

Abstract
Although the biological roles of many members of the sirtuin family of lysine deacetylases have been well characterized, a broader understanding of their role in biology is limited by the challenges in identifying new substrates. We present here an in vitro method that combines biotinylation and mass spectrometry (MS) to identify substrates deacetylated by sirtuins. The method permits labeling of deacetylated residues with amine-reactive biotin on the ϵ-nitrogen of lysine. The biotin can be utilized to purify the substrate and identify the deacetylated lysine by MS. The biotinyl-lysine method was used to compare deacetylation of chemically acetylated histones by the yeast sirtuins, Sir2 and Hst2. Intriguingly, Sir2 preferentially deacetylates histone H3 lysine 79 as compared to Hst2. Although acetylation of K79 was not previously reported in Saccharomyces cerevisiae, we demonstrate that a minor population of this residue is indeed acetylated in vivo and show that Sir2, and not Hst2, regulates the acetylation state of H3 lysine 79. The in vitro biotinyl-lysine method combined with chemical acetylation made it possible to identify this previously unknown, low-abundance histone acetyl modification in vivo. This method has further potential to identify novel sirtuin deacetylation substrates in whole cell extracts, enabling large-scale screens for new deacetylase substrates.
Sir2-like enzymes, known as sirtuins, are a family of conserved NAD+-dependent lysine deacetylases (1–3) that comprise one of four classes of lysine deacetylases (KDACs, also known as histone deacetylases, or HDACs) (4). Sirtuins regulate many different biological processes, in part by targeting specific substrates for deacetylation. Humans have seven sirtuins, SirT1-7 (5, 6), which differ in their subcellular localization and play distinct roles (reviewed in ref. 7) in processes including DNA repair (8–10), fat mobilization (11), and apoptosis (12, 13). The founding member of the sirtuin family, Sir2, was discovered in Saccharomyces cerevisiae as an enzyme involved in transcriptional silencing at the heterochromatic mating-type loci (14), telomeres (15), and rDNA repeats (16, 17). This silencing is accomplished through deacetylation of acetyl-lysines on histones, giving rise to transcriptional repression (18–20). Sir2 specifically deacetylates histone H3 K4, H3 K56, and H4 K16 in vivo (1, 21). Both H3 K4 and H4 K16 are located on the flexible N-terminal tail of the histone, and H3 K56 is located in the core of histone H3 at the entry-exit points of DNA on the nucleosome.
S. cerevisiae possesses four Sir2 homologues (Hst1–4), all of which also deacetylate histones and overlap with Sir2 in specificity for particular histone residues. Hst1–4 regulate sporulation, control of genome integrity, and other processes (2, 22–27) via their histone deacetylation activity. Although Hst1–4 and Sir2 all target histone substrates, they appear to have distinct preferences for particular acetylated lysines within each histone, which changes based on genomic context or phase in the cell cycle. While Sir2 is responsible for deacetylating H3 K4 in heterochromatin, Hst1 is the main deacetylase for this residue in euchromatin (21). Hst3 and Hst4 are responsible for regulating the global level of acetylation on histone H3 K56, while Sir2 deacetylates this residue at the telomeric and HM loci (26, 28). In addition, Sir2 deacetylates H4 K16 at telomeric heterochromatin to maintain the boundary with euchromatin (29, 30). Unlike the other four yeast sirtuins that localize to the nucleus, Hst2 is largely cytoplasmic, but translocates into the nucleus to deacetylate histone H4 K16 during mitosis (31, 32). To date, no non-histone substrates have been identified for Hst1–4, although Sir2 has been shown to specifically deacetylate at least one non-histone protein, Pck1, in vivo (33). In contrast with the yeast sirtuins, a number of non-histone substrates have been identified for sirtuins from other organisms, most notably for the human sirtuin SirT1 (reviewed in ref. 34).
As acetylation appears to be as frequent as phosphorylation (35, 36), it is likely that sirtuins have many more substrates than those currently known. A challenge in the study of sirtuins, as well as of other lysine deacetylases, is in identifying their substrates. To date, a number of sirtuin substrates have been identified by genetic or biochemical approaches. For example, the interaction between mammalian SirT1 and p53 was first identified by coimmunoprecipitation, which led to further experiments to confirm acetylated p53 as a substrate of SirT1 (12, 13). While these approaches have been successful, they rely on sufficiently tight binding between enzyme and substrate, and many enzymes interact weakly and transiently with their substrate. Recently, a peptide array-based high-throughput method was used to successfully identify mitochondrial SirT3 substrates (37). This methodology relied upon the synthesis of peptides containing an acetyl-lysine analog that increases the affinity of sirtuins for the substrate. Because of the very large number of peptides that would be required to cover all mitochondrial proteins, the study was limited to previously identified sites of acetylation.
We set out to develop a method for identifying sirtuin substrates that relied upon direct identification of deacetylation sites and could be adapted to high-throughput studies. The major obstacle in developing this type of method is that it is difficult to identify substrates of enzymes that remove modifications from their substrates. In addition, acetylated substrates expressed in low abundance or present in a small percentage of the population are likely to be overlooked, as are transient sites of acetylation, due to the dynamic nature of this modification.
We present here a method to study sirtuin deacetylation substrates in vitro that makes it possible to identify deacetylation substrates while simultaneously mapping specific deacetylation sites. The method involves chemical acetylation of protein substrates, which provides all surface-exposed lysines as potential substrates and also serves to block all nonsubstrate lysine residues in a subsequent chemical modification step. Following incubation with a sirtuin in vitro, the deacetylated lysines are tagged with a modified biotin that specifically reacts with the unmodified lysines. The biotinylated lysines can be detected by streptavidin blotting or mass spectrometry (MS) and can be used to isolate substrates from complex mixtures. A second round of MS is then used to identify the substrate and map the biotinyl-lysine residues. The method can be used on specific substrates or complex mixtures.
We present an application of the biotinyl-lysine method to compare the relative in vitro specificity of two yeast sirtuins, Sir2 and Hst2, for acetylated histones. We find that Sir2 preferentially deacetylates K79 of histone H3, a residue methylated in a large proportion of histones in yeast (38), but not previously known to be acetylated. A previous study comparing the modification status of histone H3 K79 between four organisms found evidence of a small fraction of acetylated K79 in a human cell line but not yeast (39). We found that H3 K79 is indeed acetylated in vivo in yeast in low abundance, which explains why this modification was not previously observed. We also provide evidence that Sir2, and not Hst2, deacetylates this residue in yeast, consistent with the silencing defects that have been observed for H3 K79 substitutions and Sir2 deletions (38, 40–42). Our finding validates the use of the biotinyl-lysine method and highlights its utility in the discovery of low-abundance acetyl modifications.
Results
The Biotinyl-Lysine Method.
We sought to develop a chemical method that could be used to directly identify substrates that have been deacetylated. Lysine deacetylases remove the acetyl group from the ϵ-nitrogen of lysine, leaving the free side-chain primary amine. Lysine side chains and unmodified amino termini, which also have primary amines, react readily with an N-hydroxysuccinimide (NHS) ester of biotin, resulting in a covalently attached biotin. This compound can therefore be used to label unmodified lysines, which makes it possible to chemically tag lysines that have been deacetylated and thereby identify deacetylase substrates. There are, however, two challenges in using this approach to study deacetylation of naturally acetylated proteins. First, proteins typically contain multiple lysine residues that are never modified in vivo, and which therefore would be biotinylated along with any deacetylated lysine. This would lead to poor signal-to-noise, due to difficulties in detecting small changes in overall biotinylation. The second challenge is that a given acetylation site is likely acetylated in only a subset of the proteins in the cell, making it especially difficult to detect low-abundance acetylation sites. To overcome these challenges, we chemically acetylated all free amines prior to incubating the substrate proteins with the deacetylase enzyme (Fig. 1, step I). In addition to ensuring that all possible exposed lysine residues are acetylated, this also prevents biotinylation of nonsubstrate lysines.
Fig. 1.

Overview of the biotinyl-lysine reaction scheme for detection of sirtuin-mediated deacetylation. (I) Lysines and N termini are acetylated with acetic anhydride. (II) Sirtuin-mediated NAD+-dependent deacetylation removes acetyl groups to regenerate unmodified lysines. (III) Deacetylated lysines are biotinylated with sulfo-NHS-biotin.
An overview of the method is shown in Fig. 1. Purified proteins or protein extracts are first treated with acetic anhydride as described in Methods (Fig. 1, step I). This results in the acetylation of virtually all lysines and N termini of proteins in the reaction. Acetylated proteins are then incubated with a sirtuin (or other deacetylase), resulting in the selective removal of acetyl modifications and the regeneration of unmodified lysine side chains (Fig. 1, step II). The unmodified lysines are then modified with biotin linked to an amine-reactive NHS moiety (Fig. 1, step III). Biotinylated proteins can then be detected by blotting or by MS.
Detection of Biotinylated Histones by Blotting.
We first conducted a series of experiments with Hst2 to confirm that the steps outlined in Fig. 1 could be carried out with adequate efficiency. Using purified histones, we determined if (i) NHS-biotin would react with the substrates, (ii) chemical acetylation could completely block biotinylation of all free amines in the sample, and (iii) incubation of the substrate protein with a deacetylase would expose free lysines that could then be biotinylated. Histones were acetylated with acetic anhydride, which was then removed by dialysis, incubated with Hst2 in the presence or absence of NAD+, and then biotinylated and separated by denaturing gel electrophoresis. To detect biotinylated proteins, samples were transferred to nitrocellulose and analyzed by Western blotting with streptavidin-HRP and chemiluminescence. As shown in Fig. 2, histones could be modified by NHS-biotin, but chemical acetylation completely blocked free amines (compare Fig. 2, lanes 1 and 2). The addition of both Hst2 and NAD+, which is required for the NAD+-dependent deacetylation reaction, enabled a subset of histones to be biotinylated (Fig. 2, lane 4). Hst2 was also biotinylated, because the enzyme was not chemically acetylated and therefore had free amines that could react with NHS-biotin. The presence of extra bands in lane 1 that are not labeled in lane 4 suggests that these are either cross-linked aggregates of histones or impurities of other proteins from the histone preparation that are not deacetylated and biotinylated by Hst2.
Fig. 2.

Biotinylated proteins detected by blotting with streptavidin-HRP. (Lane 1) Unmodified histones were labeled with biotin at free lysines and N termini. (Lanes 2 and 3) Acetylated histones were not labeled because they did not have any free amines. (Lane 4) Acetylated histones in the presence of sirtuin and NAD+ were deacetylated, and these free lysines were biotinylated, allowing them to be detected by the streptavidin-HRP.
Identification of Deacetylated and Biotinylated Residues by MALDI MS.
We utilized the biotinyl-lysine method combined with mass spectrometry to identify histone lysines that were deacetylated by the Sir2 and Hst2 deacetylases. Purified yeast histones were first chemically acetylated, then deacetylated by either Sir2 or Hst2 for 360 min. Deacetylated histones were biotinylated and separated by denaturing gel electrophoresis and stained with Coomassie in order to visualize the bands corresponding to the histone proteins. Histone bands were excised from the gel and digested with trypsin. While trypsin usually cleaves C-terminal to lysine and arginine residues, all of the lysines in our experiments are either acetylated or biotinylated and thus cannot be digested by trypsin. Thus, histones are only cleaved at arginine residues in both the acetylated control samples and the deacetylated, biotinylated reactions. This simplifies comparison of control and deacetylated samples by creating a set of uniform peptides between the two reactions; if the deacetylated lysines were not biotinylated, trypsin would cleave after these residues, generating a different set of peptides than in the control sample.
In order to identify deacetylated lysines, tryptic peptides were subjected to matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS). MALDI-TOF MS, used without chromatographic separation of the peptides, reveals in a single mass spectrum a series of peaks with a mass-to-charge ratio (m/z) corresponding to the tryptic peptides. Because the sample is known in this case, it is possible to identify the histone peptide corresponding to each peak by first calculating all possible peptide masses for tryptic digests of each histone, including acetyl or biotin modifications on lysines. This task is simplified by the fact that ionized peptides from MALDI are mostly singly charged; thus, m/z ratios of peaks observed in MALDI-TOF are representative of the mass of the peptide.
A schematic diagram illustrating the changes in molecular weight and peak shifts of acetylated and deacetylated, biotinylated peptides is shown in Fig. 3. For each lysine, 42 Da was added to the peptide molecular weight to account for addition of the acetyl moiety. Expected mass shifts of deacetylated and biotinylated lysines were based on removal of the 42 Da acetyl group and addition of a 226 Da biotin moiety, resulting in a net increase of 184 Da on the molecular weight of the peptide for each lysine, a mass shift clearly visible with MALDI-TOF MS. Calculations for peptides with multiple lysines took into account the possibility that not all lysines were deacetylated and biotinylated. An example of the calculated masses for histone H3 tryptic peptides containing lysines is given in Table 1.
Fig. 3.

Schematic depicting changes in molecular weight and mass/charge peak shifts in MALDI-TOF as a result of deacetylation and biotinylation. The molecular weight of a peptide including a lysine is increased by 42 Da by acetylation. Deacetylation removes this 42 Da modification, and biotinylation adds 226 Da. Deacetylated, biotinylated lysines therefore result in a net change of 184 Da compared to acetylated lysines.
Table 1.
Expected masses of acetylated or deacetylated, biotinylated histone H3 tryptic peptides
| AAs | Peptide sequence | Mass | No. Ks | +1 Ac | +2 Ac | +3 Ac | −1 Ac | −2 Ac | −3 Ac |
| +1 Biotin | +2 Biotin | +3 Biotin | |||||||
| 3–8 | TKQTAR | 703.79 | 1 | 745.80 | 930.09 | ||||
| 9–17 | KSTGGKAPR | 901.03 | 2 | 943.04 | 985.05 | 1,169.34 | 1,353.62 | ||
| 18–26 | KQLASKAAR | 972.15 | 2 | 1,014.16 | 1,056.17 | 1,240.46 | 1,424.74 | ||
| 27–40 | KSAPSTGGVKKPHR | 1,449.67 | 3 | 1,491.68 | 1,533.69 | 1,575.70 | 1,759.99 | 1,944.27 | 2,128.56 |
| 41–49 | YKPGTVALR | 1,004.19 | 1 | 1,046.20 | 1,230.49 | ||||
| 54–63 | FQKSTELLIR | 1,234.46 | 1 | 1,276.47 | 1,460.75 | ||||
| 64–69 | KLPFQR | 787.96 | 1 | 829.97 | 1,014.25 | ||||
| 73–83 | EIAQDFKTDLR | 1,335.48 | 1 | 1,377.49 | 1,561.77 | ||||
| 84–116 | FQSSAIGALQESVEAYLVSLFEDTNLAAIHAKR | 3,580.00 | 1 | 3,622.01 | 3,806.30 | ||||
| 117–128 | VTIQKKDIKLAR | 1,412.73 | 3 | 1,454.75 | 1,496.76 | 1,538.77 | 1,723.05 | 1,907.34 | 2,091.62 |
Mass spectra of histone samples treated with Sir2 or Hst2 in the absence and presence of NAD+ were used as control and deacetylation reactions, respectively. A sample mass spectrum from a control reaction with Hst2 and histones but lacking NAD+ (Fig. 4A) compared to a deacetylation reaction with Hst2, NAD+, and histones (Fig. 4B) illustrates mass shifts of peptides that undergo deacetylation and biotinylation. Deacetylation resulted in the disappearance of an acetylated peptide peak along with the appearance of its biotinylated counterpart with a molecular weight shift of 184 Da. The m/z peaks were matched by mass to predicted tryptic peptides generated in silico. For example, the peptide signal at m/z 1,377.7 in the control sample (Fig. 4A) is significantly decreased in the deacetylated sample (Fig. 4B), and a peptide that has an m/z of 1,561.6, an increase of 184 Da from the 1,377.6 peptide, appears. As shown in Table 1, the 1,377.7 and 1,561.6 peaks match to the acetylated and biotinylated forms of the histone H3 peptide including residues 73–83, EIAQDFK79TDLR, respectively. The control spectrum also includes a peak at 1,245.6 m/z (Fig. 4A), which corresponds to the mass of an acetylated form of a histone H2A peptide containing residues 19–30, SAK21(Ac)AGLTFPVGR. In Fig. 4B, the mass of this peptide is shifted by 184 Da to m/z 1,429.6, indicating deacetylation and biotinylation at K21 (SAK21(biotin)AGLTFPVGR). This analysis was applied to the entire spectrum to identify all deacetylated and biotinylated histone peptides (Hst2 results summarized in Table 2).
Fig. 4.

Sample MALDI mass spectrum of Hst2-deacetylated histones. (A) MALDI-TOF spectrum of histone peptide peaks in a control reaction lacking NAD+. (B) MALDI mass spectrum of a deacetylation reaction containing Hst2, NAD+, and histones shows mass shifting of peptides that are deacetylated and biotinylated in comparison to A.
Table 2.
Histone peptides from Hst2-deacetylated, biotinylated samples identified by MALDI and ESI MS
| Histone | Residues | Amino acid sequence | Biotinylated lysines | MS method | Observed m/z (ESI) | Charge state | Mass accuracy (ppm) |
| H2A | 19–30 | SAKAGLTFPVGR | 21 | E, M | 715.3904 | +2 | 7.9 |
| 73–78 | DNKKTR | 75, 76 | M | 1,213.43* | +1 | 131.8 | |
| H2B | 76–95 | IATEASKLAAYNKKSTISAR | 89 | E | 1,217.1530 | +2 | 7.3 |
| 76–95 | IATEASKLAAYNKKSTISAR | 88, 89 | E | 1,309.1870 | +2 | 7.2 | |
| 103–119 | LILPGELAKHAVSEGTR | 111 | E, M | 1,009.0550 | +2 | 6.3 | |
| H3 | 3–8 | TKQTAR | 4 | E | 465.7469 | +2 | 4.3 |
| 18–26 | KQLASKAAR | 18, 23 | E | 712.8834 | +2 | 6.5 | |
| 27–40 | KSAPSTGGVKKPHR | 27 | E | 587.3176 | +3 | 6.3 | |
| 27–40 | KSAPSTGGVKKPHR | 27, 36 | E | 972.5061 | +2 | 5.4 | |
| 53–63 | RFQKSTELLIR | 56 | E | 808.9556 | +2 | 6.0 | |
| 54–63 | FQKSTELLIR | 56 | E, M | 730.9045 | +2 | 6.0 | |
| 64–49 | KLPFQR | 64 | M | 1,014.43* | +1 | 128.1 | |
| 73–83 | EIAQDFKTDLR | 79 | E, M | 781.3920 | +2 | 5.6 | |
| 84–116 | FQSSAIGALQESVEAYLVSLFEDTNLAAIHAKR | 115 | E | 1,268.9910 | +3 | 8.3 | |
| 117–128 | VTIQKKDIKLAR | 121, 122, 125 | E, M | 1,046.0750 | +2 | 7.2 | |
| H4 | 4–17 | GKGGKGLGKGGAKR | 5, 8, 12, 16 | E | 1,088.0530 | +2 | 8.2 |
| 20–23 | KILR | 20 | M | 755.37* | +1 | 119.1 | |
| 20–35 | KILRDNIQGITKPAIR | 20 | E | 1,052.6120 | +2 | 5.1 | |
| 41–45 | GGVKR | 44 | M | 742.31* | +1 | 134.7 | |
| 56–67 | AVLKSFLESVIR | 59 | E, M | 794.4569 | +2 | 8.6 | |
| 60–78 | SFLESVIRDDSVTYTEHAKR | 77 | E | 822.0876 | +3 | 7.7 | |
| 68–78 | DSVTYTEHAKR | 77 | E, M | 766.8680 | +2 | 8.2 | |
| 79–92 | KTVTSLDVVYALKR | 79 | E | 931.0237 | +2 | 8.0 | |
| 79–92 | KTVTSLDVVYALKR | 79, 91 | E, M | 1,023.0570 | +2 | 7.1 |
K, biotinylated lysine; K, acetylated lysine; E, electrospray ionization on the LTQ/Orbitrap mass spectrometer; M, MALDI on the AXIMA TOF2 mass spectrometer.
*Observed only by MALDI.
Identification of Deacetylated and Biotinylated Residues by Tandem MS.
Although MALDI-TOF MS makes it possible to rapidly monitor peptide mass shifts resulting from deacetylation and biotinylation, this approach is only feasible for a sample containing a maximum of a few known proteins because of the calculations necessary to identify peptides and the complexity in the mass spectra. In addition, peptides containing multiple lysines pose a difficulty; if only some of the lysines are deacetylated, the specific deacetylated lysine residue cannot be determined by MALDI-TOF MS. To overcome this ambiguity and identify the actual residues that were deacetylated and biotinylated, the histone samples were subjected to high-performance liquid chromatography followed by nanoelectrospray ionization tandem MS (nanoLC ESI MS/MS, LC MS/MS or ESI). Data were searched by MASCOT to identify and score peptide sequence assignments.
LC MS/MS identified sites of histone deacetylation by both Sir2 and Hst2. Fig. 5A shows a sample tandem mass spectrum of the doubly charged, histone H3 peptide acetylated at K79, EIAQDFK79(Ac)TDLR, from a control sample incubated with only Hst2. Fig. 5B shows the results for the same peptide incubated with Hst2 in the presence of NAD+. In this reaction, the acetyl group on K79 was removed and replaced with biotin (EIAQDFK79(biotin)TDLR), resulting in a 92 Da mass increase of the precursor ion and a 184 Da increase in mass between the y4 and y5 ions.
Fig. 5.

Sample tandem mass spectra of the doubly charged, histone H3 peptide (aa 73–83), EIAQDFK79TDLR. (A) Mass spectrum of the control sample of histone incubated with only Hst2 shows this peptide with acetylated K79. (B) Mass spectrum of the histone H3 peptide biotinylated at K79. The sample was treated with Hst2 in the presence of NAD+, resulting in the replacement of the acetyl group on K79 with a biotin tag. There is therefore a 92 Da mass increase of the precursor ion and a 184 Da increase in mass between the y4 and y5 ions.
A total of 24 tryptic peptides with 28 biotinylated lysines were identified by peptide mass matching from MALDI-TOF MS data or MASCOT searches of nanoLC ESI MS/MS data (Hst2 results summarized in Table 2). When possible, mass accuracies are listed for LC MS/MS identifications, which were within 4–9 parts per million (ppm). For peptides that were only identified by MALDI-TOF, mass accuracies are listed for the MALDI data. MALDI-TOF identifications in all cases had an error of 120–135 ppm, within the range of TOF analyzers, which have a reported mass accuracy of approximately 50 ppm (43). Although the MALDI data are not as accurate as the LC MS/MS data, we included the MALDI identifications because the samples contain known proteins, where all molecular species are calculable.
We found that, given the long deacetylation incubation times, almost every histone lysine identified by MS could be deacetylated by both Sir2 and Hst2. There were a few residues, however, that were not identified as being deacetylated and biotinylated by either Sir2 or Hst2. For example, K37 on histone H3 was never observed to be deacetylated, although the neighboring K36 was. Other histone H3 peptides containing K42 and K64, and a histone H4 peptide containing K31, were also never observed to be biotinylated by either MALDI-TOF MS or nanoLC MS/MS. The acetylated forms of these peptides were observed, however, suggesting that these residues are also not deacetylated by Sir2 or Hst2.
Time Course Analysis of Histone Deacetylation.
To investigate the relative preferences of Sir2 and Hst2 for particular acetylated lysine residues on histones, we used a series of incubation times in order to identify residues that might be deacetylated more rapidly than others. Histones were incubated with Sir2 or Hst2 for 10, 30, 60, 180, or 360 min, and then reactions were quenched with sodium borohydride, which reduces the remaining NAD+ to NADH and stops the deacetylation reaction. The reactions were then biotinylated and prepared for MALDI-TOF MS or LC MS/MS as described above. In some cases, the MALDI-TOF data were simpler to analyze because there was no chromatographic separation of peptides and only the one, singly ionized form of each peptide, whereas multiple charged states of peptides had to be considered for the LC MS/MS data; however, LC MS/MS data were more accurate and eliminated the need to match peaks to calculated peptide masses.
Over the time course, the signal intensities for the acetylated peptides decreased with concomitant increases in the peak intensities of their biotinylated counterparts as the deacetylated lysines became available for reaction with NHS-biotin (Fig. 6A). Tracking the peak intensity changes of individual deacetylated, biotinylated peptides over time provided information about differences in preference for the various lysines on histones as well as relative deacetylation preferences for both Hst2 and Sir2. We found that the majority of histone lysine residues were deacetylated by Sir2 and Hst2 at a similar rate. For example, MALDI data showed that both Sir2 and Hst2 deacetylate the histone H4 peptide containing K20, K20ILR, at a similar rate (Fig. 6B), indicating that the two sirtuins show comparable activity at this residue in vitro.
Fig. 6.

Histone H3 K79 is preferentially deacetylated by Sir2 in comparison to Hst2. (A) MALDI mass spectra of time courses of Sir2-mediated histone deacetylation demonstrating the decrease in peak intensity of acetylated peptides along with increases in peak intensities of their biotinylated counterparts. (B) Histone samples deacetylated by Sir2 or Hst2 demonstrate a similar rate of appearance of a MALDI-TOF peptide peak at 755.5 m/z, corresponding to the biotinylated histone H4 peptide K20(biotin)ILR. (C) Comparison of the appearance of a 1,561.8 m/z peptide from MALDI-TOF corresponding to the histone H3 peptide EIAQDFK79(biotin)TDLR. (D) A graph showing the area of biotinylated K79 peaks at each timepoint in C reveals a preference of Sir2 deacetylation activity (red squares) over Hst2 (green circles) for this peptide. (E) Areas of peaks from Hst2 and Sir2 LC MS/MS data of biotinylated H3 K79 peptide (EIAQDFK79(biotin)TDLR) also shows a Sir2 preference for K79 (red squares) over Hst2 (green circles), supporting the MALDI-TOF data in D. Each datapoint represents the area of biotinylated peptide from extracted ion chromatograms (XICs) of MS1 data at the various timepoints.
To look for differences in specificity between Sir2 and Hst2, we searched the spectra for differences in the rate of appearance of biotinylated peptides following deacetylation by Sir2 or Hst2. Most notably, we found that the core histone residue, H3 K79, was preferentially deacetylated by Sir2 (Fig. 6 C and D). This can be seen by comparing the rate of appearance of a 1,561.8 m/z peptide, which corresponds to the biotinylated histone H3 K79-containing peptide (EIAQDFK79(biotin)TDLR) between histones deacetylated by Sir2 and Hst2. This peptide appears within 10 min of deacetylation by Sir2, but only shows a small peak for Hst2-mediated deacetylation by 60 min (Fig. 6 C and D). This peptide did not appear in a spectrum of histones that were incubated with no enzyme but in the presence of NAD+ even after 360 min (Fig. S1); therefore the observed biotinylated H3 peptides in the Sir2- and Hst2-treated samples represents a deacetylated peptide.
LC MS/MS results also confirmed that Sir2 deacetylates histone H3 K79 faster than Hst2 in vitro. Extracted ion chromatograms (XICs) of the LC MS/MS data, which selectively display the HPLC elution profiles of a particular mass based on MS1 data, demonstrated that the area of the biotinylated H3 K79 peak increased at a faster rate for Sir2 than Hst2 (Fig. 6E). In comparison to the previously identified Sir2 substrate histone H3 K56, H3 K79 appeared to be deacetylated at a similar rate by this enzyme (Fig. S2). To determine a relative rate, the area of the peak of biotinylated peptide at each timepoint was plotted as a percent of the final timepoint (180 min). Because peptides of different sequence are not chemically equivalent, their ionization efficiencies also vary, and therefore peak intensities cannot be directly compared. Although we did not make true quantitative comparisons using this method, as this requires normalization with internal standards, qualitatively it appears that H3 K79 is deacetylated at a rate similar to H3 K56 relative to their normalized abundances. Because the histones in the experiment were acetylated chemically, these results established the in vitro preference of Sir2 for H3 K79 but left open the question of its relevance in vivo.
In Vivo Acetylation Status of Histone H3 K79.
The marked in vitro preference for histone H3 K79 deacetylation by Sir2 as compared to Hst2 led us to examine whether these observed differences could reflect an in vivo role for this activity. Histone H3 K79 had not previously been observed to be acetylated in yeast cells, but is approximately 90% methylated in vivo by the Dot1 methyltransferase (38). If H3 K79 acetylation were a transient modification or present in a minor population of total histone H3, this would explain why this modification may not have been observed in previous studies (38, 44–46).
To investigate whether H3 K79 is ever acetylated in yeast and can therefore be a target of Sir2, we explored the acetylation status of H3 K79 in a variety of mutant yeast strains containing deletions of Dot1, Sir2, or Hst2 that could potentially affect the acetylation of H3 K79. Because methylation competes with acetylation of lysine residues, deletion of both the Dot1 methyltransferase and the deacetylase that targets H3 K79 would be expected to increase the relative number of histones containing acetylated H3 K79. Histone H3 was isolated from six yeast strains, wild type (BY4741), Δdot1, Δhst2, Δhst2 Δdot1, Δsir2, and Δsir2 Δdot1, and these samples were subjected to nanoLC MS/MS. The spectra were searched by MASCOT in order to identify both the methylation and acetylation status of histone H3 K79. As expected, nanoLC MS/MS analysis of all strains containing a functional Dot1 allele indicated the presence of unmodified, mono-, di-, or trimethylated histone H3 K79, while deletion of Dot1 in any strain resulted in the complete loss of all forms of methylated K79. Fig. 7A shows MS1 data of the triply charged mono-, di-, and trimethylated H3 K79-containing peptide (EIAQDFK79(Me1,2,3)TDLR) obtained from wild-type yeast. The unmodified peptide is not shown as it is cleaved after K79 by trypsin to the peptide, EIAQDFK79, and elutes at a different retention time with different m/z ratios than the longer methylated K79 peptides. Strains bearing the deletions Δdot1 or Δhst2 Δdot1 contained the unmodified K79 peptide, but did not contain detectable acetylated H3 K79 peptide. However, histones purified from the strain lacking both Dot1 and Sir2 (Δsir2 Δdot1) yielded triply charged peaks corresponding to the m/z of an acetylated histone H3 K79 peptide (MS1 spectrum in Fig. 7B). This indicates that histone H3 K79 is indeed acetylated in vivo in S. cerevisiae.
Fig. 7.

Mass spectra of endogenous histone H3 peptides from wild-type and Δsir2 Δdot1 yeast strains. (A) MS1 spectrum of triply charged peaks from wild-type histone H3 peptides shows mono-, di-, and trimethylated forms of H3 K79. (B) MS1 spectrum of triply charged peaks from the sir2 dot1 double knockout. The methylated forms of H3 K79 are absent in this strain; however, a new peak corresponding to the mass of an acetylated form of H3 K79 is observed. (C) MS1 spectrum of doubly charged histone H3 peptides from the wild-type strain. (D) MS1 spectrum of doubly charged histone H3 peaks from the sir2 dot1 double knockout. (E) MS2 spectrum of the doubly charged, trimethylated form of the H3 K79 peptide (EIAQDFK79(Me3)TDLRA) from wild type. (F) MS2 spectra of a doubly charged H3 K79 peptide from the Δsir2 Δdot1 strain. The mass accuracy of the b9 ion of this spectrum confirms that the modification on K79 is acetylation and not trimethylation. (G) Extracted ion chromatogram (XIC) of H3 peptides from wild-type yeast that eluted at an m/z corresponding to the trimethylated K79 peptide, EIAQDFK79(Me3)TDLR (Left). These peptides had a retention time of 26.1 min. The unmodified peptide, EIAQDFK79, eluted at 23 min (Right). (H) XIC of H3 peptides from Δsir2 Δdot1 that eluted at an m/z corresponding to the acetylated K79 peptide, EIAQDFK79(Ac)TDLR (Left). These peptides had a retention time of 28.8 min. The control unmodified peptide, EIAQDFK79 , also eluted at 23 min (Right), as in G.
The doubly charged molecular ions for the trimethylated and acetylated peptides were also observed for the wild type and Δsir2 Δdot1 strains, respectively (Fig. 7 C and D). In addition, MS2 spectra obtained for these species resulted in fragment ions consistent with their purported sequences, with accurate mass measurements of the respective b9 ions used to distinguish them (Fig. 7 E and F). Moreover, the trimethyl-lysine peptide was always observed with corresponding mono- and dimethylated peptides (Fig. 7 A and C), the latter two of which were not visible in the Δsir2 Δdot1 histone H3 mass spectrum (Fig. 7 B and D). Taken together, these results indicate that H3 K79 is acetylated in S. cerevisiae, and that Sir2 may be the physiologically relevant deacetylase for this residue.
We considered whether the peak assigned as acetylated H3 K79 could in fact have been trimethylated by another methyltransferase. While trimethyl- and acetyl-lysine have nearly identical mass (differ by only 0.03639 Da), it is possible to distinguish between these modifications using the high mass accuracy of the Orbitrap, which is reported as 2–5 ppm (47). The predicted monoisotopic m/z of the triply charged acetylated K79 peptide is 459.9051 (corresponding to the mass of the 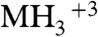 ion of EIAQDFK79(Ac)TDLR). We observed an m/z of 459.9035 for the K79 peptide in the Δsir2 Δdot1 strain, an error of 3.48 ppm of the observed m/z from the predicted acetylated m/z. If the observed m/z is compared with that calculated for the trimethylated K79 peptide (m/z 459.9173), the resulting error is 30.01 ppm, 10-fold higher than the error for the acetylated version of the peptide (Fig. 7B).
ion of EIAQDFK79(Ac)TDLR). We observed an m/z of 459.9035 for the K79 peptide in the Δsir2 Δdot1 strain, an error of 3.48 ppm of the observed m/z from the predicted acetylated m/z. If the observed m/z is compared with that calculated for the trimethylated K79 peptide (m/z 459.9173), the resulting error is 30.01 ppm, 10-fold higher than the error for the acetylated version of the peptide (Fig. 7B).
The mass accuracy of the doubly charged peaks also supports the identification of acetylated H3 K79 in the double sir2 dot1 knockout. Specifically, assignment of the observed 689.3559 peak from MS1 (Fig. 7C) to acetyl K79 (calculated m/z 689.3541) results in an error of 2.61 ppm, whereas assigning it to trimethyl K79 (calculated m/z 689.3723) would result in an error of 23.79 ppm. The mass accuracy within the MS2 spectra of the peptides from both wild type and the double sir2 dot1 knockout also supports this conclusion. The MS2 spectrum of the doubly charged molecular ion of the peptide from wild type revealed a b9 ion at m/z 1,090.5431, an error of 1.47 ppm for the trimethyl isoform (calculated m/z 1,090.5415) and 34.8 ppm from the acetylated isoform (calculated m/z 1,090.5051; Fig. 7E). In contrast, the MS2 spectrum of the doubly charged ion of the peptide from the Δsir2 Δdot1 mutant revealed a b9 ion at m/z 1,090.5055, an error of 0.37 ppm from the calculated b9 ion of an acetylated species and 33.01 ppm from the calculated b9 mass for the trimethylated species (Fig. 7F). Thus, the mass accuracy of the Orbitrap for both the doubly and triply charged ions, including MS2 molecular ions, confirms that the modified H3 K79 peptide observed in the Δsir2 Δdot1 strain is acetylated and not trimethylated.
Finally, the peptide in question displayed significantly different chromatography retention times during HPLC separation than the trimethylated H3 K79 peptide, suggesting that these two peptides are chemically different. Extracted ion chromatograms (XICs) show the differing retention times of the trimethylated or acetylated H3 K79 peptides between the wild type and double sir2 dot1 knockout, respectively (Fig. 7 G and H). The acetylated H3 K79 peptide displayed a significantly different retention time, 29 min, (Fig. 7H, Left) than trimethylated K79, which eluted at 26 min (Fig. 7G, Left) from strains with an intact DOT1 gene. As an internal control, the smaller, unmodified K79 peptide (EIAQDFK79) eluted at 23 min in both samples (Fig. 7 G and H, Right). Taken together, these observations definitively support the conclusion that the newly observed peptide corresponds to acetylated, and not trimethylated, histone H3 K79.
We attempted to quantify the proportion of acetylated H3 K79 in vivo using the method of Smith et al. for quantifying acetylation at specific lysine sites using treatment with deuteroacetic anhydride (48, 49). Deuteroacetic anhydride acts similarly to acetic anhydride, resulting in the deuteroacetylation of unmodified lysines, making them chemically equivalent to the endogenously acetylated lysines but differentiated in MS by the 3 dalton difference in mass due to replacement of three hydrogens with three deuterium atoms. Our laboratory (Cotter) has used this approach successfully for assessing the relative amount of acetylated histone H3 K56 in various yeast deletion mutants (26) and for global lysine modification analyses including both acetylation and methylation (50). We therefore attempted to quantify the amount of histone H3 that is acetylated at K79 in a double sir2 dot1 knockout by treating purified histones with deuterated acetic anhydride, followed by trypsin digestion and analysis by LC MS/MS. However, we were unable to observe endogenously acetylated H3 K79 in the presence of the considerably more intense signal from the deuteroacetylated peptide (Fig. S3 A and B). Based upon the signal intensities of the deuteroacetylated K79 peak compared to endogenously acetylated K79 (from the same sample but not treated with deuteroacetic anhydride), we estimate at least 2–3 orders of magnitude difference in the abundances of these analogues. This suggests that the reason we are not able to observe the endogenously acetylated H3 K79 peptide in the same MS1 spectrum as the deuteroacetylated form is due to the automatic gain control in the Orbitrap, which allows a limited number of ionized peptides to be analyzed. Thus, attempting to observe both of these similar species with a difference in intensity of this magnitude is beyond the dynamic range of the instrument (51), and likely places the endogenously acetylated peptide signal below the level of detection.
Discussion
We have developed the biotinyl-lysine method as an approach to studying sirtuin substrates, and have demonstrated the utility of the method by comparing histone deacetylation by Sir2 and Hst2. Because the approach relies upon chemically acetylated substrates, the method is particularly useful in identifying low-abundance or transient acetylation sites. An important caveat of the method we describe is that the complete chemical modification of every lysine in a substrate protein could interfere with proper enzyme-substrate interactions, thereby blocking the enzymatic removal of the modification. This could occur if another lysine residue is sufficiently close to the “true” substrate lysine, in which case that particular substrate will not be detected. Conversely, it is also important to verify independently that an in vitro substrate is actually acetylated in vivo. In the example described in this paper, artificially enriching a rare modification was an advantage, but it is important to analyze native samples to rule out the possibility of false positives.
Importantly, the biotinyl-lysine method led to the identification of H3 K79 as an acetylated residue in vivo that appears to be regulated by Sir2 deacetylation. Acetylation at this residue has never been observed in yeast; instead, approximately 90% of yeast H3 is reported to be methylated on K79 by the Dot1 methyltransferase (38). Interestingly, low levels of H3 K79 acetylation have been reported for human histone H3 (39), although the significance of this observation was unclear. Our finding that acetylated H3 K79 is a substrate for yeast Sir2 raises the possibility that its human homologue, SIRT1, may regulate the acetylation state of this residue in human histone H3.
Intriguingly, while Dot1 methylates 90% of histone H3 K79, the remaining 10% of H3 K79 that is hypomethylated localizes to heterochromatin, specifically at the telomeres, rDNA repeats, and HM loci, and correlates with Sir2 localization (38, 52, 53). Catalytic mutants of Sir2 exhibit loss of silencing (1, 54, 55) and increased methylation at all three loci (52), and reduced Sir2 association at telomeres, suggesting that either Sir2 localization or activity is important for maintaining the hypomethylated state of K79 in heterochromatin (52). Although previous reports suggested that deletion of dot1 resulted in loss of silencing at the HM and telomeric loci, two recent studies showed that these phenotypes were reporter-dependent (42, 56). One study demonstrated that deletion of dot1 has no effect on HM silencing and only causes loss of silencing at four out of 32 telomeric chromosome ends, but that mutation of K79 results in a mating defect due to loss of silencing at an HM locus (42). In fact, several mutations of H3 K79 (K79A, P, Q, E, or R) have been shown to affect silencing at the rDNA repeats, telomeric, and HM loci (38, 40–42). All but one K79 mutation results in loss of silencing at the heterochromatic locus tested, recapitulating Sir2 mutant phenotypes. Notably, mutation of H3 K79 had a greater effect on Sir2 localization in the heterochromatin than a dot1 deletion (38). These data suggested that the methylation status of K79 is not required for silencing, but leaves open the possibility that another K79 modification, such as acetylation, could be playing a role in silencing.
Our finding that H3 K79 is acetylated in vivo and that Sir2 deacetylates this residue accounts for the silencing defects due to the substitutions at H3 K79 and deletions of Sir2. We hypothesize that H3 K79 is actually transiently acetylated in heterochromatic regions to help recruit Sir2 to these loci for proper silencing, whereas H3 K79 is methylated by Dot1 in actively transcribed regions. In this way, the interplay between Sir2 and Dot1 activity and the modification status of H3 K79 would help to establish or maintain the boundary between heterochromatin and euchromatin. A possible mechanism could be that acetylated H3 K79 helps to recruit Sir2 to heterochromatin, and that Sir2, which forms a complex with Sir3 and Sir4, prevents methylation by remaining associated with the heterochromatin and sterically hindering access to these loci by Dot1. The presence of H3 K79 acetylation at highly specific loci, which contain a very small percentage of the overall histone proteins in the cell, could explain why this modification had not been previously observed in yeast. Moreover, corresponding studies had not been done with strains lacking both the dot1 and sir2 genes, in which this modification is enriched enough to observe by MS.
Although we have focused our studies on the yeast sirtuins and histones, the biotinyl-lysine method could be adapted to identify substrates of sirtuins or other deacetylase enzymes in cell extracts. The method could also be modified to study other sirtuin deacylation activities, including depropionylation and debutyrylation (57, 58). These modifications have thus far been identified as bona fide modifications in histones and in the p53 tumor suppressor protein in mammalian cells (59, 60). Our protocol could be modified to study these activities, for example, by propionylating or butyrylating substrates with propionic or butyric anhydride.
The biotinyl-lysine method has the potential for broad applications in studying other lysine demodifying enzymes beyond deacetylases. Lysine is subject to other reversible modifications such as methylation and ubiquitination, and identifying the substrates for demethylases and deubiquitinating enzymes is similarly of great utility. Our method could be modified to study demethylase substrates by chemically methylating substrates instead of acetylating them (61). Perhaps more significantly, the method could, in principle, be adapted to screen for substrates of deubiquitinating enzymes using endogenously ubiquitinated proteins, with chemical acetylation or methylation to block all unmodified lysines while retaining native ubiquitination sites. Given the prevalence of many different types of lysine modifications in biology, the method we describe here provides a fresh approach to identifying substrates of enzymes that remove lysine modifications.
Methods
Sirtuin Expression and Purification.
Recombinant Hst2 and Sir2 were expressed as 10xHis-tagged fusion proteins from the Novagen pET21-d(+) vector in BL21(DE3) Escherichia coli. Proteins were affinity purified using nickel resin via the C-terminal histidine fusion tag and dialyzed into a final buffer containing 20 mM HEPES, pH 7.5, 150 mM NaCl, 10% glycerol, and 1 mM dithiothreiotol (DTT).
Histone Acetylation.
Recombinant S. cerevisiae histones were expressed and reconstituted as dimers, tetramers, and octamers as previously described (ref. 62; and web protocol from the Tsukiyama Laboratory, http://labs.fhcrc.org/tsukiyama/protocols/expression.pdf) with modifications. Briefly, histones were expressed and purified from inclusion bodies in E. coli. During the refolding process, all four histones were mixed, and then histone dimers, tetramers, and octamers were separated from high molecular weight aggregates by size exclusion chromatography. Histone octamers were then dialyzed into the following buffer for acetylation: 50 mM sodium bicarbonate, pH 8.0, 2 M sodium chloride, 1 mM EDTA, 0.5 mM DTT, and 100 μM PMSF (Buffer A). Histone octamers were acetylated with a single addition of 10–50 mM acetic anhydride (Fisher Scientific) on ice while maintaining a pH of 8–8.5 with a dilute stock of sodium hydroxide, then dialyzed back into Buffer A. Finally, histone octamers were step dialyzed into the following deacetylation buffer: 50 mM HEPES, pH 7.5, 250 mM sodium chloride, 1 mM EDTA, 0.5 mM DTT, and 100 μM PMSF (Buffer B). Although histone octamers are stabilized by acetylation, the reduction in salt concentration to 250 mM sodium chloride is likely to result in the dissociation of histone octamers to H2A/H2B and H3/H4 histone dimers over time.
Deacetylation and Biotinylation Reactions.
Deacetylation reactions contained 50 μM acetylated histones, 250 nM Sir2 or Hst2, and 500 μM NAD+ in Buffer B. Reactions were incubated at 20 °C for various lengths of time, and then quenched by addition of 1 M sodium borohydride to a final concentration of 50 mM to convert all remaining NAD+ to NADH. Samples were then biotinylated with 10 mM sulfo-NHS-biotin (Pierce or ProteoChem) for 3 h at 20 °C. Biotinylation reactions were quenched with addition of 4x LDS sample buffer and the labeled proteins were separated by SDS-PAGE. For blot analysis, the proteins were transferred to nitrocellulose membranes, probed with streptavidin conjugated to horseradish peroxidase (Invitrogen), and detected by enhanced chemiluminescence (Pierce).
Tryptic Digestion.
For MS analysis, histone bands were excised from Coomassie-stained polyacrylamide protein gels. The gel bands were destained, dried, and incubated with trypsin overnight at 37 °C as described previously (63). Finally, the peptide extract was dried in preparation for either MALDI-TOF MS or nanoLC MS/MS.
MALDI-TOF MS.
Peptide extracts were resuspended in 50% acetonitrile. One microliter of each peptide solution was applied to a MALDI plate and allowed to air dry. Then 1.0 μL of α-cyano-hydroxycinnamic acid (10 mg/mL) in 70∶30 acetonitrile: 0.1% trifluoroacetic acid was added. All samples were analyzed on a Shimadzu AXIMA-TOF2 mass spectrometer in reflectron mode with a 337 nm N2 laser. Spectra were acquired as an average of 100 profiles. The instrument was externally calibrated using a four-point calibration with Bradykinin (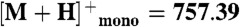 ), Angiotensin II (
), Angiotensin II (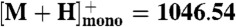 ),
), 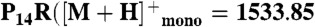 ), and ACTH fragment 18–39 (
), and ACTH fragment 18–39 (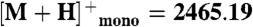 ).
).
NanoLC ESI MS/MS.
Tryptic peptides were resuspended in 0.1% formic acid and loaded onto a 10 cm reverse phase C18 column (5 μm particle size) and eluted over 40 min with a 3–40% acetonitrile gradient.
For the LC MS/MS data in Figs. 5, 6, and Fig. S2 using recombinant acetylated histones, mass spectra were acquired in data-dependent mode on a Thermo LTQ-Orbitrap. Full MS scans were acquired in the Orbitrap at a resolution of 30,000. The top five most intense peaks were selected for MS2, which were acquired in the LTQ.
For the data in Fig. 7 and Fig. S3 identifying endogenously acetylated histone H3 K79, the Thermo LTQ-Orbitrap mass spectrometer was operated in selected reaction monitoring mode (SRM). Initially, a precursor scan was performed in the Orbitrap with a resolution of 60,000 and a narrow mass range from 425–725 Da. Peptides with m/z values corresponding to the doubly and triply charged masses of the various modified forms of H3 K79 were isolated and fragmented in the LTQ, followed by detection of the fragment ions with Fourier transform instrumentation using a mass range of 200–2,000 Da and a resolution of 30,000. For each SRM event, the isolation window was 2 Da, and each fragmentation event was carried out for 30 ms at a normalized collision energy of 35% and an activation Q of 0.25.
MASCOT Database Searching.
All LC MS/MS data were searched using MASCOT. Mass spectra were searched against the Swissprot database using S. cerevisiae as the selected taxonomy. The peptide precursor mass tolerance was set at ± 0.1 Da, whereas the MS/MS fragment ions mass tolerance was set at ± 0.8 Da. All spectra were searched with acetylation(K), biotinylation(K), and oxidation(M) as variable modifications. Following MASCOT database searches, only peptide hits with e-values less than 0.05 were considered significant.
Yeast Strains.
hst2 and sir2 deletions were obtained from the Yeast Knock-out Collection. DOT1 was deleted in the wild-type parental strain BY4741 and the Δhst2 and Δsir2 derivatives via PCR-based gene replacement with the hphMX4 cassette from the pAG32 plasmid. dot1 knockouts were validated by junction PCR. S. cerevisiae histone H3 was cloned into pRS415 as a FLAG-tagged, 10xHis fusion protein along with histone H4 and transformed into the yeast strains for constitutive expression. Strains and their genotypes are listed in Table 3.
Table 3.
List of yeast strains and genotypes
| Strain | Genotype* |
| wild-type (BY4741) | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 |
| Δdot1 | BY4741; Δdot1∷hphMX4 |
| Δhst2 | BY4741; Δhst2∷kanMX4 |
| Δhst2 Δdot1 | BY4741; Δhst2∷kanMX4 Δdot1::hphMX4 |
| Δsir2 | BY4741; Δsir2∷kanMX4 |
| Δsir2 Δdot1 | BY4741; Δsir2∷kanMX4 Δdot1∷hphMX4 |
*All strains contain a pRS415 plasmid for expressing histone H3/H4.
Histone H3 Purification.
Yeast strains expressing histone H3 were grown to an optical density of approximately 1 (late log phase), harvested, and frozen at −80 °C. Histones were purified as previously described (64), with modifications. Briefly, cells were lysed with an SDS buffer containing the sirtuin inhibitors nicotinamide (5 mM) and sodium butyrate (50 mM). The lysed supernatant was diluted into a urea buffer for denaturation and to reduce the concentration of SDS, and then passed through a nickel column to bind the histones via the N-terminal 10xHis fusion tag. Histone H3 was eluted with a urea buffer containing 400 mM imidazole. The eluate was diluted with 4x Laemmli’s sample buffer and separated by SDS-PAGE. The histone bands were excised, trypsin-digested, and subjected to nanoLC MS/MS.
Histone H3 Deuteroacetylation.
Endogenous histone H3 was purified and prepared for tryptic digestion as above, with the addition of a treatment with deuterated acetic anhydride after destaining of the gel bands, as previously described (26, 48). Briefly, gel bands were incubated with 50 μL deuterated acetic acid (d4) and 10 μL deuterated acetic anhydride (d6) (Sigma) for 5 h at room temperature. The gel bands were then washed with 200 mM ammonium bicarbonate and water, dehydrated, and incubated with trypsin as mentioned previously.
Supplementary Material
Acknowledgments.
We thank Hidekazu Takahashi for the Hst2 and Sir2 expression vectors and Jeff McKnight and Greg Bowman for providing us with yeast histones. Supported by grants from the National Science Foundation (MCB-0920082) (C.W.) and the National Institutes of Health (U54-RR-020839) (R.J.C., J.B., and C.W.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Author Summary on page 5919 (volume 109, number 16).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1121471109/-/DCSupplemental.
References
- 1.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 2.Smith JS, et al. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proc Natl Acad Sci USA. 2000;97:6658–6663. doi: 10.1073/pnas.97.12.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landry J, et al. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci USA. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun. 1999;260:273–279. doi: 10.1006/bbrc.1999.0897. [DOI] [PubMed] [Google Scholar]
- 6.Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 7.Haigis MC, Sinclair DA. Mammalian sirtuins: Biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell. 1999;97:621–633. doi: 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- 9.McAinsh AD, Scott-Drew S, Murray JA, Jackson SP. DNA damage triggers disruption of telomeric silencing and Mec1p-dependent relocation of Sir3p. Curr Biol. 1999;9:963–966. doi: 10.1016/s0960-9822(99)80424-2. [DOI] [PubMed] [Google Scholar]
- 10.Mills KD, Sinclair DA, Guarente L. MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell. 1999;97:609–620. doi: 10.1016/s0092-8674(00)80772-2. [DOI] [PubMed] [Google Scholar]
- 11.Picard F, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo J, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 13.Vaziri H, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 14.Rine J, Herskowitz I. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics. 1987;116:9–22. doi: 10.1093/genetics/116.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aparicio OM, Billington BL, Gottschling DE. Modifiers of position effect are shared between telomeric and silent mating-type loci in S.cerevisiae. Cell. 1991;66:1279–1287. doi: 10.1016/0092-8674(91)90049-5. [DOI] [PubMed] [Google Scholar]
- 16.Smith JS, Boeke JD. An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev. 1997;11:241–254. doi: 10.1101/gad.11.2.241. [DOI] [PubMed] [Google Scholar]
- 17.Bryk M, et al. Transcriptional silencing of Ty1 elements in the RDN1 locus of yeast. Genes Dev. 1997;11:255–269. doi: 10.1101/gad.11.2.255. [DOI] [PubMed] [Google Scholar]
- 18.Isenberg I. Histones. Annu Rev Biochem. 1979;48:159–191. doi: 10.1146/annurev.bi.48.070179.001111. [DOI] [PubMed] [Google Scholar]
- 19.Sealy L, Chalkley R. Modification of histones immediately following synthesis. Arch Biochem Biophys. 1979;197:78–82. doi: 10.1016/0003-9861(79)90221-2. [DOI] [PubMed] [Google Scholar]
- 20.Braunstein M, Rose AB, Holmes SG, Allis CD, Broach JR. Transcriptional silencing in yeast is associated with reduced nucleosome acetylation. Genes Dev. 1993;7:592–604. doi: 10.1101/gad.7.4.592. [DOI] [PubMed] [Google Scholar]
- 21.Guillemette B, et al. H3 lysine 4 is acetylated at active gene promoters and is regulated by H3 lysine 4 methylation. PLoS Genet. 2011;7:e1001354. doi: 10.1371/journal.pgen.1001354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Froyd CA, Rusche LN. The duplicated deacetylases Sir2 and Hst1 subfunctionalized by acquiring complementary inactivating mutations. Mol Cell Biol. 2011;31:3351–3365. doi: 10.1128/MCB.05175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li M, et al. Thiamine biosynthesis in Saccharomyces cerevisiae is regulated by the NAD+-dependent histone deacetylase Hst1. Mol Cell Biol. 2010;30:3329–3341. doi: 10.1128/MCB.01590-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie J, et al. Sum1 and Hst1 repress middle sporulation-specific gene expression during mitosis in Saccharomyces cerevisiae. EMBO J. 1999;18:6448–6454. doi: 10.1093/emboj/18.22.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brachmann CB, et al. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 1995;9:2888–2902. doi: 10.1101/gad.9.23.2888. [DOI] [PubMed] [Google Scholar]
- 26.Celic I, et al. The sirtuins Hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr Biol. 2006;16:1280–1289. doi: 10.1016/j.cub.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 27.Maas NL, Miller KM, DeFazio LG, Toczyski DP. Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol Cell. 2006;23:109–119. doi: 10.1016/j.molcel.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 28.Xu F, Zhang Q, Zhang K, Xie W, Grunstein M. Sir2 deacetylates histone H3 lysine 56 to regulate telomeric heterochromatin structure in yeast. Mol Cell. 2007;27:890–900. doi: 10.1016/j.molcel.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kimura A, Umehara T, Horikoshi M. Chromosomal gradient of histone acetylation established by Sas2p and Sir2p functions as a shield against gene silencing. Nat Genet. 2002;32:370–377. doi: 10.1038/ng993. [DOI] [PubMed] [Google Scholar]
- 30.Suka N, Luo K, Grunstein M. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat Genet. 2002;32:378–383. doi: 10.1038/ng1017. [DOI] [PubMed] [Google Scholar]
- 31.Vaquero A, et al. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006;20:1256–1261. doi: 10.1101/gad.1412706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson JM, Le VQ, Zimmerman C, Marmorstein R, Pillus L. Nuclear export modulates the cytoplasmic Sir2 homologue Hst2. EMBO Rep. 2006;7:1247–1251. doi: 10.1038/sj.embor.7400829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin YY, et al. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell. 2009;136:1073–1084. doi: 10.1016/j.cell.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang T, Kraus WL. SIRT1-dependent regulation of chromatin and transcription: Linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim Biophys Acta. 2010;1804:1666–1675. doi: 10.1016/j.bbapap.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim SC, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 36.Choudhary C, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 37.Smith BC, Settles B, Hallows WC, Craven MW, Denu JM. SIRT3 substrate specificity determined by peptide arrays and machine learning. ACS Chem Biol. 2011;6:146–157. doi: 10.1021/cb100218d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109:745–756. doi: 10.1016/s0092-8674(02)00759-6. [DOI] [PubMed] [Google Scholar]
- 39.Garcia BA, et al. Organismal differences in post-translational modifications in histones H3 and H4. J Biol Chem. 2007;282:7641–7655. doi: 10.1074/jbc.M607900200. [DOI] [PubMed] [Google Scholar]
- 40.Ng HH, et al. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 2002;16:1518–1527. doi: 10.1101/gad.1001502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park JH, Cosgrove MS, Youngman E, Wolberger C, Boeke JD. A core nucleosome surface crucial for transcriptional silencing. Nat Genet. 2002;32:273–279. doi: 10.1038/ng982. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi YH, et al. Dot1 and histone H3K79 methylation in natural telomeric and HM silencing. Mol Cell. 2011;42:118–126. doi: 10.1016/j.molcel.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLuckey SA, Wells JM. Mass analysis at the advent of the 21st century. Chem Rev. 2001;101:571–606. doi: 10.1021/cr990087a. [DOI] [PubMed] [Google Scholar]
- 44.Jiang L, et al. Global assessment of combinatorial post-translational modification of core histones in yeast using contemporary mass spectrometry. LYS4 trimethylation correlates with degree of acetylation on the same H3 tail. J Biol Chem. 2007;282:27923–27934. doi: 10.1074/jbc.M704194200. [DOI] [PubMed] [Google Scholar]
- 45.Drogaris P, Wurtele H, Masumoto H, Verreault A, Thibault P. Comprehensive profiling of histone modifications using a label-free approach and its applications in determining structure-function relationships. Anal Chem. 2008;80:6698–6707. doi: 10.1021/ac800739d. [DOI] [PubMed] [Google Scholar]
- 46.Ngubo M, Kemp G, Patterton HG. Nano-electrospray tandem mass spectrometric analysis of the acetylation state of histones H3 and H4 in stationary phase in Saccharomyces cerevisiae. BMC Biochem. 2011;12:34. doi: 10.1186/1471-2091-12-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu Q, et al. The Orbitrap: A new mass spectrometer. J Mass Spectrom. 2005;40:430–443. doi: 10.1002/jms.856. [DOI] [PubMed] [Google Scholar]
- 48.Smith CM, et al. Mass spectrometric quantification of acetylation at specific lysines within the amino-terminal tail of histone H4. Anal Biochem. 2003;316:23–33. doi: 10.1016/s0003-2697(03)00032-0. [DOI] [PubMed] [Google Scholar]
- 49.Smith CM. Quantification of acetylation at proximal lysine residues using isotopic labeling and tandem mass spectrometry. Methods. 2005;36:395–403. doi: 10.1016/j.ymeth.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 50.Hersman E, Nelson DM, Griffith WP, Jelinek C, Cotter RJ. Analysis of histone modifications from tryptic peptides of deuteroacetylated isoforms. Int J Mass Spectrom. 2011;312:5–16. doi: 10.1016/j.ijms.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Makarov A, Denisov E, Lange O, Horning S. Dynamic range of mass accuracy in LTQ Orbitrap hybrid mass spectrometer. J Am Soc Mass Spectrom. 2006;17:977–982. doi: 10.1016/j.jasms.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 52.Ng HH, Ciccone DN, Morshead KB, Oettinger MA, Struhl K. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: A potential mechanism for position-effect variegation. Proc Natl Acad Sci USA. 2003;100:1820–1825. doi: 10.1073/pnas.0437846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Altaf M, et al. Interplay of chromatin modifiers on a short basic patch of histone H4 tail defines the boundary of telomeric heterochromatin. Mol Cell. 2007;28:1002–1014. doi: 10.1016/j.molcel.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tanny JC, Dowd GJ, Huang J, Hilz H, Moazed D. An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell. 1999;99:735–745. doi: 10.1016/s0092-8674(00)81671-2. [DOI] [PubMed] [Google Scholar]
- 55.Tanny JC, Moazed D. Coupling of histone deacetylation to NAD breakdown by the yeast silencing protein Sir2: Evidence for acetyl transfer from substrate to an NAD breakdown product. Proc Natl Acad Sci USA. 2001;98:415–420. doi: 10.1073/pnas.031563798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rossmann MP, Luo W, Tsaponina O, Chabes A, Stillman B. A common telomeric gene silencing assay is affected by nucleotide metabolism. Mol Cell. 2011;42:127–136. doi: 10.1016/j.molcel.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garrity J, Gardner JG, Hawse W, Wolberger C, Escalante-Semerena JC. N-lysine propionylation controls the activity of propionyl-CoA synthetase. J Biol Chem. 2007;282:30239–30245. doi: 10.1074/jbc.M704409200. [DOI] [PubMed] [Google Scholar]
- 58.Smith BC, Denu JM. Acetyl-lysine analog peptides as mechanistic probes of protein deacetylases. J Biol Chem. 2007;282:37256–37265. doi: 10.1074/jbc.M707878200. [DOI] [PubMed] [Google Scholar]
- 59.Chen Y, et al. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol Cell Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cheng Z, et al. Molecular characterization of propionyllysines in non-histone proteins. Mol Cell Proteomics. 2009;8:45–52. doi: 10.1074/mcp.M800224-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim Y, et al. Large-scale evaluation of protein reductive methylation for improving protein crystallization. Nat Methods. 2008;5:853–854. doi: 10.1038/nmeth1008-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luger K, Rechsteiner TJ, Richmond TJ. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol Biol. 1999;119:1–16. doi: 10.1385/1-59259-681-9:1. [DOI] [PubMed] [Google Scholar]
- 63.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2007;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 64.Masumoto H, Hawke D, Kobayashi R, Verreault A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436:294–298. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
