

Abstract
Synaptic train stimulation (10 Hz × 25 s) in hippocampal slices results in a biphasic response of NAD(P)H fluorescence indicating a transient oxidation followed by a prolonged reduction. The response is accompanied by a transient tissue PO2 decrease indicating enhanced oxygen utilization. The activation of mitochondrial metabolism and/or glycolysis may contribute to the secondary NAD(P)H peak. We investigated whether extracellular lactate uptake via monocarboxylate transporters (MCTs) contributes to the generation of the NAD(P)H response during neuronal activation. We measured the effect of lactate uptake inhibition [using the MCT inhibitor α-cyano-4-hydroxycinnamate (4-CIN)] on the NAD(P)H biphasic response, tissue PO2 response, and field excitatory post-synaptic potential in hippocampal slices during synaptic stimulation in area CA1 (stratum radiatum). The application of 4-CIN (150–250 μmol/L) significantly decreased the reduction phase of the NAD(P)H response. When slices were supplemented with 20 mmol/L lactate in 150–250 μmol/L 4-CIN, the secondary NAD(P)H peak was restored; whereas 20 mmol/L pyruvate supplementation did not produce a recovery. Similarly, the tissue PO2 response was decreased by MCT inhibition; 20 mmol/L lactate restored this response to control levels at all 4-CIN concentrations. These results indicate that lactate uptake via MCTs contributes significantly to energy metabolism in brain tissue and to the generation of the delayed NAD(P)H peak after synaptic stimulation.
Keywords: hypoglycemia, mitochondria, monocarboxylate transporters, α-cyano-4-hydroxycinnamate
NAD(P)+ is fluorescent in its reduced form (NAD(P)H) when excited by UV light at 360 nm (Aubin 1979). Studying changes in NAD(P)H fluorescence in brain tissue allows for real time monitoring of mitochondrial oxidative metabolism (Kann et al. 2003; Foster et al. 2005), as well as CNS activity (Mironov and Richter 2001). In hippocampal slices brief synaptic stimulation of the Shaffer collaterals in the CA1 region results in a rapid decrease in NAD(P)H fluorescence followed by a prolonged NAD(P)H fluorescence elevation (Schuchmann et al. 2001; Shuttleworth et al. 2003). This stimulus-induced NAD(P)H biphasic change has been described in the brain slice preparation of the hippocampus (Shuttleworth et al. 2003; Foster et al. 2005), the cerebral cortex (Lipton 1973), and the brainstem (Mironov and Richter 2001).
The application of both α-amino-3-hydroxy-5-methylis-oxazole-4-propionic acid (AMPA) and NMDA glutamate receptor inhibitors prior to synaptic stimulation dramatically decreased both components of the response (Shuttleworth et al. 2003; Brennan et al. 2006). These results suggest that the NAD(P)H signal depends upon neuronal post-synaptic ionotropic glutamate receptor activation (Shuttleworth et al. 2003). However, results from other studies have suggested that the signal may depend on metabolic neuron–glia interactions mediated by glutamate (Poitry et al. 2000; Kasischke et al. 2004).
Several investigators have proposed that the initial decrease in NAD(P)H following synaptic stimulation is due to mitochondrial oxidation of NAD(P)H to NAD(P)+ and that this process occurs predominantly in neurons (Shuttleworth et al. 2003; Kasischke et al. 2004; Foster et al. 2005). Neuronal depolarization because of synaptic stimulation (Schuchmann et al. 2001) or glutamate application (Shuttleworth et al. 2003) results in rapid oxygen utilization (Foster et al. 2005) and the oxidation of reduced cofactors such as NAD(P)H and FADH in the electron transport chain to produce ATP.
However, the cellular mechanisms that may contribute to the secondary increase of NAD(P)H fluorescence are still controversial. Imaging studies, using dissociated dorsal root ganglion neurons (Duchen 1992) and organotypic hippocampal slice cultures double labeled with cytosolic and mitochondrial Ca2+ indicators (Kann et al. 2003), have revealed that the NAD(P)H overshoot is correlated with an increase in mitochondrial Ca2+ accumulation during stimulation (which was decreased in the absence of Ca2+) (Duchen 1992; Kann et al. 2003). The authors have proposed that mitochondrial Ca2+ accumulation, as a result of neuronal depolarization, leads to the activation of Ca2+-dependent dehydrogenases in the tricarboxylic acid (TCA) cycle and a subsequent NAD(P)H increase (Duchen 1992; Kann et al. 2003). In contrast, data from a study using multiphoton microscopy and spatial three-dimensional processing in the acute hippocampal slice suggested that the reduction phase of the NAD(P)H response occurs primarily in astrocytes (Kasischke et al. 2004). These authors proposed that the activation of astrocytic glycolysis after neuronal stimulation is responsible for the net production of NAD(P)H prior to the conversion of pyruvate to lactate (in which NAD(P)H is oxidized NAD(P)+).
Both glutamate application and intense neuronal activation promote glycolytic lactate release from the glia, which can be taken up by neurons via specialized monocarboxylate transporters (MCTs) (Elekes et al. 1996; Schurr et al. 1999a; Pellerin and Magistretti 2003). Lactate can be used as an energy substrate and can support neuronal activity during or after substrate deprivation (Schurr et al. 1988; Sakurai et al. 2002; Schurr 2006) or during intense neuronal activation (Schurr et al. 1999b) because it can be rapidly converted into pyruvate by lactate dehydrogenase (LDH) without requiring ATP. As lactate can be an effective energy source for neurons, we hypothesize that lactate uptake and metabolism contribute to the NAD(P)H response following neuronal stimulation.
In order to investigate whether cellular lactate uptake is important for the secondary NAD(P)H peak, we have monitored the effect of MCT blocker α-cyano-4-hydroxy-cinnamate (4-CIN) on NAD(P)H fluorescence changes, tissue oxygen levels, and field excitatory post-synaptic potential (fEPSP) following synaptic stimulation in hippocampal slices. Our studies indicate that blocking lactate uptake with 4-CIN (150–250 μmol/L) significantly decreases the reduction phase of the NAD(P)H response after synaptic stimulation, without affecting the early NAD(P)H oxidation phase, indicating that lactate uptake into neurons via MCT may play an important role in this phase of the NAD(P)H signal. In addition, our data support the hypothesis that lactate uptake from an extracellular pool provides a rapid pathway for pyruvate and NAD(P)H generation for oxidative metabolism after neuronal stimulation.
Materials and methods
Tissue slice preparation
Hippocampal tissue slices were prepared from male Fischer 344 rats (75–150 g; Harlan, Indianapolis, IN, USA). All animal use was approved by the Duke University Animal Care and Use Committee. The rats were anesthetized with halothane (Abbott Laboratories, North Chicago, IL, USA) in an anesthesia induction chamber until respirations ceased. The rat was decapitated and the brain was removed from the skull and placed in ice-cold artificial CSF (ACSF) oxygenated with 95% O2/5% CO2 for 2 min. The ACSF solution consisted of (in mmol/L): NaCl 124, KCl 3.0, NaH2PO4 1.25, NaHCO3 24, CaCl2 2.00, MgSO4 2.00, and dextrose 10, pH 7.4. The hippocampus was rapidly dissected and transverse slices (400 μm) were cut on a manual tissue chopper. Slices were transferred immediately to an oxygenated holding chamber maintained at 22°C, and allowed to recover for 2 h. Slices were then transferred to a recording chamber and submerged (∼1 mm) in ACSF buffer which was continuously perfused (1.5 mL/min) and aerated with 95% O2–5% CO2 (Fayuk et al. 2002). The temperature in the chamber was kept at 36.5–37°C for all experimental conditions.
Synaptic stimulation
The Shaffer collateral/commissural pathway was stimulated with a bipolar electrode situated in the stratum radiatum of the CA1 hippocampal region. Stimulus current was adjusted using single pulses (100 μs 0.1–0.3 Hz) to produce a fEPSP of nearly 50% of maximal amplitude. fEPSPs were recorded using glass microelectrodes filled with 0.2 mol/L NaCl (4–8 mΩ) placed in the stratum radiatum. An extended synaptic stimulation was used to generate the NAD(P)H biphasic response which consisted of a 25 s stimulus train (100 μs pulses at 10 Hz) at the same amplitude.
To investigate the relative contribution of various metabolic pathways to the NAD(P)H biphasic response hippocampal slices were exposed to pharmacological manipulation or modified buffers. A stimulus train under control conditions (i.e. ACSF containing 10 mmol/L glucose) was performed in each slice prior to pharmacological manipulation.
To assess the role of lactate uptake in regard to the NAD(P)H signals, we added the MCT competitive antagonist 4-CIN (Halestrap et al. 1974; Broer et al. 1997, 1999) (Sigma, St Louis, MO, USA). Lactate (20 mmol/L) or pyruvate (20 mmol/L) (Sigma) was added to the ACSF in the presence of 4-CIN or during hypoglycemia (20 mmol/L of lactate or pyruvate is an equicaloric concentration of glucose 10 mmol/L). In these experiments, the concentration of NaCl was adjusted to maintain osmolarity. In addition, during any pharmacological manipulation the pH of each solution was monitored and adjusted to pH 7.4 when necessary.
Ionotropic and metabotropic glutamate receptor antagonists were used in a separate series of experiments. The NMDA receptor antagonist D-(–)-2-amino-5-phosphonopentanoic (APV) acid (100 μmol/L) was applied with kainate/AMPA receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) (50 μmol/L) to block ionotropic glutamate receptors. A combination of (RS)-1-aminoindan-1,5-dicarboxylic acid (AIDA) (100 μmol/L) and (RS)-1-amino-5-phosphonoindan-1-carboxylic acid (APICA) (50 (μmol/L) was used to block group I and II metabotropic glutamate receptors; while (RS)-a-methylserine-o-phosphate (MSOP) (100 μmol/L) was used to block Group III metabotropic glutamate receptors (Tocris, St Louis, MO, USA).
NAD(P)H fluorescence imaging
Changes in NAD(P)H fluorescence in hippocampal slices were monitored using a 290–370 nm excitation filter and a 420 nm long pass filter for the emission (Omega Optical, Brattleboro, VT, USA) as previously described by our laboratory (Foster et al. 2005). The light source was a Lambda DG-4 (Sutter Instruments, Novato, CA, USA) equipped with a stabilized xenon arc lamp. Slices were epiilluminated with an incident angle of 45° and imaged through a Nikon upright microscope (UM-2) with a compound lens (4×, NA 0.13) (Nikon Inc., Melville, NY, USA). Slices were imaged using a linear, cooled 12-bit CCD camera (Cooke Instruments Sensicam QE, Auburn Hills, MI, USA) with 1280 × 1040 digital spatial resolution. Because of the low level of fluorescence emission for this fluorophore, NAD(P)H images were acquired every 5 s as 8 × 8 binned images (effective spatial resolution is 160 × 130 pixels). These imaging specifications resulted in stable digital images with high-quality signal/noise ratios, as well as shorter exposure time (∼300 ms) to avoid tissue damage. The images were stored on a computer as 12-bit files (4096 intensity level). Each binned pixel corresponded to a slice region of 144 μm2. Only those slices with a stable baseline, ≤ 5% NAD(P)H fluorescence change in the initial baseline period (10 min), and a fluorescence intensity between 1000 and 2000 optical density levels, were used for data analysis. Changes in NAD(P)H were expressed as the percentage changes in fluorescence over the baseline [(ΔF/F) × 100].
When slices were incubated with either 4-CIN or low glucose (2.5 mmol/L), there was a decline in the NAD(P)H baseline fluorescence over time that was steeper than control slices. The decline of baseline fluorescence over a period of 15 min was 7% in control slices and ∼12%, 14%, and 25% in the presence of 150, 250, and 500 μmol/L 4-CIN, respectively. This decline was probably related to a decrease in the NADH/NAD+ ratio, as previously observed by other investigators after variation of substrate availability or hypoglycemia (Garofalo et al. 1988; Scholz et al. 1995). As the decline in NAD(P)H fluorescence was reversible after the slices were returned to the control buffer it is unlikely that it represents a loss of the total NAD(P)H pool. The slow decrease of fluorescence was corrected using a curve fitting program and linear regression (Prism; Graph Pad, San Diego, CA, USA). Data were analyzed for statistical significance using anova followed by Tukey's multicomparison test and are shown as mean ± SEM.
Oxygen recording
A Clark-style oxygen microelectrode (737gc, tip diameter 10 μm; Diamond General, Ann Arbor, MI, USA) was used to measure brain tissue PO2 (Foster et al. 2005). The electrode consisted of a glass-insulated Ag/AgCl reference anode with guard cathode. The electrode was connected to a polarographic amplifier (Chemical Microsensor II; Diamond General) and the cathode (noble metal type) was polarized at −800 mV in normal saline at 36°C for up to 12 h before use. A two-point calibration (in mA) was performed following polarization by inserting the electrode in normal saline solution (at 36°C) equilibrated with either 95% O2–5% CO2 or 95% N2–5% CO2–0% O2 (medical grade).
Calibrations were repeated after every slice to determine the PO2 values, calibrated to mmHg. Electrode drift was generally linear over the course of an experiment. The ampere values obtained from the two calibration points in 95% and 0% O2 during an experiment varied by 3.7 ± 2.7% and 8.8 ± 7.4% h−1, respectively. Following calibration, the oxygen electrode was positioned in the stratum radiatum in proximity to the recording electrode and was then manually lowered into the tissue at 50 μm intervals using a micrometer to a depth at which the PO2 was at the minimum (nadir) (Foster et al. 2005). The amplitude of the PO2 transient as a result of synaptic stimulation was calculated by the equation: ΔPO2 = [PO2 (baseline) – PO2 (stim)].
Results
Synaptic stimulation and effect of glutamate receptor antagonists
The stimulation of Schaffer collaterals (10 Hz, 25 s, ∼50% of the maximum fEPSP) resulted in a reproducible NAD(P)H biphasic response, recorded in stratum radiatum of the CA1 region (Fig. 1a). This response consisted of an initial decrease of NAD(P)H autofluorescence intensity which peaked within ∼10 s (representing the NAD(P)H oxidation phase). This phase was immediately followed by a NAD(P)H fluorescence increase beyond baseline which peaked at ∼45 s and is indicative of an increase in the reduced state of the pyridine nucleotide (reduction phase) (Schuchmann et al. 2001) (Fig. 1b). The typical control values in the stratum radiatum for the oxidation and reduction phases were −3.1 ± 0.3% and 3.8 ± 0.2%, respectively (n = 43).
Fig. 1.

Series of images taken before, during, and after the stimulus train. (a) Un-subtracted image indicates the region of interest (ROI) within the stratum radiatum (SR) of the CA1 region. The recording electrode is indicated by the white asterisk. Scale bar = 500 μm. Note: The ROI is situated between the stimulating and recording electrodes. Stratum pyramidale (SP) and stratum oriens (SO) are also indicated. (b) Control: NAD(P)H biphasic response consisted of a brief decrease in NAD(P)H fluorescence (oxidation phase) (10 s), followed by a more prolonged NAD(P)H fluorescence increase (reduction phase) (45 s). (c) α-Cyano-4-hydroxycinnamate (4-CIN): Effect of monocarboxylate transporter inhibition by 4-CIN on the NAD(P)H biphasic response. Slices were incubated with 4-CIN 15 min prior to the stimulus train. In the presence of monocarboxylate transporter blocker 4-CIN (150 μmol/L), the early oxidation (10 s) was not affected, while the secondary NAD(P)H fluorescence peak was significantly decreased (45–80 s).
Previous studies have demonstrated that the NAD(P)H biphasic response following extended train stimulation is dependent on post-synaptic ionotropic glutamate receptor activation (Shuttleworth et al. 2003). However, the degree of sensitivity to glutamate receptor antagonists varies depending on both the specific type of glutamate receptor antagonist applied and the duration and intensity of the stimulation (Shuttleworth et al. 2003; Kasischke et al. 2004; Brennan et al. 2006). Therefore, we tested the sensitivity of the NAD(P)H biphasic response to the application of ionotropic glutamate receptor antagonists under our experimental conditions. Consistent with previous studies we have found that both phases of the response are significantly reduced after the application of NMDA and AMPA/kainate receptor antagonists. The application of APV (50 μmol/L) in combination with CNQX (50 μmol/L) significantly attenuated both phases of the NAD(P)H biphasic response to synaptic stimulation [oxidation phase −2.8 ± 0.3% for control vs. −0.39 ± 0.1% (APV + CNQX); n = 8, ***p < 0.001; reduction phase 3.73 ± 0.3% for control vs. 0.36 ± 0.14% (APV + CNQX); n = 8, ***p < 0.001 by ANOVA and Tukey's multicomparison test). In contrast, the application of metabotropic glutamate receptor antagonist AIDA/RS-APICA or MSOP did not affect the NAD(P)H response [oxidation phase −2.8 ± 0.3% for control vs. −3.13 ± 0.7% (AIDA/RS-APICA); n = 3, p > 0.05; −2.7. ± 0.35% (MSOP); n = 4, p > 0.05; reduction phase 3.73 ± 0.3% for control vs. 1.88 ± 0.16 (AIDA/RS-APICA); n = 3, p > 0.05; 3.03. ± 0.51% (MSOP); n = 4, p > 0.05 by anova and Tukey's multicomparison test].
Effect of monocarboxylate transport inhibitor 4-CIN on the NAD(P)H biphasic response
During intense stimulation in the brain, it is possible that in addition to glucose uptake cells may also take up lactate from the extracellular pool and use it as an energy substrate (Schurr et al. 1988; Pellerin and Magistretti 2003). To investigate the contribution of lactate uptake to the NAD(P)H biphasic response we measured the effect of different concentrations of the MCT inhibitor 4-CIN on the amplitude of the NAD(P)H biphasic fluorescence response. This inhibitor has been shown to block the uptake of lactate in hippocampal slices (Schurr et al. 1997, 1999a), but not the extrusion of lactate from glia (Volk et al. 1997). We perfused hippocampal slices with ACSF containing 4-CIN for 15 min prior to the synaptic stimulation. In the presence of 150 μmol/L 4-CIN the amplitude of the NAD(P)H overshoot was significantly reduced (Fig. 1c) by ∼70% (3.8 ± 0.2% for control, n = 43, vs. 1.29 ± 0.22% for 4-CIN 150 μmol/L, n = 6). When we raised the concentration of 4-CIN to 250 μmol/L the reduction phase of the NAD(P)H response was completely suppressed (Fig. 2a). In contrast, the oxidation phase was not significantly affected by 4-CIN at these concentrations (−3.06 ± 0.17% for control, n = 43, vs. −3.785 ± 0.75% for 150 μmol/L 4-CIN, n = 6, and −2.32 ± 0.4% for 250 μmol/L 4-CIN, n = 6; NS). In some cases, after the application of 150–250 μmol/L 4-CIN the NAD(P)H oxidation phase was a little bit larger and/or longer lasting (Figs 2a and 3), although these changes did not reach significance; this trend might be because of the oxidation phase not being masked by the appearance of the large NAD(P)H overshoot (Shuttleworth et al. 2003). When the concentration of 4-CIN was raised to 500 μmol/L we observed a significant decrease of the oxidation phase by ∼75%. The effects of different 4-CIN concentrations, compared with their paired control, on the biphasic response are summarized in Fig. 3.
Fig. 2.
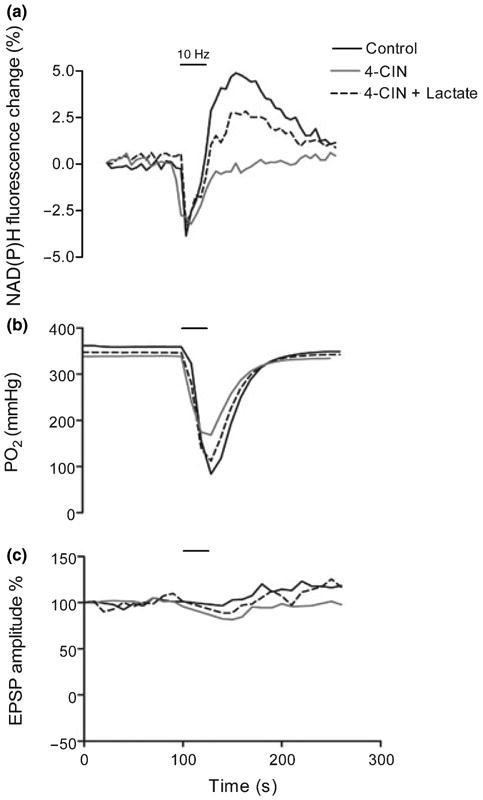
Representative traces showing changes in NAD(P)H fluorescence, tissue PO2, and excitatory post-synaptic potential (EPSP) amplitude after brief synaptic stimulation. Slices were incubated with α-cyano-4-hydroxycinnamate (4-CIN; 15 min) with or without lactate (10 min) prior to the stimulus train. (a) In the presence of 4-CIN (250 μmol/L) the late NAD(P)H peak was reduced, but was restored by lactate supplementation. (b) The transient decrease in tissue PO2 occurring during the stimulus train was reduced by 30% with 4-CIN and restored to control values by lactate supplementation. (c) 4-CIN and lactate did not significantly affect field EPSP amplitude. [Correction added after online publication (18/10/07): at the top of the figure 25 Hz was changed to 10 Hz].
Fig. 3.
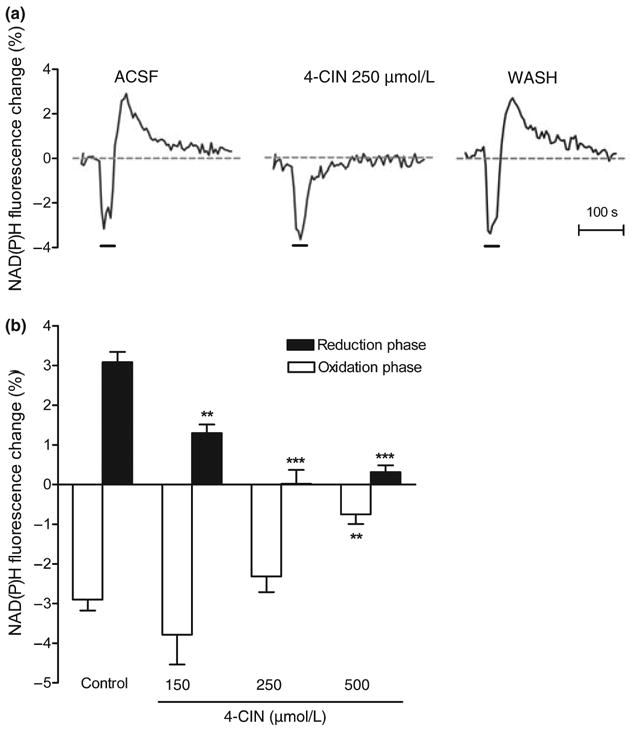
Monocarboxylate transport inhibition by α-cyano-4-hydroxycinnamate (4-CIN) results in the decrease of the NAD(P)H biphasic response. Various concentrations of 4-CIN were applied to the hippocampal slice for 15 min before the stimulus train. (a) Representative traces of the NAD(P)H biphasic response before, during, and after the application of monocarboxylate transporter inhibitor 4-CIN (250 μmol/L). (b) The effect of 4-CIN on the NAD(P)H biphasic response is concentration dependent. The application of 150–250 μmol/L 4-CIN significantly decreased the reduction phase of the NAD(P)H response while the oxidation phase was not affected. At higher concentrations (i.e. 500 μmol/L) both reduction and oxidation were significantly decreased. Data are the mean ± SEM of 5–18 slices/condition. ***p < 0.001 and **p < 0.01 versus control (ANOVA and Tukey's multicomparison test).
Effect of monocarboxylate transport inhibition by 4-CIN on the tissue PO2 response during synaptic stimulation
The application of 4-CIN may result in a severe restriction of pyruvate availability and uptake by mitochondria by both limiting the lactate transport at the plasma membrane and blocking pyruvate uptake at mitochondrial MCT (Halestrap 1975; McKenna et al. 2001). Therefore, we monitored the tissue PO2 response in hippocampal slices to investigate the effect of MCT inhibition by 4-CIN on oxygen consumption. Previous studies have shown that synaptic stimulation induces a rapid intracellular (mitochondria) O2 uptake, leading to a transient decrease in PO2, and therefore a net decrease in extracellular oxygen levels (Foster et al. 2005; Offenhauser et al. 2005). In control slices, the tissue PO2 decreased as soon as the stimulation was initiated but, after the end of the stimulation PO2 returned to baseline levels within ∼70 s (Fig. 2b). In the presence of 150 μmol/L 4-CIN we observed a similar oxygen transient following the stimulus train, although compared with the control condition there was a ∼30% decrease of the tissue PO2 response during synaptic stimulation. A similar effect was observed in the presence 250 μmol/L 4-CIN. The higher concentration of 4-CIN (500 μmol/L) led to a 70% reduction of the PO2 transient (Fig. 4b). The effect of 4-CIN on the oxygen transient was completely reversible and after 15 min washout the amplitude of the tissue PO2 response was restored to control levels (Fig. 4a). The application of 4-CIN for 15 min did not result in any significant changes in fEPSP amplitudes at any concentration used in this study (105.5% ± 1.8 for ACSF vs. 143.4% ± 23.87 for 4-CIN 150 μmol/L, 112.8 ± 13.19 for 4-CIN 250 μmol/L, 87.69 ± 12.68 for 4-CIN 500 μmol/L; n = 5–16, p > 0.05 by ANOVA and Tukey's multicomparison test, fEPSP amplitudes in the presence of 4-CIN are expressed as the % of baseline and compared with controls).
Fig. 4.
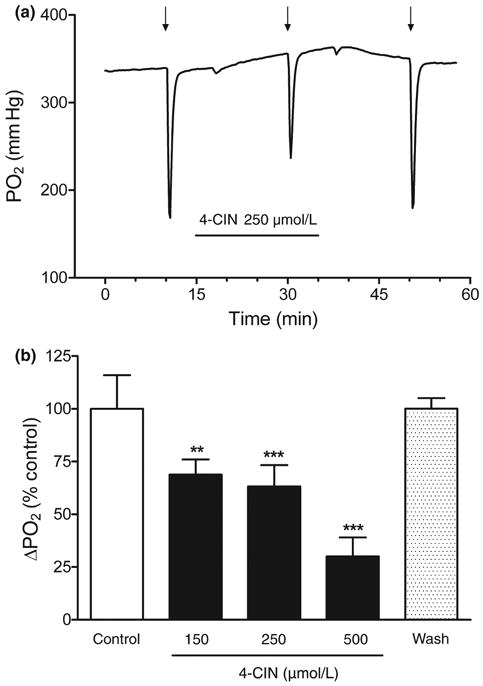
Effect of monocarboxylate transporter inhibitor α-cyano-4-hy-droxycinnamate (4-CIN) on the tissue PO2 response. Various concentrations of 4-CIN were applied to the hippocampal slice for 15 min before the stimulus train. Twenty minutes after 4-CIN was removed from the perfusion buffer the stimulus train was repeated to demonstrate that the effect of 4-CIN on the tissue PO2 response was reversible. (a) Representative traces of the tissue PO2 response before, during, and after the application of monocarboxylate transporter inhibitor 4-CIN (250 μmol/L). In the presence of 4-CIN, the PO2 tissue response during a stimulus train is reduced. The arrows (↓) indicate when the stimulus train started. (b) The effect of 4-CIN, on the tissue PO2 response, was concentration dependent. Data are the mean ± SEM of 4–12 slices/condition. ***p < 0.001 and **p < 0.01 versus control (ANOVA and Tukey's multicomparison test).
The effect of lactate supplementation on the NAD(P)H biphasic response in the presence of 4-CIN
The previous experiments suggested that the reduction phase of the NAD(P)H response is primarily dependent upon lactate uptake via MCT. Therefore, we tested whether lactate supplementation could overcome the competitive block of 4-CIN at the cell membrane transporters and restore the NAD(P)H overshoot. We first addressed the possibility that lactate supplementation alone may increase the amplitude of the NAD(P)H biphasic response and therefore allow the recovery of the response by a different mechanism. In control experiments the addition of lactate (10 and 20 mmol/L) to the control buffer without 4-CIN did not have any significant effect on the amplitude of either phase of the NAD(P)H biphasic response (Fig. 5a).
Fig. 5.
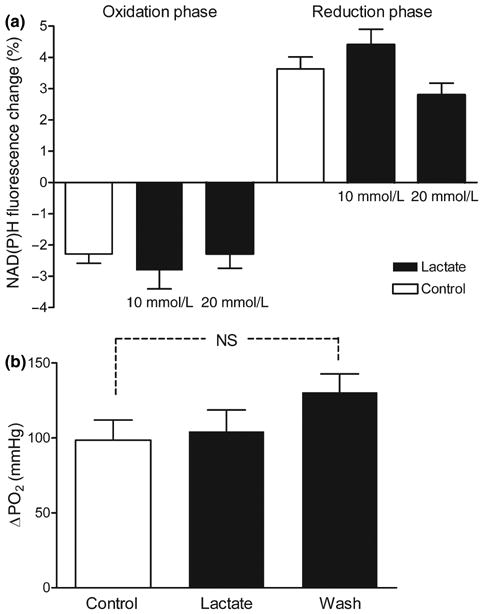
Hippocampal slices were supplemented with lactate (10–20 mmol/L) 10 min before the stimulus train (10 mmol/L glucose). Lactate supplementation alone did not change the amplitude of the NAD(P)H biphasic response (a) or the amplitude of tissue PO2 response (b). Data are the mean ± SEM of four to six slices, NS p > 0.05 (ANOVA and Tukey's multicomparison test).
Lactate (20 mmol/L) was added to the ACSF buffer in the presence of various 4-CIN concentrations for 10 min before the stimulus train (4-CIN was added 15 min before the stimulus train). Lactate supplementation restored the reduction phase of the NAD(P)H response to control levels in the presence of 150 μmol/L 4-CIN (3.8 ± 0.2% for control, n = 43, vs. 3.14 ± 0.25% for 150 μmol/L 4-CIN + 20 mmol/L lactate, n = 9) (Fig. 5a). In the presence of 250 μmol/L 4-CIN, lactate supplementation also resulted in a partial but significant recovery of the reduction phase (Figs 2a and 5a) (0.021 ± 0.34% for 250 μmol/L 4-CIN, n = 7, vs. 1.358 ± 0.2% for 250 μmol/L 4-CIN + 20 mmol/L lactate, n = 14). In the presence of 500 μmol/L 4-CIN, the reduction phase did not recover with lactate supplementation. However, the early NAD(P)H oxidation phase, which was significantly decreased only with 500 μmol/L 4-CIN, recovered significantly with lactate supplementation (−0.75 ± 0.25% for 500 μmol/L 4-CIN, n = 5, vs. −2.75 ± 0.59% for 500 μmol/L 4-CIN + 20 mmol/L lactate, n = 4) (Fig. 6a).
Fig. 6.
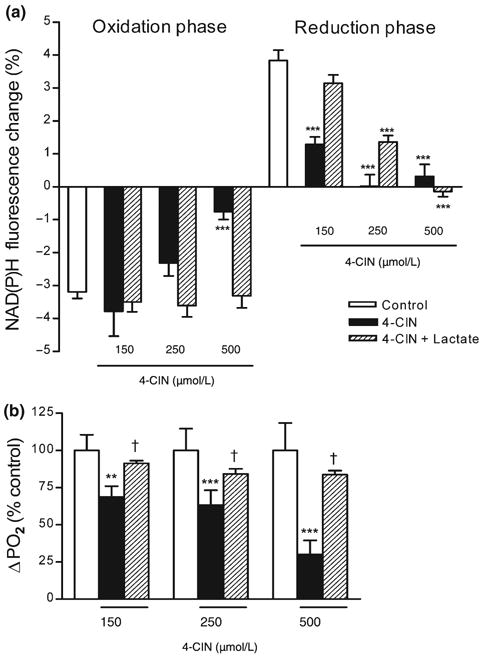
Effect of lactate supplementation on the NAD(P)H biphasic response and the tissue PO2 response in the presence of α-cyano-4-hydroxycinnamate (4-CIN). After synaptic stimulation hippocampal slices were supplemented with lactate (20 mmol/L) 10 min before the stimulus train in the presence of monocarboxylate transporter inhibitor 4-CIN. (a) Lactate restored the reduction phase of the NAD(P)H response in the presence of 4-CIN at lower concentrations. The early NAD(P)H oxidation phase was decreased only in the presence of 500 μmol/L 4-CIN and was restored to control levels by lactate supplementation. Data are the mean ± SEM of 5-35 slices. ***p < 0.001 versus control, †p < 0.05 versus 4-CIN (anova and Tukey's multicomparison test). (b) Lactate supplementation restored tissue PO2 response to control levels at all 4-CIN concentrations. Data are the mean ± SEM of 4-13 slices. **p < 0.01, ***p < 0.001 versus control, †p < 0.05 versus 4-CIN (anova and Tukey's multicomparison test).
To determine if the ability of lactate to restore the reduction phase of the NAD(P)H response was exclusively because of an increase of intracellular pyruvate concentration, after LDH-mediated conversion of lactate to pyruvate, we incubated hippocampal slices with pyruvate (20 mmol/L) in the presence of 4-CIN. Unlike lactate, pyruvate supplementation did not restore the reduction phase of the NAD(P)H response (0.021 ± 0.34% for 250 μmol/L 4-CIN, n = 20, vs. −0.173 ± 0.3% for 250 μmol/L 4-CIN + 20 mmol/L pyruvate, n = 4; p > 0.05 by ANOVA and Tukey's multicomparison test). Interestingly, pyruvate supplementation, similarly to lactate, did result in the recovery of the NAD(P)H oxidation phase in the presence of 500 μmol/L 4-CIN (−0.75 ± 0.25% for 500 μmol/L 4-CIN, n = 5, vs. −3.72 ± 0.43% for 500 μmol/L 4-CIN + 20 mmol/L pyruvate, n = 4; **p < 0.01 by ANOVA and Tukey's multicomparison test).
It is possible that incubation with 20 mmol/L lactate may result in a larger lactate uptake by astroglial cells which express predominantly low affinity MCTs (MCT1 lactate Km 3.5–8 mmol/L) (Broer et al. 1997, 1999), MCT4 Km 35 mmol/L (Dimmer et al. 2000), compared with neurons which express predominantly high affinity MCTs (MCT2 lactate Km 0.7 mmol/L) (Broer et al. 1999). Although, lactate has been proposed to being oxidized predominantly by neuronal cells (Bouzier et al. 2000; Itoh et al. 2003), a recent report has indicated that glial cells may oxidize up to 50% of lactate available in the brain (Zielke et al. 2007).
Therefore, to identify if astrocytic oxidative metabolism is involved in the recovery of the biphasic response (when slices are supplemented with either 20 mmol/L lactate or pyruvate) we have also perfused hippocampal slices with acetate (20 mmol/L) in the presence of 500 μmol/L 4-CIN. Acetate is metabolized only by glia cells within the TCA cycle after selective uptake (Waniewski and Martin 1998; Lebon et al. 2002). Supplementation with acetate did not restore the NAD(P)H oxidation phase in the presence of 500 μmol/L 4-CIN (−0.75 ± 0.25% for 500 μmol/L 4-CIN, n = 5, vs. −0.29 ± 0.5 for 500 μmol/L 4-CIN ± 20 mmol/L acetate, n = 4; p ≥ 0.05 by ANOVA and Tukey's multicomparison test), indicating only a minor role for glial oxidative metabolism in these circumstances.
The effect of lactate supplementation on the tissue PO2 response during synaptic stimulation in the presence of 4-CIN
We have also determined the affect of lactate (20 mmol/L) supplementation on the tissue PO2 response simultaneously to the NAD(P)H signal. The application of lactate (20 mmol/L) to control slices did not cause any significant changes to the amplitudes of the tissue PO2 response during synaptic stimulation (Fig. 5b). In contrast, in the presence of 4-CIN, the application of lactate (20 mmol/L) restored the amplitudes of the tissue PO2 response to control levels. The effect of lactate on the PO2 transient was similar in the presence of all 4-CIN concentrations (Figs 2b and 6b).
Effect of hypoglycemia and lactate/pyruvate substitution on NAD(P)H biphasic response
Synaptic activity stimulates glucose uptake and glucose utilization by both neurons and astrocytes (Sokoloff 1999). Therefore, we investigated the effect of hypoglycemia (2.5 mmol/L glucose) on the NAD(P)H biphasic response. We chose moderate hypoglycemia because under these conditions the synaptic responses were maintained, although the amplitude of the fEPSP was reduced by ∼50% after 30 min (100% ± 12.09 for 10 mmol/L glucose vs. 50.71% ± 5.78 for 2.5 mmol/L glucose; n = 5, p > 0.05, t-test). In addition, the amplitude of fEPSPs recovered completely when the slices were returned to 10 mmol/L glucose after 60 min of exposure to hypoglycemia. In contrast, when slices were perfused with ACSF containing 0 mmol/L glucose (at ∼37°C) we observed a rapid and complete loss of the field potential (Sadgrove et al. 2007), which was irreversible. Thirty min after hippocampal slices were exposed to hypoglycemia (2.5 mmol/L glucose), there was a significant decrease by 60% in the reduction phase of the NAD(P)H response while the oxidation phase was not affected (Fig. 7).
Fig. 7.
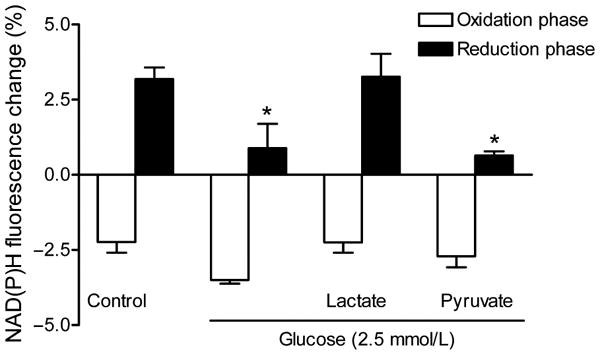
Effect of hypoglycemia and lactate/pyruvate substitution on the NAD(P)H biphasic response. Slices were incubated with 2.5 mmol/L glucose for 30 min before the stimulus train and were returned to 10 mmol/L glucose after 60 min of exposure to hypoglycemia. In some cases, slices were supplemented with pyruvate or lactate (20 mmol/L) during hypoglycemia. Data are the mean ± SEM of 4–13 slices. *p < 0.05 versus control (ANOVA and Tukey's multicomparison test).
We next evaluated whether the metabolic intermediates lactate or pyruvate were able to restore the NAD(P)H biphasic response during hypoglycemia. Pyruvate supplementation (20 mmol/L) was unable to restore the reduction phase of the NAD(P)H response to synaptic stimulation. However, lactate supplementation restored the reduction phase to control levels (ACSF 3.18 ± 0.38%, n = 13, vs. 2.5 mmol/L glucose + lactate 3.268 ± 0. 37%, n = 4) (Fig. 7). In addition, lactate supplementation prevented the decrease of the fEPSP amplitude during hypoglycemia (50.71% ± 5.78 for 2.5 mmol/L glucose vs. 123.3% ± 15.69 for 2.5 mmol/L glucose + 20 mmol/L lactate; n = 5, *p < 0.05, t-test).
Discussion
Optical imaging techniques used to monitor changes in NAD(P)H reflect real time changes in brain cell metabolic state. However, various metabolic pathways may underlie the biphasic NAD(P)H changes following intense brain stimulation (Shuttleworth et al. 2003; Kasischke et al. 2004; Brennan et al. 2006). In this study, we have investigated for the first time the role of lactate uptake via MCT in the generation of the NAD(P)H biphasic response and tissue PO2 response following synaptic stimulation.
In the present study, low concentrations of 4-CIN selectively suppressed the reduction phase of the NAD(P)H response, whereas the NAD(P)H oxidative phase was not significantly altered and fEPSPs were maintained. The MCT inhibitor 4-CIN has been shown to inhibit cellular lactate uptake in various brain preparations (Phillis et al. 2001) as demonstrated by the increased lactate levels after the application of 4-CIN in cortical superfusate from 2 to 14 mg/dL (Phillis et al. 2001) and in hippocampal slices after hypoxia (Schurr et al. 1997). Therefore, it is likely that cellular lactate uptake from the extracellular space and subsequent metabolism has a role in the generation of the NAD(P)H response.
α-Cyano-4-hydroxycinnamate is a competitive inhibitor of various MCT isoforms including MCT1, MCT2, and MCT4, all of which have been described in the CNS (Bergersen et al. 2002). However, several studies have shown that 4-CIN is a more potent inhibitor of lactate uptake via MCT2 (IC50 24 μmol/L) (Broer et al. 1999), expressed predominantly in neurons (Pierre et al. 2000; Bergersen et al. 2001; Rafiki et al. 2003), than via MCT1 and MCT4 (IC50 425 and 350 μmol/L, respectively) (Broer et al. 1999; Dimmer et al. 2000), expressed predominantly in astroglia (Gerhart et al. 1997; Rafiki et al. 2003).
Therefore, we would expect that at the lower concentrations of 4-CIN (150–250 μmol/L) the significant decrease of the reduction phase of the NAD(P)H response observed in our experiments is due predominantly to the inhibition of lactate transport into neurons via MCT2, rather than in glia via MCT1 and MCT4. This is consistent with previous observations suggesting that lactate transport and oxidation of exogenous lactate occurs predominantly in neurons (Larrabee 1995; Bouzier et al. 2000; Pellerin and Magistretti 2003; Bouzier-Sore et al. 2006; Aubert et al. 2007). However, without the application of detailed single cell imaging we cannot rule out that part of the signal may also originate from astrocytes.
Mitochondrial NADH turnover in the electron transport chain is intimately related to oxygen consumption. Therefore, we measured tissue PO2 in order to elucidate the relationship between oxygen consumption, the NAD(P)H biphasic response, and the effect of MCT inhibition by 4-CIN in brain tissue. Previous studies performed in vivo have detected an early increase in oxygen utilization during neuronal activation using functional magnetic resonance imaging (Malonek and Grinvald 1996; Grinvald et al. 2000) and Clark-style polaro-graphic oxygen microelectrode measurements (Thompson et al. 2003; Offenhauser et al. 2005).
Similarly, we have found that synaptic stimulation (95% ambient oxygen) caused a transient decrease in PO2 which is temporally correlated with the NAD(P)H oxidation phase (Foster et al. 2005). At low concentrations of 4-CIN we have observed a 30% decrease in the tissue oxygen uptake compared with control slices whereas the amplitude of the early NAD(P)H decrease was not changed. In contrast, when we raised the concentration of 4-CIN to 500 μmol/L both the tissue PO2 response and NAD(P)H oxidation phase were decreased by 70%.
Studies have shown that in isolated liver mitochondria 4-CIN is a powerful inhibitor of mitochondrial pyruvate uptake (Halestrap et al. 1974; Halestrap and Denton 1975). Therefore, it is possible that 4-CIN prevents pyruvate, originating from both glycolysis and lactate metabolism, from being utilized in the mitochondria.
Interestingly, in our study, 4-CIN at low concentrations did not significantly affect the NAD(P)H oxidation phase or the fEPSP. Our findings are consistent with previous studies that have demonstrated that in acute hippocampal slices, in the presence of 500 μmol/L 4-CIN plus 10 mmol/L glucose, both fEPSPs (Izumi et al. 1997; Tanaka et al. 2004) and ATP levels were maintained (Cox et al. 1985). From the results of these studies investigators have concluded that when used in brain preparations the inhibition of mitochondrial pyruvate uptake does not appear to be the primary effect of 4-CIN (Cater et al. 2001; Ogawa et al. 2005). Moreover, bioradiography experiments in brain slices have shown that the application of 500 μmol/L 4-CIN did not affect mitochondrial aerobic metabolism in neurons or astrocytes (Tanaka et al. 2004; Ogawa et al. 2005).
In a study using both neuronal and astrocytic cultures investigators found that 4-CIN decreased the rate of both lactate and glucose oxidation (McKenna et al. 2001). However, in neurons the rate of lactate oxidation was more potently suppressed than the rate of glucose oxidation (to 12% and 42% of control values, respectively) by 250 μmol/L 4-CIN. These data confirmed the preferential surface MCT2 antagonism of 4-CIN over the mitochondrial pyruvate transporter in neurons (McKenna et al. 2001). Therefore, the inhibition of lactate transport into neurons would be largely responsible for the effect of 4-CIN that we observed on both the decrease of oxygen utilization and the decrease of the reduction phase of the NAD(P)H response, especially at the lowest concentration of the inhibitor (150 μmol/L 4-CIN).
The reduction phase of the NAD(P)H response was restored in the presence 20 mmol/L lactate, probably because of the competition of added lactate with 4-CIN at the surface MCT2. The ability of lactate to overcome the block by 4-CIN at MCT was concentration dependent, as the reduction phase of the NAD(P)H response recovered to 80% of control with 150 μmol/L 4-CIN and 30% with 250 μmol/L 4-CIN. But, in the presence of higher concentrations of 4-CIN (i.e. 500 μmol/L) 20 mmol/L lactate could not overcome the 4-CIN block and there was no recovery of the NAD(P)H overshoot. In contrast, the amplitude of tissue PO2 response was restored to control levels by lactate supplementation at all 4-CIN concentrations. Lactate supplementation also restored the NAD(P)H oxidation phase which was suppressed by 500 μmol/L 4-CIN. Likewise, pyruvate supplementation restored the oxidation phase (500 μmol/L 4-CIN), but not the reduction phase, of the NAD(P)H response.
Lactate can contribute significantly to the mitochondrial NADH pool by providing additional pyruvate for the TCA cycle. In addition, the LDH-mediated conversion of lactate to pyruvate provides reducing equivalents (cytosolic NADH) in the first step of its metabolism. Because cytosolic NADH cannot directly enter the mitochondria, reducing equivalents are transferred to the mitochondria via the malate–aspartate shuttle, which will contribute to generation of NADH in the mitochondria (Wiesner et al. 1988; McKenna et al. 2006). This last mechanism may explain the ability of lactate compared with pyruvate to restore the reduction phase of the NAD(P)H response.
Because there is no dose–response relationship for the recovery of the tissue PO2 response and the NAD(P)H oxidation phase, we have considered the possibility that lactate (at high concentrations) accumulates within cells by other mechanisms that are not sensitive to MCT inhibition by 4-CIN. For example, lactate and pyruvate can enter cells by free diffusion of the undissociated form, especially at high concentration of these monocarboxylates (Poole and Halestrap 1993; Juel 1997). Hale-strap and collaborators reported a 4-CIN-insensitive lactate influx in liver cell exposed to 10 mmol/L lactate; however, the rate of this flux was slower than MCT-mediated transport (Edlund and Halestrap 1988; Poole and Halestrap 1993; Jackson and Halestrap 1996).
Therefore, the exposure to high extracellular lactate concentrations could have resulted lactate accumulation in both neurons and glia, even in the presence of 500 μmol/L 4-CIN. A high intracellular lactate/pyruvate ratio will drive the LDH reaction towards pyruvate generation (Lipton 1973; O'Brien et al. 2007) which will enter the mitochondria, determining the recovery of both the tissue PO2 response and the early NAD(P)H oxidation phase. This possibility is supported by the finding that direct pyruvate supplementation also resulted in the recovery of the early oxidation phase at 500 μmol/L 4-CIN. Slow lactate or pyruvate accumulation occurring with the supplementation in the presence of 500 μmol/L 4-CIN likely supports a pool of mitochondrial metabolic intermediates, which are likely utilized early during the train stimulation, fueling oxidative phosphorylation. However, it seems that a rapid lactate transport via MCT2 (Poole and Halestrap 1993) is necessary for the generation of the NAD(P)H overshoot.
Results from in vivo studies using an enzyme-based lactate sensor (Hu et al. 1997), proton magnetic resonance spectroscopy (Mangia et al. 2003), and mathematical modeling studies (Aubert et al. 2007) have all indicated that there is increase of lactate uptake during intense neuronal activity; however, the exact kinetic of lactate transport in relationship to the NAD(P)H and PO2 response remain to be established (Korf 2006).
Previous studies have suggested that brain activation stimulates glucose uptake and glycolysis, both in neurons and astrocytes (Chih and Roberts 2003). Following moderate hypoglycemia (i.e. 2.5 mmol/L glucose for 30 min) fEPSPs were partially depressed (50%) (Kamal et al. 1999; Sakurai et al. 2002), and the NAD(P)H overshoot was significantly decreased (Sadgrove et al. 2007). These results are in contrast to a recent study that found that a short exposure to hypoglycemia (0 mmol/L, up to 17 min) with or without the glycolysis inhibitors 2-deoxyglucose and iodoacetic acid did not affect the amplitude of the NAD(P)H response (Brennan et al. 2006). However, the experimental conditions in this study were different because the slices were maintained at 25°C and were supplemented with pyruvate. In addition, the adenosine receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine was added to the solution to prevent adenosine induced loss of synaptic transmission (Brennan et al. 2006). Energy deprivation as a result of hypoglycemia causes the breakdown of intracellular ATP, which may result in an increase of adenosine, and therefore an inhibitory effect on synaptic transmission (Martin et al. 1994). The loss of synaptic transmission also causes inhibition of the NAD(P)H biphasic response (Brennan et al. 2006). The hypoglycemia-induced loss of synaptic transmission is preventable, however, with substrate substitution such as lactate (Fowler 1993) and pyruvate (Izumi et al. 1997). Similar to the 4-CIN results, the NAD(P)H overshoot recovered with lactate but not pyruvate supplementation.
Glucose utilization in neurons plays a critical role in supporting synaptic function and mitochondrial oxidative metabolism demonstrated by the observation that in the presence of 4-CIN both fEPSPs and the NAD(P)H oxidative phase were maintained. Our findings also indicate that lactate uptake from the extracellular pool and its metabolism significantly contribute to the reduction phase of the NAD(P)H response and metabolic intermediates from lactate metabolism are utilized by mitochondria for oxidative metabolism.
Several authors have also shown that 4-CIN has no effect on anaerobic glycolysis (Schurr et al. 1997) and glucose uptake remains constant in the presence of 4-CIN in brain cellular preparations in vitro (McKenna et al. 2001) and in brain slices ex vivo (Ogawa et al. 2005). Therefore, if an increase of glycolysis alone was responsible for the NAD(P)H overshoot, we would have expected the NAD(P)H peak to be maintained as a result of the application of 4-CIN rather than being suppressed. In the case of lactate supplementation (during either 4-CIN or hypoglycemia), the elevated intra-cellular lactate concentrations would presumably slow the rate of glycolysis by end-product inhibition (Itoh et al. 2003).
Although, the NAD(P)H signal does not mirror glucose uptake and astrocytic glycolysis activation after neuronal stimulation, these processes are key events sustaining CNS metabolism and may contribute to the delayed increase in extracellular lactate levels (De Bruin et al. 1990; Fellows et al. 1993).
Acknowledgments
We are grateful to Professor G. G. Somjen and Professor C. A. Piantadosi for critical reading of the manuscript. This work was supported by grants from NINDS (R21 NS45304 and R01 NS051856) and a VA Merit Review award.
Abbreviations used
- 4-CIN
α-cyano-4-hydroxycinnamate
- ACSF
artificial CSF
- AIDA
(RS)-1-aminoindan-1,5-dicarboxylic acid
- AMPA
α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid
- APICA
(RS)-1-amino-5-phosphonoindan-1-carboxylic acid
- APV
d-(–)-2-amino-5-phosphonopentanoic
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- fEPSP
field excitatory post-synaptic potential
- LDH
lactate dehydrogenase
- MCT
monocarboxylate transporter
- MSOP
(RS)-α-methylserine-o-phosphate
- TCA
tricarboxylic acid
References
- Aubert A, Pellerin L, Magistretti PJ, Costalat R. A coherent neurobiological framework for functional neuroimaging provided by a model integrating compartmentalized energy metabolism. Proc Natl Acad Sci USA. 2007;104:4188–4193. doi: 10.1073/pnas.0605864104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubin JE. Autofluorescence of viable cultured mammalian cells. J Histochem Cytochem. 1979;27:36–43. doi: 10.1177/27.1.220325. [DOI] [PubMed] [Google Scholar]
- Bergersen L, Waerhaug O, Helm J, Thomas M, Laake P, Davies AJ, Wilson MC, Halestrap AP, Ottersen OP. A novel postsynaptic density protein: the monocarboxylate transporter MCT2 is co-localized with delta-glutamate receptors in postsynaptic densities of parallel fiber-Purkinje cell synapses. Exp Brain Res. 2001;136:523–534. doi: 10.1007/s002210000600. [DOI] [PubMed] [Google Scholar]
- Bergersen L, Rafiki A, Ottersen OP. Immunogold cyto-chemistry identifies specialized membrane domains for monocarboxylate transport in the central nervous system. Neurochem Res. 2002;27:89–96. doi: 10.1023/a:1014806723147. [DOI] [PubMed] [Google Scholar]
- Bouzier AK, Thiaudiere E, Biran M, Rouland R, Canioni P, Merle M. The metabolism of [3-(13)C]lactate in the rat brain is specific of a pyruvate carboxylase-deprived compartment. J Neurochem. 2000;75:480–486. doi: 10.1046/j.1471-4159.2000.0750480.x. [DOI] [PubMed] [Google Scholar]
- Bouzier-Sore AK, Voisin P, Bouchaud V, Bezancon E, Franconi JM, Pellerin L. Competition between glucose and lactate as oxidative energy substrates in both neurons and astrocytes: a comparative NMR study. Eur J Neurosci. 2006;24:1687–1694. doi: 10.1111/j.1460-9568.2006.05056.x. [DOI] [PubMed] [Google Scholar]
- Brennan AM, Connor JA, Shuttleworth CW. NAD(P)H fluorescence transients after synaptic activity in brain slices: predominant role of mitochondrial function. J Cereb Blood Flow Metab. 2006;26:1389–1406. doi: 10.1038/sj.jcbfm.9600292. [DOI] [PubMed] [Google Scholar]
- Broer S, Rahman B, Pellegri G, Pellerin L, Martin JL, Verleysdonk S, Hamprecht B, Magistretti PJ. Comparison of lactate transport in astroglial cells and monocarboxylate transporter 1 (MCT 1) expressing Xenopus laevis oocytes. Expression of two different monocarboxylate transporters in astroglial cells and neurons. J Biol Chem. 1997;272:30096–30102. doi: 10.1074/jbc.272.48.30096. [DOI] [PubMed] [Google Scholar]
- Broer S, Broer A, Schneider HP, Stegen C, Halestrap AP, Deitmer JW. Characterization of the high-affinity monocarboxylate transporter MCT2 in Xenopus laevis oocytes. Biochem J. 1999;341:529–535. doi: 10.1042/0264-6021:3410529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cater HL, Benham CD, Sundstrom LE. Neuroprotective role of monocarboxylate transport during glucose deprivation in slice cultures of rat hippocampus. J Physiol (Lond) 2001;531:459–466. doi: 10.1111/j.1469-7793.2001.0459i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih CP, Roberts EL., Jr Energy substrates for neurons during neural activity: a critical review of the astrocyte-neuron lactate shuttle hypothesis [see comment] J Cereb Blood Flow Metab. 2003;23:1263–1281. doi: 10.1097/01.WCB.0000081369.51727.6F. [DOI] [PubMed] [Google Scholar]
- Cox DW, Drower J, Bachelard HS. Effects of metabolic inhibitors on evoked activity and the energy state of hippocampal slices superfused in vitro. Exp Brain Res. 1985;57:464–470. doi: 10.1007/BF00237833. [DOI] [PubMed] [Google Scholar]
- De Bruin LA, Schasfoort EM, Steffens AB, Korf J. Effects of stress and exercise on rat hippocampus and striatum extracellular lactate. Am J Physiol. 1990;259:R773–R779. doi: 10.1152/ajpregu.1990.259.4.R773. [DOI] [PubMed] [Google Scholar]
- Dimmer KS, Friedrich B, Lang F, Deitmer JW, Broer S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem J. 2000;350(Pt. 1):219–227. [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Ca(2+)-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochem J. 1992;283:41–50. doi: 10.1042/bj2830041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlund GL, Halestrap AP. The kinetics of transport of lactate and pyruvate into rat hepatocytes. Evidence for the presence of a specific carrier similar to that in erythrocytes. Biochem J. 1988;249:117–126. doi: 10.1042/bj2490117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elekes O, Venema K, Postema F, Dringen R, Hamprecht B, Korf J. Evidence that stress activates glial lactate formation in vivo assessed with rat hippocampus lactography. Neurosci Lett. 1996;208:69–72. doi: 10.1016/0304-3940(96)12553-2. [DOI] [PubMed] [Google Scholar]
- Fayuk D, Aitken PG, Somjen GG, Turner DA. Two different mechanisms underlie reversible, intrinsic optical signals in rat hippocampal slices. J Neurophysiol. 2002;87:1924–1937. doi: 10.1152/jn.00231.2001. [DOI] [PubMed] [Google Scholar]
- Fellows LK, Boutelle MG, Fillenz M. Physiological stimulation increases nonoxidative glucose metabolism in the brain of the freely moving rat. J Neurochem. 1993;60:1258–1263. doi: 10.1111/j.1471-4159.1993.tb03285.x. [DOI] [PubMed] [Google Scholar]
- Foster KA, Beaver CJ, Turner DA. Interaction between tissue oxygen tension and NADH imaging during synaptic stimulation and hypoxia in rat hippocampal slices. Neuroscience. 2005;132:645–657. doi: 10.1016/j.neuroscience.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Fowler JC. Glucose deprivation results in a lactate preventable increase in adenosine and depression of synaptic transmission in rat hippocampal slices. J Neurochem. 1993;60:572–576. doi: 10.1111/j.1471-4159.1993.tb03187.x. [DOI] [PubMed] [Google Scholar]
- Garofalo O, Cox DW, Bachelard HS. Brain levels of NADH and NAD+ under hypoxic and hypoglycaemic conditions in vitro. J Neurochem. 1988;51:172–176. doi: 10.1111/j.1471-4159.1988.tb04851.x. [DOI] [PubMed] [Google Scholar]
- Gerhart DZ, Enerson BE, Zhdankina OY, Leino RL, Drewes LR. Expression of monocarboxylate transporter MCT1 by brain endothelium and glia in adult and suckling rats. Am J Physiol. 1997;273:E207–E213. doi: 10.1152/ajpendo.1997.273.1.E207. [DOI] [PubMed] [Google Scholar]
- Grinvald A, Slovin H, Vanzetta I. Non-invasive visualization of cortical columns by fMRI [comment] Nat Neurosci. 2000;3:105–107. doi: 10.1038/72045. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. The mitochondrial pyruvate carrier. Kinetics and specificity for substrates and inhibitors. Biochem J. 1975;148:85–96. doi: 10.1042/bj1480085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Denton RM. The specificity and metabolic implications of the inhibition of pyruvate transport in isolated mitochondria and intact tissue preparations by alpha-cyano-4-hydroxycinnamate and related compounds. Biochem J. 1975;148:97–106. doi: 10.1042/bj1480097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap AP, Brand MD, Denton RM. Inhibition of mitochondrial pyruvate transport by phenylpyruvate and alpha-ketoisocaproate. Biochim Biophys Acta. 1974;367:102–108. doi: 10.1016/0005-2736(74)90140-0. [DOI] [PubMed] [Google Scholar]
- Hu Y, Wilson GS, Hu Y, Wilson GS. A temporary local energy pool coupled to neuronal activity: fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. J Neurochem. 1997;69:1484–1490. doi: 10.1046/j.1471-4159.1997.69041484.x. [DOI] [PubMed] [Google Scholar]
- Itoh Y, Esaki T, Shimoji K, Cook M, Law MJ, Kaufman E, Sokoloff L. Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc Natl Acad Sci USA. 2003;100:4879–4884. doi: 10.1073/pnas.0831078100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Benz AM, Katsuki H, Zorumski CF. Endogenous monocarboxylates sustain hippocampal synaptic function and morphological integrity during energy deprivation. J Neurosci. 1997;17:9448–9457. doi: 10.1523/JNEUROSCI.17-24-09448.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson VN, Halestrap AP. The kinetics, substrate, and inhibitor specificity of the monocarboxylate (lactate) transporter of rat liver cells determined using the fluorescent intracellular pH indicator, 2′,7′-bis(carboxyethyl)-5(6)-carboxyfluorescein. J Biol Chem. 1996;271:861–868. doi: 10.1074/jbc.271.2.861. [DOI] [PubMed] [Google Scholar]
- Juel C. Lactate-proton cotransport in skeletal muscle. Physiol Rev. 1997;77:321–358. doi: 10.1152/physrev.1997.77.2.321. [DOI] [PubMed] [Google Scholar]
- Kamal A, Spoelstra K, Biessels GJ, Urban IJA, Gispen WH. Effects of changes in glucose concentration on synaptic plasticity in hippocampal slices. Brain Res. 1999;824:238–242. doi: 10.1016/s0006-8993(99)01215-9. [DOI] [PubMed] [Google Scholar]
- Kann O, Schuchmann S, Buchheim K, Heinemann U. Coupling of neuronal activity and mitochondrial metabolism as revealed by NAD(P)H fluorescence signals in organotypic hippo-campal slice cultures of the rat. Neuroscience. 2003;119:87–100. doi: 10.1016/s0306-4522(03)00026-5. [DOI] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis [see comment] Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- Korf J. Is brain lactate metabolized immediately after neuronal activity through the oxidative pathway? J Cereb Blood Flow Metab. 2006;26:1584–1586. doi: 10.1038/sj.jcbfm.9600321. [DOI] [PubMed] [Google Scholar]
- Larrabee MG. Lactate metabolism and its effects on glucose metabolism in an excised neural tissue. J Neurochem. 1995;64:1734–1741. doi: 10.1046/j.1471-4159.1995.64041734.x. [DOI] [PubMed] [Google Scholar]
- Lebon V, Petersen KF, Cline GW, Shen J, Mason GF, Dufour S, Behar KL, Shulman GI, Rothman DL. Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci. 2002;22:1523–1531. doi: 10.1523/JNEUROSCI.22-05-01523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P. Effects of membrane depolarization on nicotinamide nucleotide fluorescence in brain slices. Biochem J. 1973;136:999–1009. doi: 10.1042/bj1360999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malonek D, Grinvald A. Interactions between electrical activity and cortical microcirculation revealed by imaging spectroscopy: implications for functional brain mapping. Science. 1996;272:551–554. doi: 10.1126/science.272.5261.551. [DOI] [PubMed] [Google Scholar]
- Mangia S, Garreffa G, Bianciardi M, Giove F, Di Salle F, Maraviglia B. The aerobic brain: lactate decrease at the onset of neural activity. Neuroscience. 2003;118:7–10. doi: 10.1016/s0306-4522(02)00792-3. [DOI] [PubMed] [Google Scholar]
- Martin RL, Lloyd HG, Cowan AI. The early events of oxygen and glucose deprivation: setting the scene for neuronal death? Trends Neurosci. 1994;17:251–257. doi: 10.1016/0166-2236(94)90008-6. [DOI] [PubMed] [Google Scholar]
- McKenna MC, Hopkins IB, Carey A. Alpha-cyano-4-hydroxycinnamate decreases both glucose and lactate metabolism in neurons and astrocytes: implications for lactate as an energy substrate for neurons. J Neurosci Res. 2001;66:747–754. doi: 10.1002/jnr.10084. [DOI] [PubMed] [Google Scholar]
- McKenna MC, Waagepetersen HS, Schousboe A, Sonnewald U. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: current evidence and pharmacological tools. Biochem Pharmacol. 2006;71:399–407. doi: 10.1016/j.bcp.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Mironov SL, Richter DW. Oscillations and hypoxic changes of mitochondrial variables in neurons of the brainstem respiratory centre of mice. J Physiol (Lond) 2001;533:227–236. doi: 10.1111/j.1469-7793.2001.0227b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien J, Kla KM, Hopkins IB, Malecki EA, McKenna MC. Kinetic parameters and lactate dehydrogenase isozyme activities support possible lactate utilization by neurons. Neurochem Res. 2007;32:597–607. doi: 10.1007/s11064-006-9132-9. [DOI] [PubMed] [Google Scholar]
- Offenhauser N, Thomsen K, Caesar K, Lauritzen M. Activity-induced tissue oxygenation changes in rat cerebellar cortex:interplay of postsynaptic activation and blood flow. J Physiol (Lond) 2005;565:279–294. doi: 10.1113/jphysiol.2005.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa M, Watabe H, Teramoto N, Miyake Y, Hayashi T, Iida H, Murata T, Magata Y. Understanding of cerebral energy metabolism by dynamic living brain slice imaging system with [18F]FDG. Neurosci Res. 2005;52:357–361. doi: 10.1016/j.neures.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Food for thought: challenging the dogmas [see comment] [comment] J Cereb Blood Flow Metab. 2003;23:1282–1286. doi: 10.1097/01.WCB.0000096064.12129.3D. [DOI] [PubMed] [Google Scholar]
- Phillis JW, Ren J, O'Regan MH. Studies on the effects of lactate transport inhibition, pyruvate, glucose and glutamine on amino acid, lactate and glucose release from the ischemic rat cerebral cortex. J Neurochem. 2001;76:247–257. doi: 10.1046/j.1471-4159.2001.00050.x. [DOI] [PubMed] [Google Scholar]
- Pierre K, Pellerin L, Debernardi R, Riederer BM, Magistretti PJ. Cell-specific localization of monocarboxylate transporters, MCT1 and MCT2, in the adult mouse brain revealed by double immunohistochemical labeling and confocal microscopy. Neuro-science. 2000;100:617–627. doi: 10.1016/s0306-4522(00)00294-3. [DOI] [PubMed] [Google Scholar]
- Poitry S, Poitry-Yamate C, Ueberfeld J, MacLeish PR, Tsaco-poulos M. Mechanisms of glutamate metabolic signaling in retinal glial (Muller) cells. J Neurosci. 2000;20:1809–1821. doi: 10.1523/JNEUROSCI.20-05-01809.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole RC, Halestrap AP. Transport of lactate and other monocarboxylates across mammalian plasma membranes. Am J Physiol. 1993;264:C761–C782. doi: 10.1152/ajpcell.1993.264.4.C761. [DOI] [PubMed] [Google Scholar]
- Rafiki A, Boulland JL, Halestrap AP, Ottersen OP, Bergersen L. Highly differential expression of the monocarboxylate transporters MCT2 and MCT4 in the developing rat brain. Neuroscience. 2003;122:677–688. doi: 10.1016/j.neuroscience.2003.08.040. [DOI] [PubMed] [Google Scholar]
- Sadgrove MP, Beaver CJ, Turner DA. Effects of relative hypoglycemia on LTP and NADH imaging in rat hippocampal slices. Brain Res. 2007;1165:30–39. doi: 10.1016/j.brainres.2007.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai T, Yang B, Takata T, Yokono K. Synaptic adaptation to repeated hypoglycemia depends on the utilization of monocarboxylates in Guinea pig hippocampal slices [Erratum appears in Diabetes 2002; 51(11), 3350] Diabetes. 2002;51:430–438. doi: 10.2337/diabetes.51.2.430. [DOI] [PubMed] [Google Scholar]
- Scholz TD, Laughlin MR, Balaban RS, Kupriyanov VV, Heineman FW. Effect of substrate on mitochondrial NADH, cytosolic redox state, and phosphorylated compounds in isolated hearts. Am J Physiol. 1995;268:H82–H91. doi: 10.1152/ajpheart.1995.268.1.H82. [DOI] [PubMed] [Google Scholar]
- Schuchmann S, Kovacs R, Kann O, Heinemann U, Buchheim K. Monitoring NAD(P)H autofluorescence to assess mitochondrial metabolic functions in rat hippocampal-entorhinal cortex slices. Brain Res Brain Res Protoc. 2001;7:267–276. doi: 10.1016/s1385-299x(01)00080-0. [DOI] [PubMed] [Google Scholar]
- Schurr A. Lactate: the ultimate cerebral oxidative energy substrate? J Cereb Blood Flow Metab. 2006;26:142–152. doi: 10.1038/sj.jcbfm.9600174. [DOI] [PubMed] [Google Scholar]
- Schurr A, West CA, Rigor BM. Lactate-supported synaptic function in the rat hippocampal slice preparation. Science. 1988;240:1326–1328. doi: 10.1126/science.3375817. [DOI] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Miller JJ, Rigor BM. Brain lactate is an obligatory aerobic energy substrate for functional recovery after hypoxia: further in vitro validation. J Neurochem. 1997;69:423–426. doi: 10.1046/j.1471-4159.1997.69010423.x. [DOI] [PubMed] [Google Scholar]
- Schurr A, Miller JJ, Payne RS, Rigor BM. An increase in lactate output by brain tissue serves to meet the energy needs of glutamate-activated neurons. J Neurosci. 1999a;19:34–39. doi: 10.1523/JNEUROSCI.19-01-00034.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr A, Payne RS, Miller JJ, Rigor BM. Study of cerebral energy metabolism using the rat hippocampal slice preparation. Methods. 1999b;18:117–126. doi: 10.1006/meth.1999.0765. [DOI] [PubMed] [Google Scholar]
- Shuttleworth CW, Brennan AM, Connor JA. NAD(P)H fluorescence imaging of postsynaptic neuronal activation in murine hippocampal slices. J Neurosci. 2003;23:3196–3208. doi: 10.1523/JNEUROSCI.23-08-03196.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokoloff L. Energetics of functional activation in neural tissues. Neurochem Res. 1999;24:321–329. doi: 10.1023/a:1022534709672. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Nakamura F, Mizokawa S, et al. Role of lactate in the brain energy metabolism: revealed by bioradiography. Neuro-sci Res. 2004;48:13. doi: 10.1016/j.neures.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Thompson JK, Peterson MR, Freeman RD. Single-neuron activity and tissue oxygenation in the cerebral cortex [see comment] Science. 2003;299:1070–1072. doi: 10.1126/science.1079220. [DOI] [PubMed] [Google Scholar]
- Volk C, Kempski B, Kempski OS. Inhibition of lactate export by quercetin acidifies rat glial cells in vitro. Neurosci Lett. 1997;223:121–124. doi: 10.1016/s0304-3940(97)13420-6. [DOI] [PubMed] [Google Scholar]
- Waniewski RA, Martin DL. Preferential utilization of acetate by astrocytes is attributable to transport. J Neurosci. 1998;18:5225–5233. doi: 10.1523/JNEUROSCI.18-14-05225.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner RJ, Kreutzer U, Rosen P, Grieshaber MK. Subcellular distribution of malate-aspartate cycle intermediates during normoxia and anoxia in the heart. Biochim Biophys Acta. 1988;936:114–123. doi: 10.1016/0005-2728(88)90258-7. [DOI] [PubMed] [Google Scholar]
- Zielke HR, Zielke CL, Baab PJ, Tildon JT. Effect of fluorocitrate on cerebral oxidation of lactate and glucose in freely moving rats. J Neurochem. 2007;101:9–16. doi: 10.1111/j.1471-4159.2006.04335.x. [DOI] [PubMed] [Google Scholar]