

Abstract
Microscopic images of specific proteins in their cellular context yield important insights into biological processes and cellular architecture. The advent of superresolution optical microscopy techniques provides the possibility to augment EM with nanometer-resolution fluorescence microscopy to access the precise location of proteins in the context of cellular ultrastructure. Unfortunately, efforts to combine superresolution fluorescence and EM have been stymied by the divergent and incompatible sample preparation protocols of the two methods. Here, we describe a protocol that preserves both the delicate photoactivatable fluorescent protein labels essential for superresolution microscopy and the fine ultrastructural context of EM. This preparation enables direct 3D imaging in 500- to 750-nm sections with interferometric photoactivatable localization microscopy followed by scanning EM images generated by focused ion beam ablation. We use this process to “colorize” detailed EM images of the mitochondrion with the position of labeled proteins. The approach presented here has provided a new level of definition of the in vivo nature of organization of mitochondrial nucleoids, and we expect this straightforward method to be applicable to many other biological questions that can be answered by direct imaging.
Keywords: correlative microscopy, photoactivated localization microscopy, electron microscopy, cryosection, mitochondrial DNA
Fluorescent labels can mark specific proteins directly through genetic expression of fusion proteins or by postattachment of labeled antibodies (1). Genetic expression of fusion proteins provides the best specificity and a higher labeling density, with the caveat that a fusion can disrupt the functionality and/or native localization of the protein. Alternatively, labeled antibodies allow the visualization of native proteins, but localization accuracy is dependent on labeling specificity and background, and resolution can be further degraded by a lower labeling density. When these fluorescent labels exhibit certain photophysical properties such as switching, wavelength shifting, or other nonlinear responses, a variety of new microscopy techniques can extract their position at resolutions well below the diffraction limit of approximately 200 nm, down to 20 to 100 nm (2). Some of the new microscopy techniques map molecule distributions by a patterned illumination such as saturated structured illumination microscopy (3) or stimulated emission depletion (STED) microscopy (4). Other methods, including photoactivated localization microscopy (PALM) (5, 6) or stochastic optical reconstruction microscopy (7) extract fluorescent protein positions one molecule at a time. For a single fluorophore color, those images may appear as protein distributions floating in darkness with no or little further context. The localization of fluorescently labeled proteins relative to other cellular structures, e.g., membranes, requires a second fluorescent label. Although multiple-color data are desirable, often the biological constraints of compatible fluorescent pairs, additional protein fusion, and expression or postattachment labeling can be limiting, and the added protocol development can be difficult and time consuming.
Electron Microscopy (EM) offers an alternative nanometer-scale context of cellular structures, but conventional fixation and staining methods usually do not provide protein-specific information. The fixation and staining required to preserve good ultrastructure can also destroy protein fluorescence and/or increase the autofluorescence background (8), thus limiting the effectiveness of this sample preparation method for fluorescence-based superresolution microscopy. Immunolabeling EM can be used to localize specific proteins, but the use of mild fixation and staining conditions needed to preserve protein epitopes often sacrifices ultrastructure (9).
Despite these challenges, there have been encouraging developments to correlate superresolution fluorescence and EM images. Correlated fluorescence and 3D EM tomography have been extended to small copy numbers of fluorescent proteins (10). Some superresolution fluorescence images were correlated with electron micrographs by using photoactivatable fluorescent proteins (PAFPs) (11), and a new protein-specific local heavy metal stain has great potential to yield high-resolution localization based on EM only (12). Nevertheless, correlative superresolution/EM requiring maximal PAFP fluorescence preservation and minimal compromise of ultrastructure has been elusive. Likewise, extending EM/superresolution fluorescence correlation from two dimensions to three dimensions could provide a more informative biological context.
In this work, we demonstrate a method to correlate 3D superresolution fluorescence images to 3D EM data. To obtain 3D superresolution fluorescence images, we used interferometric PALM (iPALM), which is an extension of PALM (13). PALM by itself sequentially localizes each PAFP labeled protein to approximately 20 nm in the image plane and renders a superresolution image from all of these molecular coordinates. iPALM uses simultaneous multiphase interference of light emitted from each molecule to further extract a z position, defining the third dimension to approximately 10 nm resolution. The same samples that were first imaged using iPALM were then imaged by using focused ion beam (FIB) ablation scanning EM (FIB-SEM) to generate a 3D EM image (14). FIB-SEM operates in a cycle in which an ion beam ablates a few-nanometer-thick layer of sample to expose a new surface that is imaged by SEM. This cycle repeats thousands of times to form a stack of images of ever-deeper layers in the sample.
As validation for this technique, we image a mtDNA-binding protein, TFAM, fused to the PAFP mEos2. This protein fusion, and mitochondria in particular, are ideal targets because (i) the small and highly compartmentalized mitochondrion requires nanometer resolution to image, (ii) mitochondria have well studied morphology that is easily identifiable, and (iii) TFAM-mEos2 expression and localization have been well characterized.
Results
Mitochondria Are Ideal Imaging Targets for Superresolution Microscopy.
Mitochondria usually exist within cells as reticular networks several microns in length with typical widths of 250 to 500 nm. Two membranes subdivide a mitochondrion into six spatially, functionally, and compositionally distinct areas: the outer membrane, intermembrane space, inner membrane, cristae, cristae space, and the matrix, which are separated by distances of 10 to 100 nm. Thus, imaging submitochondrial structures requires microscopes and techniques capable of nanometer resolution.
The submitochondrial structure imaged in this study is the nucleoid. Mitochondrial nucleoids are discrete protein–mtDNA complexes that are the mitochondrial units of inheritance (15, 16). Mitochondrial DNA encodes 13 essential protein components of the ATP-generating oxidative phosphorylation machinery and genes for ribosomal and transfer RNAs necessary for mitochondrial mRNA translation.
To label mitochondrial nucleoids, we used the transcription factor and mtDNA-binding protein TFAM as has been done previously (17). TFAM is an ideal marker for mitochondrial nucleoids because it binds mtDNA at a high copy number (18, 19) and is not stable when mtDNA is depleted (20, 21). TFAM was fused to mEos2 (22) and maintained in a stably transfected mouse fibroblast cell line, as described previously (17). The characterization of TFAM-mEos2 expression and its negligible effect on the relevant properties of mitochondria have been described previously (17).
Preparation, Fixation, and Sectioning of Cells.
Mitochondria are found mostly in the perinuclear regions of cells and are best imaged with PALM/iPALM by sectioning cells into samples of less than 1 μm in thickness to access the interior of cells, where mitochondria reside. In addition to the sectioning requirement, protein fluorescence and EM-grade ultrastructure had to be preserved. Previous work by our group has shown that PAFP fluorescence is well preserved in sections of cells embedded in LR White resin and in Tokuyasu cryosections (23). Unfortunately, mitochondrial ultrastructure was not well defined in sections of London white resin-embedded cells when viewed by EM; however, immunolabeling EM showed that the cristae and inner and outer membranes were preserved and easily identifiable in Tokuyasu cryosections (17). Thus, we decided to perform correlative microscopy by using Tokuyasu cryosections because they are suitable for PALM imaging and show good mitochondrial ultrastructure by EM. Fig. 1A shows a schematic overview of the entire correlative process described in the subsequent sections.
Fig. 1.

Overview of correlative iPALM and EM procedure and the imaging systems. (A) Flow diagram showing the steps involved in correlating iPALM and EM images. (B) iPALM imaging setup shows the 500- to 750-nm-thick cryosection sandwiched between the two opposing objectives and coverslips. The cryosection sits on gold fiducials and remains hydrated with PBS solution. (C) Schematic shows how the ion beam mills through the sample and into the coverglass (double arrows). The coverglass has a few gold fiducials (yellow dots), and is covered with the section (gray image), coated with methylcellulose and cyanoacrylate (black), and covered with a thin layer of carbon for charge conductivity (purple).
Cells were prepared for Tokuyasu cryosectioning as described previously (17) except that the fixative agent contained 2% (wt/vol) glutaraldehyde and 5% (wt/vol) sucrose. Generally, cells are fixed for Tokuyasu cryosectioning with 4% (wt/vol) paraformaldehyde and little [0.2% (wt/vol)] or no glutaraldehyde. As Tokuyasu cryosections are often used for immunolabeling, a lower glutaraldehyde concentration likely results in less protein denaturation and/or more penetration of the antibody into the section. We found that increasing the glutaraldehyde concentration to 2% (wt/vol) did not reduce mEos2 fluorescence but improved the ultrastructure of mitochondrial membranes by EM. Although use of 0.2% (wt/vol) glutaraldehyde can provide good EM ultrastructure, we often saw poorer ultrastructure in EM after PALM imaging, possibly as a result of reversible crosslinking of paraformaldehyde that could occur during the several-hour PALM imaging sessions when the sample was in PBS solution (24). Glutaraldehyde at a 2% (wt/vol) concentration generates a prohibitive amount of autofluorescence; however, this was mitigated by a brief treatment with sodium borohydride (25) before fluorescence imaging.
After fixation and freezing, the cells expressing TFAM-mEos2 were sectioned to thicknesses of 500 or 750 nm and placed on coverslips coated with bare Au nanorods (25 × 35 nm). Small 25- to 100-nm gold particles are known to fluoresce at wavelengths used for PALM imaging and are used to generate calibration sets and correct for drift that may occur during the PALM imaging session (13). Fortuitously, gold scatters electrons strongly and is easily identifiable by EM. Therefore, the gold particles are used in the alignment and registration between the iPALM and EM datasets.
Three-Dimensional Superresolution Fluorescence Imaging of TFAM-mEos2.
iPALM uses opposed objectives to create the two interfering light paths, requiring that the sample be sandwiched between two coverslips (Fig. 1B). Otherwise, the imaging conditions and sample requirements are the same as for PALM. Differential interference contrast microscopy was used to locate intact sections. When a section had been located, cells were illuminated with low power (40 W/cm2) 488-nm light to find areas suitable for iPALM imaging. When a suitable area with several nucleoids had been located, iPALM imaging was performed as described previously (13, 17). For each frame, a small fraction (approximately 1/100,000th) of the PAFP mEos2 was switched from a green to red fluorescent state through activation with each 405-nm laser pulse. The red state of mEos2 molecules was imaged by excitation with approximately 2 kW/cm2 561-nm light. On average, the fluorescence lasts approximately 50 ms before it bleaches. In iPALM, a frame triplet captures the interferometric data along with some less sensitive focus-based signature of the z position, giving a working range of 0.75 μm in the third dimension (17). The activation, imaging, bleach cycle was repeated for approximately 40,000 frames. The data were cross-registered, drift-corrected, and processed to give 3D volumes (∼50 μm × 50 μm × 0.75 μm) of all the TFAM-mEos2 positions in the imaged area.
Three-Dimensional EM Imaging Using FIB-SEM.
After iPALM imaging, the cryosections were stained with uranyl acetate in methylcellulose. A layer of cyanoacrylate was added over the dried sections to form an approximately 1- to 5-μm layer that smoothes the topography of the cells to ensure more uniform ion milling. The 3D EM images were obtained with focused ion beam blockface ablation followed by scanning EM imaging (14). A focused beam of Ga+ ions milled a trench through the sample and into the coverslip (Fig. 1C). The upper wall of the trench was imaged from above with an SEM forming a rectangular 80 μm × 5 μm image. This cycle was repeated with a subsequent pass of the ion beam that removes another 5 nm of the sample/trench wall followed by another SEM image. A stack of approximately 6,000 frames was accumulated, giving a data volume of 80 μm × 30 μm × 0.75 μm.
Nucleoid Relationship to Mitochondrial Structures.
Fig. 2A is a slice from a correlated iPALM/EM dataset showing an overview of the aligned volume. The FIB-SEM image appears as a standard electron micrograph in grayscale with easily recognizable features such as nuclei. Overlaid on the EM image is the rendered iPALM image of TFAM-mEos2 molecules shown in a transparent red. The large field of view afforded by this method allowed us to examine many nucleoids in multiple mitochondria in different cells, revealing a diversity of nucleoid locations within mitochondria and membrane interactions.
Fig. 2.

Correlated images of TFAM-mEos2 iPALM data with EM images generated by FIB-SEM. Labels denote cellular nuclei (Nu), the mitochondrial boundary membrane (BM), including the outer and inner mitochondrial membranes, and cristae (Cr). (A) A correlated iPALM image of TFAM-mEos2 with a slice (x–y cross-section) from a FIB-SEM–generated volume showing many different cells containing nucleoids. Lettered boxes correspond to the magnified images shown in C, F, I, and L, except that I is rotated 90° with respective to its orientation in A. B, E, H, and K are slices through the FIB-SEM volume showing mitochondria with well defined membrane ultrastructure. C, F, I, and L show the aligned and overlaid TFAM-mEos2 iPALM data with the SEM images from B, E, H, and K. D, G, J, and M are slices (z–y or z–x cross-sections) through the iPALM/SEM correlated data that are perpendicular to C, F, I, and L, demonstrating the z-dimension of the data. White hatch marks in C, F, I, and L indicate the location through the image from which the perpendicular plane shown in D, G, J, and M was derived. (Scale bars: 500 nm.)
Fig. 2 B–M show 2D slices through mitochondria from the volume shown in Fig. 2A. The left panels (Fig. 2 B, E, H, and K) show the SEM-only image and the middle panels (Fig. 2 C, F, I, and L) show the overlay (x–y cross-section) of the TFAM-mEos2 signal (transparent red) over the corresponding SEM image. The right panels (Fig. 2 D, G, J, and M) are slices (z–y or z–x cross-sections) through the nucleoids that are perpendicular to the images in the middle panels. Each slice represents a sample thickness of 5 nm. From these images, one can observe a variety of nucleoid size, shape, location within mitochondria, and membrane relationship to cristae and the boundary membranes. Although there was no obvious single nucleoid location, size, or orientation to membranes that we observed, some patterns became apparent. Most often, the nucleoid occupied its own space in areas that were free of cristae (e.g., Fig. 2 C, I, and L, nucleoid 1). However, the cristae do appear to interact with nucleoids in several ways and orientations. In Fig. 2 C and I, the ends of cristae terminate into nucleoids. In the other panels (e.g., Fig. 2 F and L, nucleoid 2), the nucleoids appear to be intertwined with cristae. The orientation of nucleoids relative to cristae membranes may have functional consequences as there are compositional differences between the cristae tips, sides, and junctions (See Discussion). Movies S1, S2, S3, and S4 animate the progression through a z-series of the correlated image volumes of Fig. 2 C, F, I, and L.
Sometimes TFAM-mEos2 signal is observed outside of nucleoids and/or mitochondria. This may arise from several factors. First, as TFAM-mEos2 is translated in the cytoplasm and transported and imported into mitochondria, one should expect to see some distribution of TFAM-mEos2 outside the nucleoids. Second, the molecular localization uncertainty of PALM, 20 nm, could allow a few TFAM-mEos2 molecules located near the inner membrane appear as slightly outside the mitochondrion. Third, no imaging method is completely free of background that may give some degree of spurious signal. Finally, alignment in the z-dimension is not as rigorously controlled as in the x and y dimensions because of some degree of variability in the significant sample shrinkage induced during sample drying before FIB-SEM. This may be improved in future studies through the use of gold beads on both sides of the sample and better control of sample shrinkage via critical point drying.
Because our method is 3D, we were able to examine the 3D organization of the nucleoids within the mitochondria. We performed automated threshold segmentation of the iPALM and FIB-SEM data to generate 3D surface maps of the nucleoids and mitochondria (Fig. 3). After the automatic segmentation, we manually removed the outer membrane data to better observe the cristae and nucleoids. In Fig. 3A, the iPALM data of the nucleoid is rendered in red along with a slice from the correlated EM volume. Fig. 3B shows a close-up view of the nucleoid with a 3D map of the cristae shown in a slightly transparent yellow. This 3D view in Fig. 3B shows the tips of the cristae interacting with the nucleoid. Movie S5 shows this interaction more dynamically, and animates the progression through a z-series of the correlated iPALM/EM dataset and a rotational view of the nucleoid/cristae 3D maps.
Fig. 3.
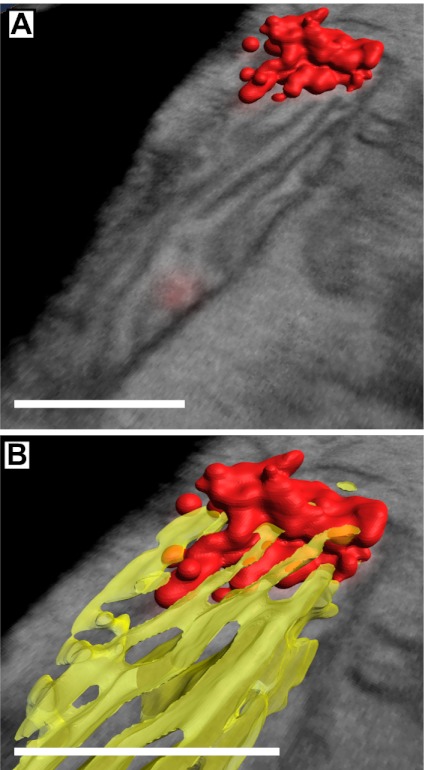
Three-dimensional map of a nucleoid and cristae membranes. Red indicates nucleoid, yellow indicates cristae. (A) Merged image of a 3D map of a nucleoid and a correlated SEM slice. (B) Closer view of the nucleoid in A with a cristae 3D map showing interaction of the cristae tips with the nucleoid. (Scale bars: 400 nm.)
Discussion
Correlation of Superresolution Fluorescence and EM Images.
Use of the Tokuyasu cryosectioning method for correlative microscopy has several advantages. One is that the time from sample fixation to PALM imaging is less than 1 d, compared with other methods used in correlative microscopy involving more conventional sample fixation and plastic embedding (10, 11), which take 4 to 8 d of processing before the sample can be sectioned. For the Tokuyasu method and staining procedure described here, membranes and membranous organelles are shown in the greatest contrast and quality. Nevertheless, artifacts such as ice crystal formation that distorts mitochondrial membranes cannot be ruled out. Alternative staining methods, including the use of ferrocyanide-reduced osmium coupled with lead citrate and uranyl acetate staining in polyvinyl alcohol, could provide contrast to different cellular structures (26). This technique will also likely be applicable to correlative PALM/EM using caged dyes, in which Tokuyasu cryosections were recently used to image nuclear DNA with nanometer resolution (27). The use of synthetic photoactivatable dyes provides the possibility to image nascent DNA (27, 28), RNA (29), protein translation (30, 31), or other metabolic processes in relation to cellular structures with nanometer precision. Identifying nucleoids in which mtDNA is replicating may aid our understanding of mtDNA replication and inheritance mechanisms and thus our understanding of mtDNA-linked diseases (32). Another option is to combine immunolabeling of sections with PALM and EM, thus providing another modality and flexibility to this technique.
Although we focused in this work on 3D techniques and iPALM and FIB-SEM in particular, the methods presented here are likely amenable to 2D analyses. For example, one could perform this procedure by using thinner sections and 2D superresolution fluorescence imaging (e.g., PALM, fluorescence-PALM, stochastic optical reconstruction microscopy, saturated structured illumination microscopy, STED) with 2D EM (scanning or transmission). It may also be possible to perform serial sectioning correlative PALM/EM to generate larger volumes from thinner sections or to image larger structures as done in array tomography using diffraction-limited microscopy (33). There are also other 3D superresolution techniques (34–36) that could be correlated to FIB-SEM or electron tomography data. The same mitochondrial nucleoid system has been studied with 2D PALM on thinner sections (∼100 nm) by using the Tokuyasu protocol (17). We confirmed here that the modified protocol as outlined in Fig. 1A also works for PALM imaging. A 2D image based on SEM should be feasible, but requires that the sections remain accessible to the electron beam, and therefore only minimal methylcellulose thickness can be tolerated so that the 1- to 2-KeV imaging electrons can penetrate. Such 2D correlative images have the advantage of a considerably simpler optical and electron microscope system, as well as a much faster and more practical image acquisition, but further protocol development is needed.
Mitochondrial Nucleoid Structure and Membrane Relationship.
Two recent studies have examined mitochondrial nucleoids by using superresolution fluorescence microscopy (17, 19). Unlike Kukat et al. (19), who found by STED microscopy that nucleoids have a uniform mean diameter of approximately 70 nm, our data presented here and elsewhere (17) show nucleoids with a variety of sizes that are organized into complex nonuniform shapes within mitochondria. Although we did not examine a large number of nucleoids in this work to build statistical significance regarding nucleoid size, our group previously found that ellipsoidal nucleoids had mean dimensions of 85 × 108 × 146 nm in the three axes, with some much larger (as large as 350 nm) (17). There are several things that may account for the observed size differences, including the fact that Kukat et al. (19) used antibody labeling, which has less molecular specificity than protein fusions. Also, we performed 3D microscopy, which may provide a better ability to examine the true 3D shape and dimensions. However, we cannot dismiss differences in sample preparation methods as a source of possible variation.
This study provides crucial data regarding the topology of nucleoids in relation to membranes. Previous superresolution data indicated that nucleoids are intimately associated with the inner mitochondrial membrane (17), but that two-color PALM data were not as definitive as the data presented here. In this study, we found multiple examples of nucleoid/cristae interaction including nucleoid/crista tip, nucleoid/crista side, and nucleoid/cristae intertwining. Other studies also seem to indicate that nucleoids interact with the inner membrane (37), possibly in ways that regulate nucleoid behavior.
The location of nucleoids relative to membranes is important for several reasons. First, the mtDNA located within nucleoids encodes essential protein components of the oxidative phosphorylation (OX-PHOS) machinery. These OX-PHOS proteins are all hydrophobic membrane proteins, and the assembly of the OX-PHOS machinery relies on the coordinated expression, import, and membrane insertion of nuclear and mtDNA-encoded proteins (38). The coordinated assembly of the OX-PHOS machinery is essential to cellular energy production, and thus defects in assembly are associated with disease (39). Second, membrane association may influence the proteins that interact with the nucleoid including those involved in mtDNA repair (40), nucleoid remodeling, and intercompartmental communication. Third, membrane interaction may be functionally important to mtDNA segregation and inheritance (37). A better understanding of how nucleoids interact with membranes may provide insight into mitochondrial-related disease mechanisms. Our current knowledge of the mtDNA genome and its mode of propagation and expression derive largely from several decades of molecular characterization of isolated mtDNA replicative intermediates and transcripts and in vitro analyses of mtDNA replication and transcription using purified macromolecular constituents. Approaches such as the one described here should lead to important insights on the nature of relevant in vivo interactions of the mitochondrial genetic system with other cellular components.
Summary and Outlook.
We described a variant of the Tokuyasu protocol to prepare biological samples for superresolution microscopy and EM. Images show the majority of the PAFPs survive the stronger fixation to give good PALM images. On the same sample, ultrastructural details such as mitochondrial cristae are preserved for EM. Nucleoids of mitochondria were thereby imaged in 3D by using iPALM and FIB-SEM to reveal the topology of the nucleoids and surrounding cristae at an unprecedented level of resolution. The methods are general and could be broadly applied to a variety of other biological questions.
Methods
Cell Culture.
The mifepristone inducible 3T3 Switch (3T3sw) mouse fibroblast cell line (Life Technologies) expresses a transcriptional activator protein with a yeast GAL4 binding domain, a progesterone ligand binding domain, and a p65 activation domain from NF-κB. The production of 3T3sw cells stably transfected with TFAM-mEos2 carrying plasmids was described previously (17). 3T3sw cells stably transfected with plasmids carrying TFAM-mEos2 were maintained in DMEM, 10% (wt/vol) normal calf serum, 1 mM sodium pyruvate, 2 mM L-alanyl-l-glutamine (GlutaMax), 50 μg/mL hygromycin B (Gibco/Invitrogen), and 200 μg/mL Zeocin. Expression of TFAM-mEos2 was induced with 200 pM mifepristone for 7 h.
Cell Fixation and Cryosectioning.
Cells were prepared for Tokuyasu cryosectioning as described previously (17) except that the fixative agent consisted of 4% (wt/vol) paraformaldehyde, 2% (wt/vol) glutaraldehyde in 100 mM sodium phosphate, pH 7.4, and 5% (wt/vol) sucrose. Cryosections were cut with a cryo immuno diamond knife (Diatome) at −80 °C in a low-temperature sectioning system (EM FC6 Cryochamber attached to an Ultracut EM UC6 ultramicrotome; Leica). Cutting speed was set at 100 mm/s. Cryosections were retrieved with drops of 1:1 2% (wt/vol) methylcellulose:2.3 M sucrose and placed on 25-mm no.-1.5 coverslips (Warner Instruments) coated with 25 × 35 nm bare Au nanorods (Nanopartz). Coverslip preparation and Au particle deposition were as described previously (13).
IPALM Imaging.
Before iPALM imaging, sections on coverslips were washed three times for 2 min with PBS solution to remove excess sucrose and methylcellulose. Sections were then treated with 0.5% sodium borohydride in 100 mM sodium phosphate buffer, pH 7.4, and washed twice for 2 min with PBS solution. An 18-mm no.-1.5 coverslip was placed on top of the other coverslip with the cryosection and sealed by using 5-min epoxy (ITW Performance Polymers) and Vaseline (Unilever). IPALM imaging of TFAM-mEos2 was performed as described previously (17).
Staining and FIB-SEM.
After iPALM imaging, the top coverslip was removed and the sections were washed three times for 2 min with PBS solution and three times for 3 min with deionized water. Staining of the sections was done by placing the coverslip with the sections on a drop of 0.5% (wt/vol) uranyl acetate in 1.8% (wt/vol) methylcellulose on ice for 20 min. Excess methylcellulose was wicked away by using filter paper, and the coverslip was air-dried at room temperature. A drop of cyanoacrylate (Elmer's Products) was added over the dried sections and centrifuged in a spin coater (Laurell Technologies WS-400-6NPP) at 1,000 rpm for 20 s. A 100-nm-thick carbon coating was applied to the specimens by using a Gatan 682 Precision Etching Coating System at 6 KeV.
A Zeiss NVision 40 system driven by a custom external scan generator and control software was used for FIB-SEM. A focused beam of Ga+ ions at 30 KeV milled an approximately 50-μm-deep and approximately 150-μm-wide trench through the sample and into the coverslip. The upper wall of the trench was imaged from above with a scanning electron microscope at 1.5 KeV, forming a rectangular 80-μm × 5-μm image. A subsequent pass of the ion beam removes another 5 nm of the sample/trench wall followed by another SEM image. This cycle of ablation and imaging is repeated until the entire area of interest is imaged.
Alignment of iPALM and 3D EM Data.
To align the iPALM and FIB-SEM datasets, the same approximately 10 to 20 fiducials were identified in each dataset (Fig. 4). The iPALM coordinates of each fiducial can be registered to less than 30 nm accuracy to their FIB-SEM coordinates as seen in a portion of the full imaged area. To achieve this accuracy over the full 80-μm × 30-μm field, a second-order polynomial transformation in x, y coordinates was adequate. Higher accuracy (<10 nm) can be achieved on a portion, e.g., half, of the imaged area or by using a third-order polynomial transformation. Alignment accuracy was confirmed by using the residual alignment errors on the mapped set of fiducials. Alignment in the third dimension was not as direct, as the sample had vertical shrinkage after optical imaging induced by dehydration and electron imaging. We estimated this shrinkage factor to be approximately 30% by registering the top boundary of the FIB-SEM stack to the top boundary of background signal in the iPALM stack. This rescaling achieved very good feature alignment.
Fig. 4.

Alignment of the iPALM and EM data by using gold fiducials. Gold particles can be seen in both EM and fluorescence images. (A) An SEM image of a portion of an imaged area at a z height cutting through the fiducial decorated surface where the fiducials are seen as tiny white dots (arrows). (B) A differential interference contrast image of the cryosection overlaid with fluorescence images of fiducials (arrows). Alignment to less than 30 nm accuracy is made by finding the fiducial centroids in each image and applying a transformation to match the fiducial coordinates. (Scale bar: 5 μm.)
Three-Dimensional Mapping.
Image segmentation and visualization was performed by using Amira (Visage Imaging).
Supplementary Material
Acknowledgments
We thank Timothy A. Brown and Ariana N. Tkachuk for the TFAM-mEos2 stable cell line and for helpful discussions and advice. We thank Yalin Wang of the Janelia Farm Research Campus electron microscopy facility for cryosectioning. This work was supported by the Howard Hughes Medical Institute.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1121558109/-/DCSupplemental.
References
- 1.Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312:217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 2.Lippincott-Schwartz J, Patterson GH. Photoactivatable fluorescent proteins for diffraction-limited and super-resolution imaging. Trends Cell Biol. 2009;19:555–565. doi: 10.1016/j.tcb.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gustafsson MG. Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci USA. 2005;102:13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klar TA, Hell SW. Subdiffraction resolution in far-field fluorescence microscopy. Opt Lett. 1999;24:954–956. doi: 10.1364/ol.24.000954. [DOI] [PubMed] [Google Scholar]
- 5.Betzig E, et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 6.Hess ST, Girirajan TP, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsien RY. The green fluorescent protein. Annu Rev Biochem. 1998;67:509–544. doi: 10.1146/annurev.biochem.67.1.509. [DOI] [PubMed] [Google Scholar]
- 9.Morphew MK. 3D immunolocalization with plastic sections. Methods Cell Biol. 2007;79:493–513. doi: 10.1016/S0091-679X(06)79019-7. [DOI] [PubMed] [Google Scholar]
- 10.Kukulski W, et al. Correlated fluorescence and 3D electron microscopy with high sensitivity and spatial precision. J Cell Biol. 2011;192:111–119. doi: 10.1083/jcb.201009037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watanabe S, et al. Protein localization in electron micrographs using fluorescence nanoscopy. Nat Methods. 2011;8:80–84. doi: 10.1038/nmeth.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shu X, et al. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 2011;9:e1001041. doi: 10.1371/journal.pbio.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shtengel G, et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc Natl Acad Sci USA. 2009;106:3125–3130. doi: 10.1073/pnas.0813131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knott G, Marchman H, Wall D, Lich B. Serial section scanning electron microscopy of adult brain tissue using focused ion beam milling. J Neurosci. 2008;28:2959–2964. doi: 10.1523/JNEUROSCI.3189-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malka F, Lombès A, Rojo M. Organization, dynamics and transmission of mitochondrial DNA: Focus on vertebrate nucleoids. Biochim Biophys Acta. 2006;1763:463–472. doi: 10.1016/j.bbamcr.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Spelbrink JN. Functional organization of mammalian mitochondrial DNA in nucleoids: History, recent developments, and future challenges. IUBMB Life. 2010;62:19–32. doi: 10.1002/iub.282. [DOI] [PubMed] [Google Scholar]
- 17.Brown TA, et al. Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol Cell Biol. 2011;31:4994–5010. doi: 10.1128/MCB.05694-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaufman BA, et al. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol Biol Cell. 2007;18:3225–3236. doi: 10.1091/mbc.E07-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kukat C, et al. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci USA. 2011;108:13534–13539. doi: 10.1073/pnas.1109263108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Legros F, Malka F, Frachon P, Lombès A, Rojo M. Organization and dynamics of human mitochondrial DNA. J Cell Sci. 2004;117:2653–2662. doi: 10.1242/jcs.01134. [DOI] [PubMed] [Google Scholar]
- 21.Larsson NG, Oldfors A, Holme E, Clayton DA. Low levels of mitochondrial transcription factor A in mitochondrial DNA depletion. Biochem Biophys Res Commun. 1994;200:1374–1381. doi: 10.1006/bbrc.1994.1603. [DOI] [PubMed] [Google Scholar]
- 22.McKinney SA, Murphy CS, Hazelwood KL, Davidson MW, Looger LL. A bright and photostable photoconvertible fluorescent protein. Nat Methods. 2009;6:131–133. doi: 10.1038/nmeth.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brown TA, Fetter RD, Tkachuk AN, Clayton DA. Approaches toward super-resolution fluorescence imaging of mitochondrial proteins using PALM. Methods. 2010;51:458–463. doi: 10.1016/j.ymeth.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tokuyasu KT, Singer SJ. Improved procedures for immunoferritin labeling of ultrathin frozen sections. J Cell Biol. 1976;71:894–906. doi: 10.1083/jcb.71.3.894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clancy B, Cauller LJ. Reduction of background autofluorescence in brain sections following immersion in sodium borohydride. J Neurosci Methods. 1998;83:97–102. doi: 10.1016/s0165-0270(98)00066-1. [DOI] [PubMed] [Google Scholar]
- 26.Takizawa T, Anderson CL, Robinson JM. A new method to enhance contrast of ultrathin cryosections for immunoelectron microscopy. J Histochem Cytochem. 2003;51:31–39. doi: 10.1177/002215540305100105. [DOI] [PubMed] [Google Scholar]
- 27.Wysocki LM, et al. Facile and general synthesis of photoactivatable xanthene dyes. Angew Chem Int Ed Engl. 2011;50:11206–11209. doi: 10.1002/anie.201104571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci USA. 2008;105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jao CY, Salic A. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc Natl Acad Sci USA. 2008;105:15779–15784. doi: 10.1073/pnas.0808480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dieterich DC, et al. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci. 2010;13:897–905. doi: 10.1038/nn.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beatty KE, et al. Fluorescence visualization of newly synthesized proteins in mammalian cells. Angew Chem Int Ed Engl. 2006;45:7364–7367. doi: 10.1002/anie.200602114. [DOI] [PubMed] [Google Scholar]
- 32.Zeviani M, Di Donato S. Mitochondrial disorders. Brain. 2004;127:2153–2172. doi: 10.1093/brain/awh259. [DOI] [PubMed] [Google Scholar]
- 33.Micheva KD, Smith SJ. Array tomography: A new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 2007;55:25–36. doi: 10.1016/j.neuron.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang B, Wang W, Bates M, Zhuang X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science. 2008;319:810–813. doi: 10.1126/science.1153529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Punge A, et al. 3D reconstruction of high-resolution STED microscope images. Microsc Res Tech. 2008;71:644–650. doi: 10.1002/jemt.20602. [DOI] [PubMed] [Google Scholar]
- 36.Juette MF, et al. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat Methods. 2008;5:527–529. doi: 10.1038/nmeth.1211. [DOI] [PubMed] [Google Scholar]
- 37.Meeusen S, Nunnari J. Evidence for a two membrane-spanning autonomous mitochondrial DNA replisome. J Cell Biol. 2003;163:503–510. doi: 10.1083/jcb.200304040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smeitink J, van den Heuvel L, DiMauro S. The genetics and pathology of oxidative phosphorylation. Nat Rev Genet. 2001;2:342–352. doi: 10.1038/35072063. [DOI] [PubMed] [Google Scholar]
- 39.Fernández-Vizarra E, Tiranti V, Zeviani M. Assembly of the oxidative phosphorylation system in humans: What we have learned by studying its defects. Biochim Biophys Acta. 2009;1793:200–211. doi: 10.1016/j.bbamcr.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 40.Stuart JA, Mayard S, Hashiguchi K, Souza-Pinto NC, Bohr VA. Localization of mitochondrial DNA base excision repair to an inner membrane-associated particulate fraction. Nucleic Acids Res. 2005;33:3722–3732. doi: 10.1093/nar/gki683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.