

Abstract
In photosystem II, oxygen evolution occurs by the accumulation of photo-induced oxidizing equivalents at the oxygen-evolving complex (OEC). The sequentially oxidized states are called the S0-S4 states, and the dark stable state is S1. Hydrogen bonds to water form a network around the OEC; this network is predicted to involve multiple peptide carbonyl groups. In this work, we tested the idea that a network of hydrogen bonded water molecules plays a catalytic role in water oxidation. As probes, we used OEC peptide carbonyl frequencies, the substrate-based inhibitor, ammonia, and the sugar, trehalose. Reaction-induced FT-IR spectroscopy was used to describe the protein dynamics associated with the S1 to S2 transition. A shift in an amide CO vibrational frequency (1664 (S1) to 1653 (S2) cm-1) was observed, consistent with an increase in hydrogen bond strength when the OEC is oxidized. Treatment with ammonia/ammonium altered these CO vibrational frequencies. The ammonia-induced spectral changes are attributed to alterations in hydrogen bonding, when ammonia/ammonium is incorporated into the OEC hydrogen bond network. The ammonia-induced changes in CO frequency were reversed or blocked when trehalose was substituted for sucrose. This trehalose effect is attributed to a displacement of ammonia molecules from the hydrogen bond network. These results imply that ammonia, and by extension water, participate in a catalytically essential hydrogen bond network, which involves OEC peptide CO groups. Comparison to the ammonia transporter, AmtB, reveals structural similarities with the bound water network in the OEC.
Keywords: amide carbonyl frequency, Amt B transporter, photosystem II, vibrational spectroscopy, water oxidation
In oxygenic photosynthesis, photosystem II (PSII) catalyzes the oxidation of water and reduction of plastoquinone (1). Each reaction center includes the transmembrane subunits D1, D2, CP43, and CP47, which bind the redox-active cofactors. After photoexcitation, a charge separation is generated between the dimeric chl donor, P680, and a bound plastoquinone, QA. 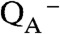 reduces a second quinone, QB, and
reduces a second quinone, QB, and 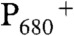 oxidizes a tyrosine residue, YZ, Y161 in the D1 polypeptide. The radical, YZ•, is a powerful oxidant (2). Under physiological conditions, YZ• oxidizes the Mn4CaO5 cluster, where water oxidation occurs (3). Four sequential photooxidations lead to the release of molecular oxygen (4). The oxygen yield fluctuates with period four. The sequentially oxidized states of the Mn cluster are called the Sn states, where n refers to the number of oxidizing equivalents stored at the OEC. S1 is the dark stable state of the OEC. A single flash given to a dark-adapted sample of PSII generates the S2 state, which corresponds to the oxidation of Mn(III) to Mn(IV) (5). The water oxidation chemistry in the S state cycle occurs on the microsecond to millisecond time scale, and oxygen is released during the S3 to S0 transition (4, 6).
oxidizes a tyrosine residue, YZ, Y161 in the D1 polypeptide. The radical, YZ•, is a powerful oxidant (2). Under physiological conditions, YZ• oxidizes the Mn4CaO5 cluster, where water oxidation occurs (3). Four sequential photooxidations lead to the release of molecular oxygen (4). The oxygen yield fluctuates with period four. The sequentially oxidized states of the Mn cluster are called the Sn states, where n refers to the number of oxidizing equivalents stored at the OEC. S1 is the dark stable state of the OEC. A single flash given to a dark-adapted sample of PSII generates the S2 state, which corresponds to the oxidation of Mn(III) to Mn(IV) (5). The water oxidation chemistry in the S state cycle occurs on the microsecond to millisecond time scale, and oxygen is released during the S3 to S0 transition (4, 6).
The structure of PSII has been reported at a resolution of 1.9 Å (3). The position of bound water molecules (approximately 1,300 per PSII monomer) was predicted in this structure. At the OEC, two waters were predicted to ligate manganese, and two were predicted to ligate calcium. This PSII structure suggests that water participates in an extensive hydrogen-bonding network around the OEC. In this work, we obtain spectroscopic evidence for the existence of this network and evaluate its role in oxygen evolution.
The PSII structure reveals that peptide carbonyl groups, for example, provided by residues D1-D170 and D1-S169, are in hydrogen-bonding distance from the network of water molecules, on the so-called donor side of the reaction center (Fig. 1A). The frequencies of these amide CO groups are expected to respond to photooxidation of manganese during the S1 to S2 transition. This step is believed to correspond to the accumulation of positive charge on the OEC (MnIII to MnIV); there is no evidence for a compensating proton transfer reaction (5). This photooxidation reaction may cause shifts in the hydrogen-bonding network, which could be important in control of catalysis (7). Peptide carbonyl infrared frequencies are known to be significantly redshifted as hydrogen bond strength increases, due to an inductive effect, which stabilizes partial negative charge on the oxygen (8, 9).
Fig. 1.

Structures of the OEC and the ammonia binding site in the transport protein, AmtB, showing peptide carbonyl interactions with a network of bound water molecules. Fig. 1A was generated from the 1.9 Å structure of PSII (3). Fig. 1B was generated from the 1.35 Å structure of AmtB (15). Dashed lines show possible interactions with carbonyls (sticks) and the nearest atom. Protein subunits are shown as ribbons. Peptide carbonyls shown in (A) are contributed by the D1 polypeptide of PSII. One side chain, N181-D1, which donates a carbonyl, is shown. Putative bound water molecules in the OEC are denoted as W1-4 in (A). Peptide carbonyls in (B) are contributed by the single polypeptide chain of AmtB.
To perturb the putative OEC hydrogen-bonding network, we used two reagents, ammonia and the sugar, trehalose. Ammonia is a substrate analog and a well-known inhibitor of photosynthetic oxygen evolution (10, 11). Substitution of ammonia or ammonium for water is expected to perturb or disrupt the hydrogen-bonding network, due to the increase in bond strength, when the NH and OH groups are compared (12, 13). Trehalose is known to increase protein stability. One mechanism has been proposed to involve the displacement of water molecules from the protein surface (14).
To define the role of peptide CO groups in ammonia binding, the OEC structure (Fig. 1A) was compared to the ammonia transporter, AmtB, from E. coli (Fig. 1B) (15, 16). This comparison suggests that peptide CO groups provide an electrostatic environment, which stabilizes ammonia/ammonium binding and a bound network of water molecules. In PSII, reaction-induced (light induced, difference) Fourier transform infrared (FT-IR) spectroscopy can be used to measure peptide CO frequencies, which are altered by the OEC oxidation reaction. On the timescale of rapid scan measurements (seconds), the difference spectrum will reflect long-lived structural rearrangements and changes in solvent polarization (17). Vibrational spectroscopy provides an incisive method with which to interrogate dynamics in complex proteins.
In this work, we report that photooxidation of Mn during the S1 to S2 transition strengthens a hydrogen-bonding network. We show that, in the presence of ammonia, the S1 to S2 transition weakens the hydrogen bond network. The effect of ammonia is reversed by addition of trehalose. From this, we conclude that the OEC hydrogen bond network plays a critical role in photosynthetic oxygen evolution.
Results
PSII core preparations were isolated from market spinach using Triton X-100 and octylthioglucoside, as previously described (18, 19). These PSII core preparations gave high rates of steady state oxygen evolution (1,100 μmol O2 (mg chl-hr)-1 at pH 6.0) both in trehalose (0.4 M) and sucrose (0.4 M) buffers. As isolated, PSII core samples (Fig. 2) exhibited bands from the CP47, CP43, D1/D2, and the extrinsic subunits, psbO, psbP (24 kDa), and psbQ (18 kDa) (19). Three resuspensions were used to transfer PSII into the appropriate buffer. After three resuspensions and incubation for 1 h at pH 7.5; i.e., the conditions used for FT-IR spectroscopy, loss of the psbP and psbQ extrinsic subunits was observed (Fig. 2). Removal of these extrinsic polypeptides allows increased access to the OEC (20), which is an advantage in our spectroscopic experiments.
Fig. 2.

Urea-SDS-PAGE analysis of PSII samples used for reaction-induced FT-IR spectroscopy. After three resuspension steps, samples were treated with 100 mM NaCl or NH4Cl at pH 6.0 or 7.5 for 1 h (0 °C). Control (Ctrl) samples contained only 15 mM NaCl. PSII samples were isolated by the procedure of (19). A photosystem II membrane preparation (BBY) was used as a control (18). The protein standards are labeled with molecular weights (kDa) on the left.
To test the effect of ammonia in this preparation, ammonia/ammonium was titrated from 0.02 to 1 M at pH 6.0 (Fig. 3B) and pH 7.5 (Fig. 3A). To mimic the FT-IR conditions, the samples were treated with the same number of buffer resuspension steps (three). The half inhibition point (pH 6.0, 170 mM ammonia; pH 7.5, 110 mM ammonia) was similar at pH 6.0 and 7.5. This result suggests that in this PSII core preparation, lacking the extrinsic peptides, the OEC binds either ammonia or the ammonium cation. A similar conclusion has been reached regarding the AmtB transporter, which can dock either ammonia or ammonium. In AmtB, the ammonium cation is deprotonated for transport across the membrane as the neutral species (15, 16).
Fig. 3.

Oxygen-evolution activity of PSII samples titrated with ammonia, either at (A) pH 7.5 or (B) pH 6.0. Before the assay, samples were treated with three resuspension steps, to mimic conditions used for FT-IR spectroscopy. Buffers containing 0.4 M sucrose (squares, solid curve) or 0.4 M trehalose (diamonds, dashed curve) were employed. See Materials and Methods for more information. The half-inhibition point is shown by the dotted lines.
Reaction-induced FT-IR spectroscopy was used to report on protein dynamics induced by the S1 to S2 transition (Fig. 4). The S1 to S2 transition occurs on the microsecond timescale (6, 21). In our experiments, a protocol, previously described (22) was employed that monitors solvent and protein relaxation events occurring after the S1 to S2 transition on the 15 s timescale. PSII was preflashed with a 532 nm flash and dark adapted for 20 min to generate the majority of centers in the S1 state. An additional 532 nm actinic flash then gives the S2 state. The difference FT-IR spectrum, associated with S2-minus-S1, was constructed. The control spectra (Fig. 4) resemble those described previously (22, 23), which have been attributed to frequency band shifts, induced for amino acid residues and amide groups near the OEC. Amplitudes and frequencies were shown to oscillate with period four. It has been concluded previously that this spectrum, acquired in the presence of the exogenous electron acceptor, potassium ferricyanide, is dominated by OEC contributions in the 1,800 - 1,200 cm-1 region (23). Potential acceptor side contributions from  and QA have been assigned by isotopic labeling of the quinones at 77 K (24, 25). Through previous global 13C labeling experiments, bands in the 1,650 cm-1 region were attributed to amide I modes (C = O) of the polypeptide backbone on the PSII donor side (26).
and QA have been assigned by isotopic labeling of the quinones at 77 K (24, 25). Through previous global 13C labeling experiments, bands in the 1,650 cm-1 region were attributed to amide I modes (C = O) of the polypeptide backbone on the PSII donor side (26).
Fig. 4.

Reaction-induced FT-IR spectra, associated with the S2-minus-S1 transition in PSII. The box highlights the amide carbonyl (CO) stretching region. The data were acquired after the addition of (A) 100 mM NaCl in 0.4 M sucrose buffer, pH 7.5 or after the addition of (B) 100 mM NaCl in 0.4 M trehalose buffer, pH 7.5. In (C), a representative baseline is shown. The spectra are averages of data obtained from (A) 14, (B) 12, and (C) 14 samples. The y-axis tick marks represent 1 × 10-4 absorbance units. See Materials and Methods for more details.
The S1 to S2 FT-IR spectrum in sucrose buffer is shown in an expanded view in Fig. 5A. In the amide CO region, a differential band with a negative 1,664 and a positive 1,653 cm-1 frequency is observed. Analysis of the amplitude suggests that this differential feature arises from approximately 1 peptide CO bond. This peptide CO frequency was used as a marker for the strength of hydrogen-bonding interactions. The observed approximately 10 cm-1 frequency decrease suggests that an increase in hydrogen bond strength accompanies the S1 to S2 transition. For example, in the matrix isolation infrared spectrum of methylacetate, the C = O band downshifted 20 cm-1 with the gain of a single hydrogen bond to water (27). Comparing Fig. 5 A and B, spectra acquired in the presence of sucrose and trehalose were similar but exhibited small frequency shifts in the amide CO 1,650 cm-1 region. This was assessed by construction of a double difference spectrum: sucrose-minus-trehalose (Fig. 5D) and comparison to a control, in which no vibrational bands are expected (Fig. 5E). This result is consistent with no direct interaction between the two different sugar molecules and protein CO groups. This conclusion is supported by previous studies of trehalose effects on protein stability (14).
Fig. 5.

The 1,800 - 1,600 cm-1 region of the reaction-induced FT-IR, S2-minus-S1 spectrum. The data in A–C are repeated from Fig. 4 A–C (boxed region). (D) is a double difference spectrum, sucrose-minus-trehalose, corresponding to the subtraction of (B) from (A). The spectrum in (E) is a control double difference spectrum, generated by subtraction of one half of the data in (B) from the other half and division by √2. Bands discussed in the text are filled. The y-axis tick marks represent 1 × 10-4 absorbance units.
Reaction-induced FT-IR experiments were conducted in the presence of 50 mM (Fig. 6A) and 100 mM (Fig. 6B) ammonia, pH 7.5, in 0.4 M sucrose buffers. Experiments were also conducted in which trehalose was substituted for sucrose; i.e., 100 mM ammonia, pH 7.5, in 0.4 M trehalose buffer (Fig. 6C). Comparison of the NaCl controls (Fig. 6 A and B, solid lines) shows a small ionic strength effect on carbonyl frequencies, although the 1,664 and 1,653 cm-1 bands are observed in both. Data from the ammonia containing samples are shown as dashed lines. At both ammonia concentrations, bands are observed at 1,678 cm-1 and 1,630 cm-1, although with less intensity at 50 mM ammonia. The substitution of trehalose for sucrose decreases the amplitude of the 1,664 and 1,653 cm-1 bands. These spectra support the conclusion that the negative 1,664 cm-1 band downshifts to 1,630 cm-1 and that the positive 1,653 cm-1 band upshifts to 1,678 cm-1 in the presence of ammonia.
Fig. 6.

The 1,800 - 1,600 cm-1 region of the reaction-induced FT-IR spectra, associated with the S2-minus-S1 transition in PSII. The data were acquired in 0.4 M sucrose buffer, pH 7.5 (A and B) or 0.4 M trehalose buffer, pH 7.5 (C). The spectra were acquired after the addition of (A, solid) 50 mM NaCl, (A, dashed) 50 mM NH3, (B and C, solid) 100 mM NaCl, or (B and C, dashed) 100 mM NH3. In (B), bands discussed in the text are labeled S1, S2 if observed in a control sample and S1A, S2A if observed in an ammonia treated sample. In (D), a representative baseline is shown. The spectra are averages of data obtained from (A) 8, (B) 14, (C) 12, and (D) 14 samples. The y-axis tick marks represent 1 × 10-4 absorbance units.
Double difference spectra were constructed: control-minus-ammonia (Fig. 7). Comparison to the control double difference spectrum, in which no vibrational bands are expected (Fig. 7D), revealed that the spectral changes in 50 mM (Fig. 7A) and 100 mM ammonia (Fig. 7B) are significant. The double difference spectra confirm that substitution of trehalose for sucrose reversed or blocked the effect of 100 mM ammonia (Fig. 7C).
Fig. 7.

Double difference spectra, showing the effect of ammonia addition on the reaction-induced FT-IR spectrum. The spectra were constructed by subtraction of data shown in Fig. 6. In (A), 50 mM NaCl addition-minus-50 mM ammonia addition, 0.4 M sucrose, in (B) 100 mM NaCl addition-minus-100 mM ammonia addition, 0.4 M sucrose, and in (C) 100 mM NaCl addition-minus-100 mM ammonia addition, 0.4 M trehalose. (D) is a control double difference spectrum, generated by subtraction of one half of the data in (B) from the other half and division by √2. Bands discussed in the text are filled. The y-axis tick marks represent 1 × 10-4 absorbance units.
Discussion
An extensive hydrogen bond network has been proposed to surround the oxygen-evolving complex in PSII. This putative network includes several water molecules, which are bound to manganese and calcium ions (3). First coordination sphere ligands to the calcium and manganese ion include carboxylate and imidazole side chains. Previous mutational analysis suggests that the activity of the OEC is influenced by a network of interactions involving residues outside this first coordination sphere (28–30). The identification of possible proton exit pathways lends support to this idea (3, 31).
Many studies have focused on the interactions of ammonia with the OEC because ammonia is isoelectronic with the substrate and is an inhibitor of steady state oxygen evolution (10). When PSII contains the extrinsic polypeptides, psbP and psbQ, there are three binding sites for ammonia. At one of these binding sites, chloride is a competitive inhibitor (32, 33). Loss of the extrinsic polypeptides (psbQ and psbP), as documented here, increases the rate of chloride exchange (34), so we conducted our experiments with a high buffer chloride concentration. NH3 has been proposed to bind to the OEC in the S2 and S3 states, but not in the S1 state (11, 35). It has been suggested previously that ammonia free base binds directly to Mn (10, 11, 36, 37). The proposal of a direct binding site to Mn is congruent with our results, in which we explored the effect of ammonia on the OEC hydrogen bond network, but not directly on the bound metal ions.
Previous reaction-induced FT-IR spectroscopic studies disagreed concerning the effect of ammonia on the spectrum and on the OEC. It was concluded either that ammonia induces a change in carboxylate ligation (38, 39) or that the ammonium cation binds to deprotonated carboxylate groups near the Mn cluster (40). In intact PSII, carboxylate groups near the OEC would be expected to be charge neutralized by the bound metal ions, so a direct interaction between ammonium and such anionic groups might be consistent with irreversible denaturation of PSII and loss of the OEC. With long incubation times, ammonia may modify posttranslationally modified amino acid residues (41) and lead to light-induced degradation reactions (42). Some of the discrepancies between the previously published FT-IR studies may have been due to the use of long ammonia incubation times, lengthy illuminations, and multiple (approximately 80) flash regiments. In our studies, we minimized the incubation time, the amount of illumination, and the concentration of ammonia to minimize such irreversible effects. Each sample was given only one preflash and one actinic flash, and the data from multiple samples were then averaged. This procedure, in which samples are used only once, promoted spectral reproducibility and sample integrity in our hands. Our experiments were conducted on a purified PSII core preparation to increase signal to noise and at high sample hydration levels, which are necessary to preserve activity. The use of these conditions precludes direct observation of water or ammonia stretching vibrations in the 3,500 cm-1 region. In PSII (BBY) membranes, these amide carbonyl spectral changes were not detectable. We attribute this to the increased antenna size and subunit complexity of the BBY preparation, which retains an extensive light harvesting complex (43).
We used the frequencies of these amide carbonyl groups as probes of the OEC hydrogen-bonding network. As assessed by their frequencies and intensity, a single peptide CO group, absorbing at 1,664 cm-1, is perturbed by photooxidation of the OEC, and downshifts to 1,653 cm-1. The addition of ammonia had differential effects on these frequencies in the S1 and S2 states. The 25 cm-1 upshift in the S2 state is consistent with a decrease in hydrogen bond strength, which might be expected from previous studies of NH⋯O = C and the OH⋯O = C bond enthalpies (12, 13). However, a much different ammonia effect was observed in the S1 state, with the CO vibrational frequency shifting down by 34 cm-1. Previously, it has been concluded that ammonia does not bind to PSII in this S state (11). Therefore, this downshift may be due to a more indirect structural or electrostatic effect on the hydrogen-bonding water network. The overall result of ammonia addition is that the S1 to S2 transition weakens a hydrogen bond network. By contrast, when ammonia is not present, this transition strengthens the network. This effect is dependent on the concentration of ammonia, suggesting that the change in hydrogen bond strength is coupled with the inhibition mechanism.
We found the interesting result that the ammonia effect was reversed in the presence of trehalose. This reversibility argues that ammonia inhibition is not caused by covalent modification or a denaturation effect. Trehalose is known to compete with water for hydrogen-bonding interactions at protein surfaces (14). Thus, the effects of trehalose are attributed to exclusion of water and ammonia from the solvation layer of the OEC.
Comparison to the ammonia/ammonium binding site in the AmtB transporter (15) is of value in interpreting our results (Fig. 8A, OEC and B, AmtB). The AmtB transporter is > 400 amino acids in length and is assembled as a trimer, with each monomer consisting of 11 transmembrane domains. Ammonia/ammonium binding occurs at a vestibule on the periplasmic side. No significant primary sequence homology exists between PSII core polypeptides and AmtB. However, the tertiary structures show that water networks in the OEC and in AmtB are each stabilized by peptide carbonyl groups. In AmtB, binding of methylamine or ammonia/ammonium displaced bound water molecules in the hydrogen-bonding network. The ammonia/ammonium binding site involves hydrogen-bonding interactions between  and the side chain oxygen of serine 219. Stabilizing pi-cation interactions with Trp 148 and Phe 107 were also inferred from the structure (Fig. 8B).
and the side chain oxygen of serine 219. Stabilizing pi-cation interactions with Trp 148 and Phe 107 were also inferred from the structure (Fig. 8B).
Fig. 8.

Similarities between the OEC and the ammonia binding site in the ammonia transport protein, AmtB. Fig. 8A was generated from the 1.9 Å structure of PSII (3). Fig. 8B was generated from the 1.35 Å structure of AmtB (15). Dashed lines show distances between atoms (Å). Amino acid residues are shown as sticks. Peptide carbonyl oxygen atoms are in red. The hydroxyl group of serine is in blue. Assigned water molecules in the OEC hydrogen-bonding network are denoted as W1-4 in (A). All other water molecules are shown as unlabeled gray spheres.
In AmtB, the carbonyl group of serine 219 hydrogen bonds with water molecules in the ordered network (Fig. 8B). Examining the OEC and AmtB networks, we identify serine 169 in the D1 polypeptide, which may play an analogous role. The carbonyl group of this amino acid is predicted to have a hydrogen-bonding interaction with W1 on Mn4 (Fig. 8A). Ammonia binding in the S2 state may involve displacement of this bound water molecule, which directly interacts with manganese.
The experiments discussed here report that photooxidation of manganese, during the S1 to S2 transition, strengthens hydrogen bonding to peptide carbonyl group(s). Disruption of this network by the water analog, ammonia, inhibits the steady state rate of water oxidation. Under these conditions, we show that the S1 to S2 transition results in a decrease in hydrogen bond strength, suggesting that the hydrogen bond network is important in catalysis. The effects of ammonia were reversed with addition of trehalose, providing additional support for the role of ordered solvent molecules in photosynthetic oxygen evolution.
Materials and Methods
PSII-enriched membranes (BBY) were isolated from market spinach as described previously (18), and PSII core preparations were performed according to the procedure outlined by Mishra et al. (19). After purification, samples were frozen (-70 °C), suspended in 400 mM sucrose, 50 mM MES-NaOH pH 6.0, 15 mM NaCl buffer. To adjust the pH to 7.5, samples were thawed, pelleted (50,000 × g, 15 min, 4 °C), and then resuspended twice in 0.4 M sucrose, 50 mM Hepes-NaOH, pH 7.5, 15 mM NaCl or in 0.4 M trehalose, 50 mM Hepes-NaOH, pH 7.5, 15 mM NaCl. The resuspended sample was frozen in small aliquots at -70 °C.
To test the activity of samples employed for FT-IR spectroscopy, oxygen-evolution activity was measured on samples treated with the same number of resuspension steps (three, see below). Recrystallized 2,6-dichlorobenzoquinone (DCBQ, recrystallized) and potassium ferricyanide were used as electron acceptors at final concentrations of 0.5 and 1 mM, respectively (44). The assay buffers were 400 mM sucrose, 50 mM MES-NaOH, pH 6.0, 15 mM NaCl or 400 mM sucrose, 50 mM Hepes-NaOH, pH 7.5, 15 mM NaCl.
To test the polypeptide content, urea-SDS-PAGE (45) was performed after the three resuspensions employed for FT-IR spectroscopy. Samples were treated with sodium chloride or ammonium chloride to mimic those conditions. For PAGE, samples were denatured (5.42 M urea; 0.125 M dithiothreitol; 4.17% sodium dodecyl sulfate) and subjected to urea-SDS-PAGE at 100 V for 2 h. The stacking gel contained 6.15% acrylamide and the resolving layer contained 13.67% acrylamide. The gel was stained with 0.05% Coomassie Brilliant Blue R-250 and destained overnight (5% methanol; 10% acetic acid).
Reaction-induced FT-IR difference spectroscopy was performed at 263 K and pH 7.5, and in the presence of 7 mM potassium ferricyanide, as previously described (17, 46–48). The resuspended sample, described above, was thawed and pelleted. The sample was resuspended in the appropriate buffer (sucrose or trehalose) for the third time. Potassium ferricyanide was added from a 100 mM stock in water, which was made up immediately before the experiment. Ammonium chloride or sodium chloride was added from 3 M buffered stock solutions to give final concentrations of 65 mM or 115 mM chloride. The sample was then centrifuged again (50,000 × g, 15 min) to produce a pellet. The total incubation time with NH4Cl or NaCl was kept constant and limited to approximately 1 h, including centrifugation, preparation of the FT-IR sample, and the dark-adaptation. The PSII pellet was spread on a CaF2 window to yield an O-H stretching absorbance (3,370 cm-1) to amide II (1,550 cm-1) absorbance ratio of > 3. Average values for O-H stretching absorbance and amide II absorbance were 1.57 ± 0.26 and 0.51 ± 0.07, respectively. Samples were sandwiched by a second CaF2 window; the edges were sealed with high vacuum grease and wrapped with parafilm to prevent sample dehydration. FT-IR data acquisition parameters were as follows: 8 cm-1 spectral resolution; four levels of zero filling; Happ-Genzel apodization function; 60 KHz mirror speed; Mertz phase correction. Samples were given a single saturating 532 nm laser preflash followed by 20 min of dark adaptation to synchronize reaction centers in the S1 state. Samples were flashed once more (actinic flash) followed by 15 s of rapid scan data collection. The S1 to S2 difference spectra were generated by ratio of single-channel data taken before and after the actinic flash. Data were normalized to an amide II intensity of 0.5 absorbance units (AU) to eliminate any small differences in sample path length. Amide II peak intensity was determined from an infrared absorption spectrum, generated through the use of an open beam background.
ACKNOWLEDGMENTS.
Supported by National Science Foundation MCB, 08-42246 (B.A.B.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
References
- 1.Nelson N, Yocum CF. Structure and function of photosystems I and II. Ann Rev Plant Biol. 2006;57:521–565. doi: 10.1146/annurev.arplant.57.032905.105350. [DOI] [PubMed] [Google Scholar]
- 2.MacDonald GM, Steenhuis JJ, Barry BA. A difference infrared spectroscopic study of chlorophyll oxidation in hydroxylamine treated photosystem II. J Biol Chem. 1995;270:8420–8428. doi: 10.1074/jbc.270.15.8420. [DOI] [PubMed] [Google Scholar]
- 3.Umena Y, Kawakami K, Shen J-R, Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Angstrom. Nature. 2011;473:55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 4.Joliot P, Kok B. Oxygen evolution in photosynthesis. In: Govindjee, editor. Bioenergetics of Photosynthesis. New York: Academic; 1975. pp. 388–412. [Google Scholar]
- 5.Haumann M, Grabolle M, Neisius T, Dau H. The first room-temperature X-ray absorption spectra of higher oxidation states of the tetra-manganese complex of photosystem II. FEBS Lett. 2002;512:116–120. doi: 10.1016/s0014-5793(02)02237-8. [DOI] [PubMed] [Google Scholar]
- 6.Dekker JP, Plijter JJ, Ouwehand L, van Gorkom HJ. Kinetics of manganese redox transitions in the oxygen-evolving apparatus of photosynthesis. Biochim Biophys Acta. 1984;767:176–179. [Google Scholar]
- 7.Keough J, Jenson DL, Zuniga A, Barry BA. Proton coupled electron transfer and redox-active tyrosine Z in the photosynthetic oxygen evolving complex. J Am Chem Soc. 2011;133:11084–11087. doi: 10.1021/ja2041139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krimm S, Bandekar J. Vibrational spectroscopy and conformation of peptides, polypeptides, and proteins. In: Anfinsen CB, Edsall JT, Richards FM, editors. Adv Protein Chem. Vol. 38. New York: Academic; 1986. pp. 181–364. [DOI] [PubMed] [Google Scholar]
- 9.Socrates G. Infrared and Raman Characteristic Group Frequencies. West Sussex: John Wiley & Son; 2001. [Google Scholar]
- 10.Sandusky PO, Yocum CF. The mechanism of amine inhibition of the photosynthetic oxygen evolving complex. FEBS Lett. 1983;162:339–343. [Google Scholar]
- 11.Boussac A, Rutherford AW, Styring S. Interaction of ammonia with the water splitting enzyme of photosystem II. Biochemistry. 1990;29:24–32. doi: 10.1021/bi00453a003. [DOI] [PubMed] [Google Scholar]
- 12.Fersht AR, et al. Hydrogen bonding and biological specificity analysed by protein engineering. Nature. 1985;314:235–238. doi: 10.1038/314235a0. [DOI] [PubMed] [Google Scholar]
- 13.Dixon DA, Dobbs KD, Valentini JJ. Amide-water and amide-amide hydrogen bond strengths. J Phys Chem B. 1994;98:13435–13439. [Google Scholar]
- 14.Jain NK, Roy I. Effect of trehalose on protein structure. Protein Sci. 2009;18:24–36. doi: 10.1002/pro.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khademi S, et al. Mechanism of ammonia transport by Amt/MEP/Rh: structure of AmtB at 1.35 Å. Science. 2004;305:1587–1594. doi: 10.1126/science.1101952. [DOI] [PubMed] [Google Scholar]
- 16.Zheng L, Kostrewa D, Berneche S, Winkler FK, Li XD. The mechanism of ammonia transport based on the crystal structure of AmtB of Escherichia coli. Proc Nat Acad Sci. 2004;101:17090–17095. doi: 10.1073/pnas.0406475101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Riso A, Jenson DL, Barry BA. Calcium exchange and structural changes during the photosynthetic oxygen evolving cycle. Biophys J. 2006;91:1999–2008. doi: 10.1529/biophysj.106.087171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berthold DA, Babcock GT, Yocum CF. A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes. FEBS Lett. 1981;134:231–234. [Google Scholar]
- 19.Mishra RK, Ghanotakis DF. Selective extraction of CP 26 and CP 29 proteins without affecting the binding of the extrinsic proteins (33, 23 and 17 kDa) and the DCMU sensitivity of a Photosystem II core complex. Photosyn Res. 1994;42:37–42. doi: 10.1007/BF00019056. [DOI] [PubMed] [Google Scholar]
- 20.Bricker TM, Frankel LK. Auxillary functions of the PsbO, PsbP, and PsbQ proteins of higher plant Photosystem II: A critical analysis. J Photochem Photobiol B. 2011;104:165–178. doi: 10.1016/j.jphotobiol.2011.01.025. [DOI] [PubMed] [Google Scholar]
- 21.Barry BA, et al. Time-resolved vibrational spectroscopy detects protein-based intermediates in the photosynthetic oxygen-evolving cycle. Proc Nat Acad Sci. 2006;103:7288–7291. doi: 10.1073/pnas.0600216103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noguchi T, Sugiura M. Flash-induced Fourier transform infrared detection of the structural changes during the S-state cycle of the oxygen-evolving complex in photosystem II. Biochemistry. 2001;40:1497–1502. doi: 10.1021/bi0023807. [DOI] [PubMed] [Google Scholar]
- 23.Hillier W, Babcock G. S-state dependent Fourier transform infrared difference spectra for the photosystem II oxygen evolving complex. Biochemistry. 2001;40:1503–1509. doi: 10.1021/bi002436x. [DOI] [PubMed] [Google Scholar]
- 24.Razeghifard MR, et al. The in vivo, in vitro, and calculated vibrational spectra of plastoquinone and the plastosemiquinone anion radical. J Phys Chem B. 1999;103:9790–9800. [Google Scholar]
- 25.Kim S, et al. Isotope-based discrimination between the infrared modes of plastosemiquinone anion radicals and neutral tyrosyl radicals in photosystem II. J Phys Chem B. 2000;104:9720–9727. [Google Scholar]
- 26.Noguchi T, Sugiura M. Analysis of flash-induced FTIR difference spectra of the S-state cycle in the photosynthetic water-oxidizing complex by uniform 15N and 13C isotope labeling. Biochemistry. 2003;42:6035–6042. doi: 10.1021/bi0341612. [DOI] [PubMed] [Google Scholar]
- 27.Maes G, Zeegers-Huyskens T. Matrix isolation infrared spectra of the complexes between methylacetate and water or hydrochloric acid. J Mol Struct. 1983;100:305–315. [Google Scholar]
- 28.Chu HA, Nguyen AP, Debus RJ. Amino acid residues that influence the binding of manganese or calcium to photosystem II. 1. The lumenal interhelical domains of the D1 polypeptide. Biochemistry. 1995;34:5839–5858. doi: 10.1021/bi00017a016. [DOI] [PubMed] [Google Scholar]
- 29.Chu HA, Nguyen AP, Debus RJ. Amino acid residues that influence the binding of manganese or calcium to photosystem II. 2. The carboxy-terminal domain of the D1 polypeptide. Biochemistry. 1995;34:5859–5882. doi: 10.1021/bi00017a017. [DOI] [PubMed] [Google Scholar]
- 30.Service RJ, Hillier W, Debus RJ. Evidence from FTIR difference spectroscopy of an extensive network of hydrogen bonds near the oxygen-evolving Mn(4)Ca cluster of photosystem II involving D1-Glu65, D2-Glu312, and D1-Glu329. Biochemistry. 2010;49:6655–666. doi: 10.1021/bi100730d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guskov A, et al. Cyanobacterial photosystem II at 2.9-A resolution and the role of quinones, lipids, channels and chloride. Nature Struc Mol Biol. 2009;16:334–342. doi: 10.1038/nsmb.1559. [DOI] [PubMed] [Google Scholar]
- 32.Sandusky PO, Yocum CF. The chloride requirement for photosynthetic oxygen evolution. Analysis of the effects of chloride and other anions on amine inhibition of the oxygen evolving complex. Biochim Biophys Acta. 1984;766:603–611. [Google Scholar]
- 33.Sandusky PO, Yocum CF. The chloride requirement for photosynthetic oxygen evolution: Factors affecting nucleophilic displacement of chloride from the oxygen-evolving complex. Biochim Biophys Acta. 1986;849:85–93. [Google Scholar]
- 34.Lindberg K, Vanngard T, Andreasson L-E. Studies of the slowly exchanging chloride in photosystem II of higher plants. Photosynth Res. 1993;38:401–408. doi: 10.1007/BF00046767. [DOI] [PubMed] [Google Scholar]
- 35.Velthuys BR. Binding of the inhibitor NH3 to the oxygen-evolving apparatus of spinach chloroplasts. Biochim Biophys Acta. 1975;396:392–401. doi: 10.1016/0005-2728(75)90145-0. [DOI] [PubMed] [Google Scholar]
- 36.Britt RD, Zimmermann J-L, Sauer K, Klein MP. Ammonia binds to the catalytic Mn of the oxygen-evolving complex of photosystem II: Evidence by electron spin-echo envelope modulation spectroscopy. J Am Chem Soc. 1989;111:3522–3532. [Google Scholar]
- 37.Dau H, et al. Structural consequences of ammonia binding to the manganese center of the photosynthetic oxygen-evolving complex: An X-ray absorption spectroscopy study of isotropic and priented Photosystem II particles. Biochemistry. 1995;34:5274–5287. doi: 10.1021/bi00015a043. [DOI] [PubMed] [Google Scholar]
- 38.Chu HA, Feng YW, Wang CM, Chiang KA, Ke SC. Ammonia-induced structural changes of the oxygen-evolving complex in photosystem II as revealed by light-induced FTIR difference spectroscopy. Biochemistry. 2004;43:10877–10885. doi: 10.1021/bi0499260. [DOI] [PubMed] [Google Scholar]
- 39.Hou LH, Wu CM, Huang HH, Chu HA. Effects of ammonia on the structure of the oxygen-evolving complex in Photosystem II as revealed by light-induced FTIR difference spectroscopy. Biochemistry. 2011;50:9248–9254. doi: 10.1021/bi200943q. [DOI] [PubMed] [Google Scholar]
- 40.Tsuno M, Suzuki H, Kondo T, Mino H, Noguchi T. Interaction and inhibitory effect of ammonium cation in the oxygen evolving center of photosystem II. Biochemistry. 2011;50:2506–2514. doi: 10.1021/bi101952g. [DOI] [PubMed] [Google Scholar]
- 41.Ouellette AJA, Anderson LB, Barry BA. Amine binding and oxidation at the catalytic site of photosynthetic water oxidation. Proc Nat Acad Sci. 1998;95:2204–2209. doi: 10.1073/pnas.95.5.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drath M, et al. Ammonia triggers photodamage of Photosystem II in the cyanobacterium Synechocystis sp. Strain PCC 6803. Plant Physiol. 2008;147:206–215. doi: 10.1104/pp.108.117218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ouellette AJA, Barry BA. Tandem mass spectrometric identification of spinach photosystem II light harvesting components. Photosynth Res. 2002;72:159–173. doi: 10.1023/A:1016132700844. [DOI] [PubMed] [Google Scholar]
- 44.Barry BA. Tyrosyl radicals in photosystem II. Methods Enzymol. 1995;258:303–319. doi: 10.1016/0076-6879(95)58053-0. [DOI] [PubMed] [Google Scholar]
- 45.Piccioni R, Bellemare G, Chua N. Methods of polyacrylamide gel electrophoresis in the analysis and preparation of plant polypeptides. In: Edelman H, Hallick RB, Chua N-H, editors. Methods in Chloroplast Molecular Biology. Amsterdam: Elsevier; 1982. pp. 985–1014. [Google Scholar]
- 46.Barry BA, Hicks C, DeRiso A, Jenson DL. Calcium ligation in photosystem II under inhibiting conditions. Biophys J. 2005;89:393–401. doi: 10.1529/biophysj.105.059667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cooper IB, Barry BA. Perturbations at the chloride site during the photosynthetic oxygen-evolving cycle. Photosynth Res. 2007;92:345–356. doi: 10.1007/s11120-007-9147-3. [DOI] [PubMed] [Google Scholar]
- 48.Cooper IB, Barry BA. Azide as a probe of proton transfer reactions in photosynthetic oxygen evolution. Biophys J. 2008;95:5843–5850. doi: 10.1529/biophysj.108.136879. [DOI] [PMC free article] [PubMed] [Google Scholar]