11.Hassinen I, Chance B. Oxidation-reduction properties of the mitochondrial flavoprotein chain. Biochem Biophys Res Commun. 1968;31:895–900. doi: 10.1016/0006-291x(68)90536-6. [DOI] [PubMed] [Google Scholar]
- 12.Jöbsis FF, Rosenthal M. Behaviour of the mitochondrial respiratory chain in vivo. Ciba Found Symp. 1978;56:149–169. doi: 10.1002/9780470720370.ch9. [DOI] [PubMed] [Google Scholar]
- 13.Schuchmann S, et al. Monitoring NAD(P)H autofluorescence to assess mitochondrial metabolic functions in rat hippocampal entorhinal cortex slices. Brain Res Brain Res Protoc. 2001;7:267–276. doi: 10.1016/s1385-299x(01)00080-0. [DOI] [PubMed] [Google Scholar]
- 14.Duchen MR. Mitochondria and calcium: from cell signalling to cell death. J Physiol. 2000;529:57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lothman E, et al. Responses of electrical potential, potassium levels and oxidative metabolism in cat cerebral cortex. Brain Res. 1975;88:15–36. doi: 10.1016/0006-8993(75)90943-9. [DOI] [PubMed] [Google Scholar]
- 16.Lakowicz JR, et al. Fluorescence lifetime imaging of free and protein-bound NADH. Proc Natl Acad Sci U S A. 1992;89:1271–1275. doi: 10.1073/pnas.89.4.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brennan AM, et al. NAD(P)H fluorescence transients after synaptic activity in brain slices: predominant role of mitochondrial function. J Cereb Blood Flow Metab. 2006;26:1389–1406. doi: 10.1038/sj.jcbfm.9600292. [DOI] [PubMed] [Google Scholar]
- 18.Kasischke KA, et al. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- 19.Harbig K, et al. In vivo measurement of pyridine nucleotide fluorescence from cat brain cortex. J Appl Physiol. 1976;41:480–488. doi: 10.1152/jappl.1976.41.4.480. [DOI] [PubMed] [Google Scholar]
- 20.LaManna JC, et al. Oxygen insufficiency during hypoxic hypoxia in rat brain cortex. Brain Res. 1984;293:313–318. doi: 10.1016/0006-8993(84)91238-1. [DOI] [PubMed] [Google Scholar]
- 21.Rosenthal M, et al. Effects of respiratory gases on cytochrome a in intact cerebral cortex: is there a critical PO2? Brain Res. 1976;108:143–154. doi: 10.1016/0006-8993(76)90170-0. [DOI] [PubMed] [Google Scholar]
- 22.Rosenthal M, et al. In situ studies of oxidative energy metabolism during transient cortical ischemia in cats. Exp Neurol. 1976;50:477–494. doi: 10.1016/0014-4886(76)90020-0. [DOI] [PubMed] [Google Scholar]
- 23.Sylvia AL, et al. Energy metabolism and in vivo cytochrome c oxidase redox relationships in hypoxic rat brain. Neurol Res. 1985;7:81–88. doi: 10.1080/01616412.1985.11739705. [DOI] [PubMed] [Google Scholar]
- 24.Fayuk D, et al. Two different mechanisms underlie reversible intrinsic optical signals in rat hippocampal slices. J Neurophysiol. 2002;87:1924–1937. doi: 10.1152/jn.00231.2001. [DOI] [PubMed] [Google Scholar]
- 25.Heinemann U, et al. Coupling of electrical and metabolic activity during epileptiform discharges. Epilepsia. 2002;43(Suppl. 5):168–173. doi: 10.1046/j.1528-1157.43.s.5.15.x. [DOI] [PubMed] [Google Scholar]
- 26.Foster KA, et al. NADH hyperoxidation correlates with enhanced susceptibility of aged rats to hypoxia. Neurobiol Aging. doi: 10.1016/j.neurobiolaging.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenthal M, Jöbsis FF. Intracellular redox changes in functioning cerebral cortex. II. Effects of direct cortical stimulation. J Neurophysiol. 1971;34:750–762. doi: 10.1152/jn.1971.34.5.750. [DOI] [PubMed] [Google Scholar]
- 28.Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev. 2001;81:1065–1096. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- 29.Sick TJ, et al. Brain potassium ion homeostasis, anoxia and metabolic inhibition in turtles and rats. Am J Physiol. 1982;243:R281–R288. doi: 10.1152/ajpregu.1982.243.3.R281. [DOI] [PubMed] [Google Scholar]
- 30.Vern BA, et al. Effects of ischemia on the removal of extracellular potassium in cat cortex during pentylenetetrazol seizures. Epilepsia. 1979;20:711–724. doi: 10.1111/j.1528-1157.1979.tb04855.x. [DOI] [PubMed] [Google Scholar]
- 31.Kreisman NR, et al. Oxidative metabolic responses during recurrent seizures are independent of convulsant, anesthetic or species. Neurology. 1983;33:861–867. doi: 10.1212/wnl.33.7.861. [DOI] [PubMed] [Google Scholar]
- 32.Rosenthal M, et al. Oxidative metabolism, extracellular potassium and sustained potential shifts in cat spinal cord in situ. Brain Res. 1979;162:113–127. doi: 10.1016/0006-8993(79)90760-1. [DOI] [PubMed] [Google Scholar]
- 33.Rosenthal M, Somjen G. Spreading depression, sustained potential shifts and metabolic activity of cerebral cortex of cats. J Neurophysiol. 1973;36:739–749. doi: 10.1152/jn.1973.36.4.739. [DOI] [PubMed] [Google Scholar]
- 34.Lewis DV, et al. Oxidative metabolism during recurrent seizures in the penicillin treated hippocampus. Electroencephalogr Clin Neurophysiol. 1974;36:347–356. doi: 10.1016/0013-4694(74)90184-9. [DOI] [PubMed] [Google Scholar]
- 35.Lewis DV, Schuette WH. NADH fluorescence and [K+]o changes during hippocampal electrical stimulation. J Neurophysiol. 1975;38:405–417. doi: 10.1152/jn.1975.38.2.405. [DOI] [PubMed] [Google Scholar]
- 36.Mayevsky A, et al. Measurement of extracellular potassium, ECoG and pyridine nucleotide levels during cortical spreading depression in rats. Brain Res. 1974;76:347–349. doi: 10.1016/0006-8993(74)90467-3. [DOI] [PubMed] [Google Scholar]
- 37.Hempel FG, et al. Redox transitions in mitochondria of cat cerebral cortex with seizures and hemorrhagic hypotension. Am J Physiol. 1980;238:H249–H256. doi: 10.1152/ajpheart.1980.238.2.H249. [DOI] [PubMed] [Google Scholar]
- 38.Villringer A, Chance B. Non-invasive optical spectroscopy and imaging of human brain function. Trends Neurosci. 1997;20:435–442. doi: 10.1016/s0166-2236(97)01132-6. [DOI] [PubMed] [Google Scholar]
- 39.Strong AJ, et al. Changes in vascular and metabolic reactivity as indices of ischaemia in the penumbra. J Cereb Blood Flow Metab. 1988;8:79–88. doi: 10.1038/jcbfm.1988.10. [DOI] [PubMed] [Google Scholar]
- 40.Strong AJ, et al. Factors influencing the frequency of fluorescence transients as markers of peri-infarct depolarizations in focal cerebral ischemia. Stroke. 2000;31:214–222. doi: 10.1161/01.str.31.1.214. [DOI] [PubMed] [Google Scholar]
- 41.Strong AJ, et al. The use of in vivo fluorescence image sequences to indicate the occurrence and propagation of transient focal depolarizations in cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:367–377. doi: 10.1097/00004647-199605000-00003. [DOI] [PubMed] [Google Scholar]
- 42.Uematsu D, et al. Alterations in cytosolic free calcium in the cat cortex during bicuculline-induced epilepsy. Brain Res Bull. 1990;24:285–288. doi: 10.1016/0361-9230(90)90217-n. [DOI] [PubMed] [Google Scholar]
- 43.Uematsu D, et al. Cytosolic free calcium and NAD/NADH redox state in the cat cortex during in vivo activation of NMDA receptors. Brain Res. 1989;482:129–135. doi: 10.1016/0006-8993(89)90549-0. [DOI] [PubMed] [Google Scholar]
- 44.Rex A, et al. Cortical NADH during pharmacological manipulation of the respiratory chain and spreading depression. J Neurosci Res. 1999;57:359–370. [PubMed] [Google Scholar]
- 45.Sonn J, Mayevsky A. Effects of brain oxygenation on metabolic, hemodynamic, ionic and electrical responses to spreading depression in the rat. Brain Res. 2000;882:212–216. doi: 10.1016/s0006-8993(00)02827-4. [DOI] [PubMed] [Google Scholar]
- 46.Lewis DV, Schuette WH. NADH fluorescence, [K+]o and oxygen consumption in cat cerebral cortex during direct cortical stimulation. Brain Res. 1976;110:523–535. doi: 10.1016/0006-8993(76)90863-5. [DOI] [PubMed] [Google Scholar]
- 47.Mayevsky A, Weiss HR. Cerebral blood flow and oxygen consumption in cortical spreading depression. J Cereb Blood Flow Metab. 1991;11:829–836. doi: 10.1038/jcbfm.1991.142. [DOI] [PubMed] [Google Scholar]
- 48.Strong AJ, Dardis R. Depolarisation phenomena in traumatic and ischaemic brain injury. In: Pickard JD, editor. Advances and Technical Standards in Neurosurgery. Vol. 30. Springer; 2005. pp. 3–49. [DOI] [PubMed] [Google Scholar]
- 49.Higuchi T, et al. Dynamic changes in cortical NADH fluorescence and direct current potential in rat focal ischemia: relationship between propagation of recurrent depolarization and growth of the ischemic core. J Cereb Blood Flow Metab. 2002;22:71–79. doi: 10.1097/00004647-200201000-00009. [DOI] [PubMed] [Google Scholar]
- 50.Hashimoto M, et al. Dynamic changes of NADH fluorescence images and NADH content during spreading depression in the cerebral cortex of gerbils. Brain Res. 2000;872:294–300. doi: 10.1016/s0006-8993(00)02509-9. [DOI] [PubMed] [Google Scholar]
- 51.Mayevsky A, et al. Factors affecting the oxygen balance in the awake cerebral cortex exposed to spreading depression. Brain Res. 1982;236:93–105. doi: 10.1016/0006-8993(82)90037-3. [DOI] [PubMed] [Google Scholar]
- 52.Takano T, et al. Spreading depression is associated with tissue hypoxia in microwatershed areas. Nat Neurosci. 2007;10:754–762. doi: 10.1038/nn1902. [DOI] [PubMed] [Google Scholar]
- 53.Shibuki K, et al. Dynamic imaging of somatosensory cortical activity in the rat visualized by flavoprotein autofluorescence. J Physiol. 2003;549:919–927. doi: 10.1113/jphysiol.2003.040709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weber B, et al. Optical imaging of the spatiotemporal dynamics of cerebral blood flow and oxidative metabolism in the rat barrel cortex. Eur J Neurosci. 2004;20:2664–2670. doi: 10.1111/j.1460-9568.2004.03735.x. [DOI] [PubMed] [Google Scholar]
- 55.Reinert KC, et al. Flavoprotein autofluorescence imaging of neuronal activation in the cerebellar cortex in vivo. J Neurophysiol. 2004;92:199–211. doi: 10.1152/jn.01275.2003. [DOI] [PubMed] [Google Scholar]
- 56.Duchen MR. Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem J. 1992;283:41–50. doi: 10.1042/bj2830041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kovács R, et al. Ca2+ signaling and changes of mitochondrial function during low Mg2+ induced epileptiform activity in organotypic hippocampal slice cultures. Eur J Neurosci. 2001;13:1311–1319. doi: 10.1046/j.0953-816x.2001.01505.x. [DOI] [PubMed] [Google Scholar]
- 58.Mironov SL, Richter DW. Oscillations and hypoxic changes of mitochondrial variables in neurons of the brainstem respiratory centre of mice. J Physiol. 2001;533:227–236. doi: 10.1111/j.1469-7793.2001.0227b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Foster KA, et al. Interaction between tissue oxygen tension and NADH imaging during synaptic stimulation and hypoxia in rat hippocampal slices. Neuroscience. 2005;132:645–657. doi: 10.1016/j.neuroscience.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 60.Kosterin P, et al. Changes in FAD and NADH fluorescence in neurosecretory terminals are triggered by calcium entry and by ADP production. J Membr Biol. 2005;208:113–124. doi: 10.1007/s00232-005-0824-x. [DOI] [PubMed] [Google Scholar]
- 61.Hayakawa Y, et al. Rapid Ca2+-dependent increase in oxygen consumption by mitochondria in single mammalian central neurons. Cell Calcium. 2005;37:359–370. doi: 10.1016/j.ceca.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 62.Buxton RB. The elusive initial dip. Neuroimage. 2001;13:953–958. doi: 10.1006/nimg.2001.0814. [DOI] [PubMed] [Google Scholar]
- 63.Offenhauser N, et al. Activity-induced tissue oxygenation changes in rat cerebellar cortex: interplay of postsynaptic activation and blood flow. J Physiol. 2005;565:279–294. doi: 10.1113/jphysiol.2005.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lindauer U, et al. No evidence for early decrease in blood oxygenation in rat whisker cortex response to functional actication. Neuroimage. 2001;13:988–1001. doi: 10.1006/nimg.2000.0709. [DOI] [PubMed] [Google Scholar]
- 65.Leão AAP. Pial circulation and spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7:391–396. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- 66.Lowry JP, et al. Measurement of brain tissue oxygen at a carbon paste electrode can serve as an index of increases in regional cerebral blood flow. J Neurosci Methods. 1997;71:177–182. doi: 10.1016/s0165-0270(96)00140-9. [DOI] [PubMed] [Google Scholar]
- 67.Kreisman NR, et al. Oxidative metabolic responses with recurrent seizures in rat cerebral cortex: role of systemic factors. Brain Res. 1981;218:175–188. doi: 10.1016/0006-8993(81)91299-3. [DOI] [PubMed] [Google Scholar]
- 68.Leniger-Follert E. Mechanisms of regulation of cerebral microflow during bicuculline-induced seizures in anaesthetized cats. J Cereb Blood Flow Metab. 1984;4:150–165. doi: 10.1038/jcbfm.1984.23. [DOI] [PubMed] [Google Scholar]
- 69.Mori K, et al. Temporal profile of changes in brain tissue extracellular space and extracellular ion (Na+, K+) concentrations after cerebral ischemia and the effects of mild cerebral hypothermia. J Neurotrauma. 2002;19:1261–1270. doi: 10.1089/08977150260338047. [DOI] [PubMed] [Google Scholar]
- 70.Gjedde A. Coupling of blood flow to neuronal excitability. In: Walz W, editor. The Neuronal Environment: Brain Homeostasis in Health and Disease. Humana Press; 2002. pp. 233–257. [Google Scholar]
- 71.Logothetis NK, Pfeuffer J. On the nature of the BOLD fMRI contrast mechanism. Magn Reson Imaging. 2004;22:1517–1531. doi: 10.1016/j.mri.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 72.Lauritzen M. Cerebral blood flow in migraine and cortical spreading depression. Acta Neurol Scand Suppl. 1987;113:1–40. [PubMed] [Google Scholar]
- 73.Lauritzen M, et al. Persistent oligemia of rat cerebral cortex in the wake of spreading depression. Ann Neurol. 1982;12:469–474. doi: 10.1002/ana.410120510. [DOI] [PubMed] [Google Scholar]
- 74.Hansen AJ, et al. Relationship between local changes in cortical flow and extracellular K+ during spreading depression. Acta Physiol Scand. 1980;109:1–6. doi: 10.1111/j.1748-1716.1980.tb06557.x. [DOI] [PubMed] [Google Scholar]
- 75.Piper RD, et al. Cortical blood flow changes during spreading depression in cats. Am J Physiol. 1991;261:H96–H102. doi: 10.1152/ajpheart.1991.261.1.H96. [DOI] [PubMed] [Google Scholar]
- 76.Wolf T, et al. Extra- and intracellular oxygen supply during cortical spreading depression in the rat. Adv Exp Med Biol. 1996;388:299–304. doi: 10.1007/978-1-4613-0333-6_38. [DOI] [PubMed] [Google Scholar]
- 77.Ayata C, et al. Pronounced hypoperfusion during spreading depression in mouse cortex. J Cereb Blood Flow Metab. 2004;24:1172–1182. doi: 10.1097/01.WCB.0000137057.92786.F3. [DOI] [PubMed] [Google Scholar]
- 78.Tomita M, et al. Initial oligemia with capillary flow stop followed by hyperemia during K+-induced cortical spreading depression in rats. J Cereb Blood Flow Metab. 2005;25:742–747. doi: 10.1038/sj.jcbfm.9600074. [DOI] [PubMed] [Google Scholar]
- 79.Kreisman NR, et al. Local tissue oxygen tension-cytochrome a,a3 redox relationship in rat cerebral cortex in vivo. Brain Res. 1981;218:161–174. doi: 10.1016/0006-8993(81)91298-1. [DOI] [PubMed] [Google Scholar]
- 80.Sick TJ, Somjen GG. Tissue slice. Application to study of cerebral ischemia. In: Ginsberg MD, Bogousslavsky J, editors. Cerebrovascular Disease: Pathophysiology, Diagnosis and Management. Blackwell Science; 1998. pp. 137–156. [Google Scholar]
- 81.Feng ZC, et al. Depth profile of local oxygen tension and blood flow in rat cerebral cortex, white matter and hippocampus. Brain Res. 1988;445:280–288. doi: 10.1016/0006-8993(88)91190-0. [DOI] [PubMed] [Google Scholar]
- 82.Rolett EL, et al. Critical oxygen tension in rat brain: a combined 31P-NMR and EPR oximetry study. Am J Physiol Regul Integr Comp Physiol. 2000;279:R9–R16. doi: 10.1152/ajpregu.2000.279.1.R9. [DOI] [PubMed] [Google Scholar]
- 83.O'Connor MJ, et al. Intracellular redox changes preceding onset of epileptiform activity in intact cat hippocampus. J Neurophysiol. 1972;35:471–483. doi: 10.1152/jn.1972.35.4.471. [DOI] [PubMed] [Google Scholar]
- 84.Chance B, Williams GR. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J Biol Chem. 1955;217:383–393. [PubMed] [Google Scholar]