

Abstract
Hepatitis C virus (HCV) infects ∼2% of the world's population. It is estimated that there are more than 500,000 new infections annually in Egypt, the country with the highest HCV prevalence. An effective vaccine would help control this expanding global health burden. HCV is highly variable, and an effective vaccine should target conserved T- and B-cell epitopes of the virus. Conserved B-cell epitopes overlapping the CD81 receptor-binding site (CD81bs) on the E2 viral envelope glycoprotein have been reported previously and provide promising vaccine targets. In this study, we isolated 73 human mAbs recognizing five distinct antigenic regions on the virus envelope glycoprotein complex E1E2 from an HCV-immune phage-display antibody library by using an exhaustive-panning strategy. Many of these mAbs were broadly neutralizing. In particular, the mAb AR4A, recognizing a discontinuous epitope outside the CD81bs on the E1E2 complex, has an exceptionally broad neutralizing activity toward diverse HCV genotypes and protects against heterologous HCV challenge in a small animal model. The mAb panel will be useful for the design and development of vaccine candidates to elicit broadly neutralizing antibodies to HCV.
Keywords: cross-neutralizing antibody, antigenic determinant, protective determinant, chronic viral infection, virus challenge
Virus neutralizing antibodies (NAbs) are potential therapeutics for preventing and treating viral infections and are valuable reagents for studying the antigenicity and function of viral surface proteins. For highly variable viruses such as influenza virus, HIV, and hepatitis C virus (HCV), antibodies that neutralize many different viral strains, known as broadly NAbs, help define conserved elements in the viral envelope spikes for rational vaccine and drug design (1, 2). The HCV envelope glycoproteins E1 and E2 form a heterodimer for virus attachment and entry and are targets for NAbs (3). A major challenge in HCV vaccine design is the extreme diversity of the virus. HCV is highly heterogeneous with six major genotypes (>30% overall sequence difference) and more than 50 subtypes (10–25% difference) (4). HCV genotypes 1, 2, and 3 are found worldwide, and genotypes 1 and 3 are the most widely distributed. Genotype 4 is the predominant genotype in Egypt (5), genotype 5 is found primarily in South Africa, and genotype 6 is widespread in Southeast Asia. The great diversity of HCV—even a single infected individual harbors a large number of quasispecies—is driven by the poor fidelity of RNA polymerase and rapid turnover of the virus. It is estimated that an individual produces as many as 1012 virions per day (6). Consequently, any antibody or drug effective against one isolate will not necessarily be useful against other isolates, perhaps not even against different quasispecies in an individual patient.
To overcome the challenge of viral diversity, conserved elements on the virus should be targeted. We, and others, have reported previously that antibodies to the CD81 receptor-binding site (CD81bs) on E2 mediate cross-neutralization of diverse HCV isolates. mAbs AP33 (7) and HCV1 (8) recognize linear epitopes between amino acids 412 and 423 of E2, whereas other mAbs recognize overlapping discontinuous E2 epitopes (9–12). A murine mAb H77.39 specific for E2 was isolated recently, and the mAb cross-neutralized HCV genotypes by blocking E2 binding to both CD81 and another HCV entry factor, scavenger receptor class B type I (SR-BI) (13). Cross-neutralizing mAbs to E1 (linear region 313–327) have also been reported (14). In addition to mAb studies, human polyclonal antibodies to the E2 linear region 412–426 were also found to neutralize HCV. However, neutralization could be inhibited with polyclonal antibodies to a downstream region, 436–446 (15). These studies highlight not only the feasibility of targeting HCV conserved epitopes but also the need for greater understanding of the antigenic properties of the E1E2 glycoprotein complex. In the absence of a molecular structure of E1E2, mAbs to diverse antigenic regions (ARs) are valuable tools in studying E1E2 folding and function.
Technological breakthroughs have greatly expedited the process of antibody discovery, which include the first-generation hybridoma generation approach, B-cell immortalization, panning of phage/yeast-display antibody libraries, and, most recently, cloning of antibodies from single antibody-producing cells (16). Despite many breakthroughs, the fundamental aim in generating mAbs with distinct and useful biological activities remains unchanged. In the case of antiviral antibodies, broadly NAbs are highly desirable, but they are often rare and difficult to isolate, particularly if the corresponding epitopes are poorly immunogenic. To explore the antigenicity of E1E2, we used a strategy termed “exhaustive panning of a phage-display antibody repertoire” to discover rare E1E2-specific mAbs (Fig. S1). Under this strategy, an antibody repertoire is interrogated repeatedly by using antigens masked with antibodies isolated from the previous rounds of selection (17, 18). In each new round of selection, only antibodies with binding properties distinct from the previous rounds are obtained, thus permitting the selection of rare antibodies in the repertoire. The selection process is completed when no new antibody specificity is recovered and that a given antibody repertoire to the panning antigen is exhausted. This strategy has resulted in the isolation of 73 antibodies recognizing five distinct ARs on the HCV E1E2 glycoproteins, with an antibody to AR4 having exceptionally broad neutralizing activity against different HCV genotypes.
Results
We have previously isolated a panel of mAbs recognizing three distinct ARs on E2 (11). We built on this knowledge and took advantage of phage-display technology to investigate whether there are other conserved neutralizing epitopes on HCV. The same HCV-immune antibody library was panned repeatedly against the E1E2 complex and E1E2 masked by antibodies isolated in the previous rounds of panning. In the panning with E1E2 heterodimer (105 kDa) masked by four IgG molecules (i.e., a total of 600 kDa), none of the bound antibodies was unique (Fig. S1). After four independent panning experiments, a total of 73 antibodies (fragment antigen-binding, Fab) to E1E2 were isolated from the antibody repertoire (Table S1). The Fab fragments were grouped based on their heavy chain sequences, and 22 Fab groups (Fab groups A–V) resulted from this panning strategy. To confirm that antibodies to new epitopes had been isolated, binding of the new antibodies (Fab groups N–V), either as crude or purified Fab fragments, to E1E2 was evaluated in competition ELISA using antibodies specific for AR1, AR2, or AR3 (Fig. S2A). The results showed that, except for Fab U1, which competed significantly with AR3-specific antibodies, the new antibodies recognize epitopes distinct from AR1, AR2, and AR3. In addition, the Fab group N cross-reacted with E1E2 derived from the six HCV genotypes (Fig. S2B), suggesting that its members may recognize conserved epitopes. To investigate the binding properties of the antibody panel, Fab fragments N4, Q2, and V1 were converted into full-length IgG1 molecules (known as mAbs AR4A, AR4B, and AR5A, respectively) for competition study among the new antibodies. The results show that the new antibodies are segregated into two clusters. Fab fragments N4, O1, P2, Q2, and S1 bind to a cluster of overlapping epitopes, whereas Fab fragments R1 and V1 bind to a different cluster (Table 1). A significant level of competition (∼50%) is also observed between Fab R1 and mAbs AR4A and AR4B, suggesting that the two antibody clusters are partially overlapping. Consequently, the clusters of overlapping epitopes, i.e., ARs, defined by this antibody panel are named AR4 and AR5 here.
Table 1.
Competition ELISA of antibodies to identify two distinct ARs on E1E2
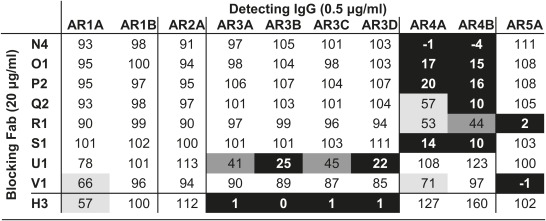 |
Residual binding level: 0–25%, black; 25–50%, dark gray; 50–75%, light gray; >75%, white. Fab fragments N4, Q2, and V1 were converted into full-length IgGs AR4A, AR4B, and AR5A, respectively. Fab H3 is the Fab fragment of mAb AR3C and is a control for competition with AR3-specific antibodies.
The general properties of the antibodies in binding to E1E2 are summarized in Table 2, and the results of individual experiments characterizing the antibodies are provided in Fig. 1, Fig. S3, and Table S2. All of the mAbs recognize discontinuous epitopes (Fig. 1A). Although mAb AR3A effectively blocked E1E2 binding to CD81, mAbs AR4A and AR5A did not interfere significantly with the binding (Fig. 1B). The epitopes were investigated further by comparing the reactivity of the antibodies to the E1E2 complex and soluble E2 (Fig. 1C). Whereas mAbs AR3A and CBH-7 (19) bind soluble E2, mAbs AR4A and AR5A bind only folded E1E2 complex. In immunoprecipitation experiments, mAbs AR4A and AR5A pulled down the wild-type E1E2 complex but not the E1E2 mutant xN196/305A, which is known not to form significant E1E2 complexes because of the loss of the first and fourth N-glycosylation sites in E1 (20) (Fig. 1D). Interestingly, the control anti-E1 or anti-E2 mAb pulled down both E1 and E2 of the xN196/305A mutant, indicating that, despite the absence of the AR4A and AR5A epitopes, some of the E1 and E2 glycoproteins were still complexed, presumably via their transmembrane domains (21). To better characterize the new epitopes, the mAbs were competed with a panel of well-characterized mAbs (Table S2). The results demonstrate that AR4A and AR4B epitopes are distinct, whereas AR5A epitope overlaps with that of mAb CBH-7 (19).
Table 2.
General properties of anti-HCV antibodies
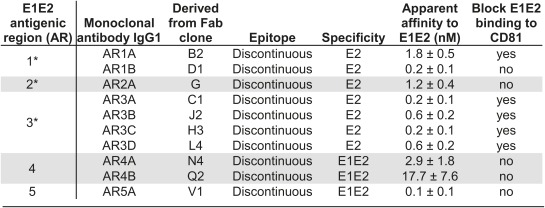 |
*The properties of mAbs to AR1–AR3 have been previously published (11) and are included here for comparison.
Fig. 1.

(A) The AR4- and AR5-specific antibodies recognize discontinuous epitopes on E1E2. E1E2 antigens (isolate H77) were denatured and reduced with SDS and DTT at 100 °C for 5 min. Native or reduced/denatured E1E2 was probed with the antibodies at the specified concentrations in ELISA. The murine mAb A4 (47), specific for E1 residues 197–207, was a positive control for reduced/denatured E1. (B) AR4 and AR5 do not overlap with CD81bs on E1E2. The effect of mAbs AR4A and AR5A on the binding of E1E2 to the large extracellular loop of CD81 (CD81-LEL) was studied by ELISA. E1E2 antigens (isolate H77) were preincubated with serially diluted mAbs AR3A, AR4A, or AR5A before being added to ELISA wells coated with CD81-LEL (fused to maltose-binding protein). Bound E1E2 was detected with biotinylated mAbs AR1B, AR2A, AR3A, or murine mAb A4. Note that the epitope recognized by mAb AR3A, but not the other three mAbs, overlaps with E1E2 CD81bs. Therefore, only mAb AR3A significantly inhibited the interaction between E1E2 and CD81-LEL. (C) Binding of mAbs AR3A, AR4A, and AR5A to lectin-captured full-length E1E2 (Left) and E2 with the transmembrane domain truncated (soluble E2; Right). The control antibodies are murine mAb A4, mAbs CBH-7 (19), and AR3A (11), recognizing discontinuous epitopes on E2, and mAb CBH-2, recognizing an epitope on the E1E2 heterodimer (35). (D) Immunoprecipitation of E1E2 complex by mAbs AR4A and AR5A. The wild-type and xN196/305A mutant E1E2 were pulled down by using mAbs AR4A and AR5A, the anti-E1 mAb IGH526 (14), the anti-E2 mAb AR3A, the isotype control anti-HIV mAb b6 (11), and protein A-conjugated agarose. The immunoprecipitants were analyzed by reducing SDS/PAGE and immunoblotting using mAb A4 for E1 (47) and mAb HCV1 for E2 (8). The amounts of E1 and E2 pulled down by the antibodies were compared with that of mAb AR3A. HC, antibody heavy chain.
Because the AR4 and AR5 antibodies bind E1E2 but not soluble E2, we initially speculated that they might recognize discontinuous epitopes on E1. However, isolated folded E1 is unavailable, and our attempts to express soluble E1 in mammalian and Drosophila systems resulted only in misfolded aggregates. Consequently, site-directed mutagenesis was applied to identify residues that are important for the formation of AR4 and AR5. Because mAbs AR4A and AR5A do not block E2–CD81 interactions, the panel of CD81bs alanine-scanning mutants constructed by Owsianka et al. would not be sufficient for the mapping (22). Therefore, we expanded this panel to include additional conserved regions of E1 and E2, excluding the transmembrane domains and cysteine residues. A total of 162 single and double alanine-scanning mutants of E1 and E2 were available for this study. Using this mutant panel, we identified mutations that are specific for mAbs AR4A and AR5A as well as a number of mutations that reduce binding of both mAbs (Table S3). The specific residues required for the binding of mAb AR4A and mAb AR5A are E2 residues D698 and R639, respectively. Notably, both D698 and R639 are extremely conserved, accounting for 2,159 and 2,158 of the 2,160 E2 sequences deposited in the Virus Pathogen Database and Analysis Resource (http://www.viprbrc.org), respectively. The E2 D698 residue is located within the membrane proximal external region (MPER), ∼20 residues upstream of the transmembrane domain. For mutations that affect the two nonoverlapping epitopes simultaneously (Y201A, T204A, N205A, D206A, R657A, D658A, and L692A), we speculate that these mutations may have a similar effect as for the xN196/305A double mutations in disrupting E1 folding and/or the formation of E1E2 complex. Further studies are needed to define their roles in E1E2 complex formation. Overall, the mapping data showed that mAb AR5A recognized an epitope on the E1E2 complex, which overlaps with that of the E2-specific mAb CBH-7 (Table S2). The previously unknown AR4A epitope is composed partly by the MPER of E2 and locates near AR5 on E1E2, overlapping with a shared epitope defined by Fab R1 (Table 1).
The antibodies were evaluated for anti-HCV activity. A panel of 16 HCV pseudotype virus particles (HCVpp) displaying E1E2 from the six major genotypes (23–25) and eight cell culture-produced HCV (HCVcc) expressing genotypes 1–6 of envelope glycoproteins (26–32) were available for neutralization studies. Table 3 summarizes the results, and the experimental details are provided in Figs. S4 and S5. Notably, the HCV neutralization assays are technically demanding, and many E1E2 genes we evaluated did not produce consistent HCVpp infectivity. To minimize variability between experiments and errors in the calculation of antibody titers attributable to background infectivity in the system, we restricted our in-house neutralization assays to HCVpp having good infectivity [signal-to-noise (S/N) ratio >10], which is particularly important for isolates that produce HCVpp with low infectivity (e.g., U.K.N2b1.1, genotype 2b). The results show that mAb AR4A cross-neutralized all isolates evaluated in the assays, with IC50 titers ranging from <1 to 38.5 μg/mL in the HCVpp assays and from 0.03 to 8.9 μg/mL in the HCVcc assays. At 90% neutralization level (IC90), mAbs AR3A, AR4A, and AR5A neutralized 38%, 63%, and 17% of the virus panel, respectively. Antibodies that did not neutralize more than 50% or 90% virus infectivity at 50 μg/mL are considered negative in this study. The mAbs inhibit the virus at both pre- and postattachment stages (Fig. 2A). The potential synergistic effect on neutralizing activity of an antibody mixture targeting multiple nonoverlapping E1E2 epitopes was also investigated (Fig. S6). The mAbs were titrated in the combinations of two and three antibodies, and the results demonstrated that a moderate synergism [combination index (CI) between 0.53 and 0.70 at IC50] was achieved in various combinations of mAbs AR3A, AR4A, and AR5A.
Table 3.
Neutralizing activity of anti-HCV antibodies
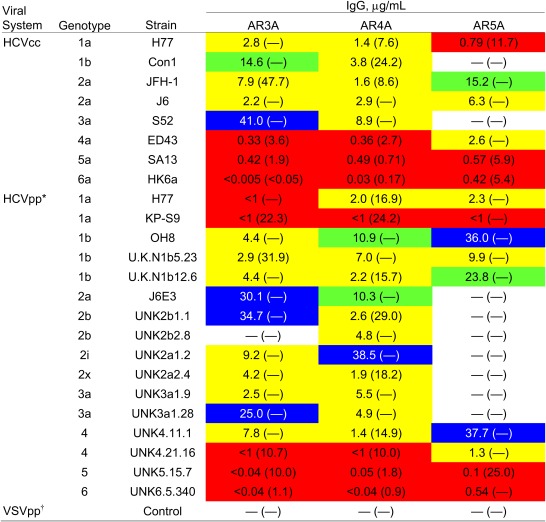 |
Serial dilutions of mAbs from 50 to below 1 μg/mL were tested for virus neutralization (Figs. S4 and S5). The antibody concentrations required for 50% and 90% (in parentheses) neutralization were calculated from the titration curves. —, mAb did not neutralize more than 50% or 90% of virus infectivity at 50 μg/mL. The IC50 titers of the mAbs are highlighted by color: <1 μg/mL, red; 1–10 μg/mL, yellow; 10–25 μg/mL, green; 25–50 μg/mL, blue.
*The panel of HCVpp shown includes HCV Envs that produce a signal at least 10-fold higher than the background signal caused by control pseudotype virus particles without HCV Env.
None of the mAbs neutralized pseudotype virus particles displaying the vesicular stomatitis virus envelope glycoprotein G (VSVpp), which were used as a control for nonspecific activity.
Fig. 2.

(A) The mAbs AR3A, AR4A, and AR5A neutralize HCV at pre- and postattachment steps. In a standard neutralization assay (Top), HCVpp was incubated with the mAbs before spinoculation onto cell monolayers, and the virus was removed after a 6-h incubation at 37 °C. To detect antibody blockage of attachment (Middle), mAbs were incubated with HCVpp before spinoculation onto cells at 8 °C for 1 h. Unbound HCVpp was washed with ice-cold media, and virus infectivity was determined as above. To detect antibody neutralization postattachment (Bottom), HCVpp was first spinoculated onto cells, and unbound virus was removed by washing cell monolayers with ice-cold media before antibodies were added to the cells and infectivity was determined after a 3-d incubation. Black bars, mAbs at 50 μg/mL; white bars, mAbs at 10 μg/mL. (B) Passive antibody protection of a genetically humanized mouse model for HCV infection. Rosa26-Fluc mice (48) (n = 5 for each group) were injected i.p. with 100 mg/kg of mAb AR3A, AR4A, a mixture of half the amount of both mAbs (AR3A/AR4A), or an isotype control IgG1 on days −3 and −2. On day −1, the animals were injected with 1011 recombinant adenoviral particles encoding human CD81, scavenger receptor class B type I (SR-BI), claudin 1, and occludin, and the animals were challenged with 2 × 107 50% tissue culture infectious dose (TCID50) of chimeric HCVcc carrying the structural proteins of the genotype 1b Con1 isolate (BiCRE-Con1/JFH1 virus; Upper) or genotype 2a J6 isolate (BiCRE-Jc1/JFH1 virus; Lower) on day 0. Bioluminescence was detected after 3 d. Data are summarized in box-and-whisker plots, and statistical significance was evaluated by one-way ANOVA (*P < 0.05; **P < 0.01; ***P < 0.001).
Next, we studied whether the mAbs could provide protection against HCV infection in vivo. We took advantage of a recently developed mouse model that is rendered susceptible to HCV infection by genetic humanization (33). Infection of the animals after challenge with recombinant HCVcc expressing Cre recombinase can be measured noninvasively by bioluminescence imaging. The model was used here to evaluate whether the broadly neutralizing mAbs AR3A and AR4A could blunt virus infection in vivo. The mouse model was first injected i.p. with single mAbs AR3A or AR4A or an AR3A/AR4A mixture of half the amount of mAbs, then challenged with HCVcc displaying the structural proteins of the genotype 1b Con1 isolate or genotype 2a Jc1 isolate. The results showed that the mAbs inhibited genotypes 1b and 2a virus infection, with mAb AR4A alone having the best antiviral activity (Fig. 2B). Notably, the mAb mixture did not provide better protection, indicating that the presence of one mAb to the virus did not enhance the antiviral activity of the other mAb in this experimental model.
Discussion
The exhaustive-panning strategy used in this study, which can be easily executed in a standard laboratory setting, facilitates the isolation of rare antibodies with interesting properties. Common to most antibody technologies, antibodies that are relatively abundant in the source materials, regardless of their biological activity, are predominantly isolated during screening procedures. These antibodies pose a major background problem in the isolation of potentially important but rare antibodies. The adaptation of high-throughput functional screens (e.g., virus neutralization assays) helps eliminate such background but requires sophisticated instrumentation support (34). This panning strategy allowed us to isolate diverse E1E2-specific antibodies using eight different heavy chain variable domain (VH) genes with their third complementarity determining region (HCDR3) lengths ranging from 5 to 25 residues (Table S1).
The mAb panel recognized five distinct clusters of epitopes (ARs) on E1E2 (Fig. S1 and Table 2). AR1, AR2, and AR3 have been described previously (11). AR1 is proximal to the CD81bs and AR3 on E2. However, AR1 is not conserved and probably not exposed on the viral surface because antibodies to AR1 bind only genotype 1a HCV and do not have significant neutralizing activity. AR2 is distal from CD81bs and is exposed on E2 because mAb AR2A can neutralize several HCV isolates. AR3 is conserved and overlaps the CD81bs. Multiple AR3-specific mAbs were found to cross-neutralize isolates of diverse HCV genotypes. AR4 and AR5 are present on the E1E2 complex and adjacent to each other with the Fab R1 epitope sitting in the middle (Table 1). Although both AR4 and AR5 are found on the E1E2 complex, they are distinct. AR5 overlaps significantly with an epitope recognized by mAb CBH-7 whose binding does not require E1 (19) (Fig. 1C and Table S2). AR4 does not overlap with the epitopes recognized by mAb CBH-7 and the E1E2-specific mAb CBH-2 (35), and part of AR4 is formed by or critically depends on the MPER of E2, involving the highly conserved D698 residue.
The binding of AR4- and AR5-specific antibodies requires proper folding of the E1E2 complex. The removal of the E1 N-glycosylation sites N196 and N305 had previously been shown to reduce E1E2 complex formation (20). Here, we showed that these two mutations, and a number of additional mutations at the conserved E1 and E2 regions, disrupt AR4 and AR5 on the E1E2 complex (Table S3). It has long been established that the transmembrane domains of E1 and E2 are important for forming the noncovalent complex (21). However, there is little information on the interaction of the ectodomains of the two glycoproteins. Based on the above results, it is clear that the ectodomains of E1 and E2 also interact and that AR4 and AR5 are formed only when the ectodomains are arranged in the proper quaternary structure. We speculate that some of the mutations are positioned at the interface of the E1 and E2 ectodomains and play a role in stabilizing the complex. The mutations that abolish AR4 and AR5 simultaneously are clustered into three regions, the E1 region 201–206 and the E2 regions 657–659 and 692. If these regions are located at the interface of the E1E2 complex, the folded E1 and E2 heterodimer will likely be orientated in a manner that the N terminus of E1 is associated with the C terminus of the E2 ectodomain.
In the study of virus neutralization (Table 3), IC90 titers of the mAbs could not be determined for a number of isolates. A caveat for interpreting neutralization data for HCVpp is that some viruses may produce background infectivity independent of the presence of E1E2. Such background infectivity introduces significant error in the determination of antibody titers, in isolates that have low infectivity. Because we have taken precautions by only using isolates that produce high S/N ratio and by subtracting background infectivity caused by pseudotype virus produced without E1E2, we consider it an unlikely explanation for not achieving 90% neutralization in the experiments. The results likely reflect a limit in antibody potency because 90% neutralization level of HCVpp-H77 was achieved by mAbs AR3A and AR5A when higher antibody concentrations were used (Fig. S6).
Interestingly, the NAbs inhibited both virus attachment and entry (Fig. 2A). Previously, Schwarz et al. showed that mAb AR3A neutralized HCV at a postattachment step by blocking the virus–CD81 interaction (36). For mAbs AR4A and AR5A, they do not target the CD81bs on E1E2 (Fig. 1B) but epitopes involving the E2 residues D698 and R639, respectively (Table S3). Because mAbs AR4A and AR5A neutralized HCV isolates from multiple genotypes in both the HCVpp and HCVcc virus systems, these relatively conserved epitopes are likely exposed on free virus particles. Virus coated with the NAbs will be less capable of binding to receptors on cells because of steric effects. The data also suggest that these epitopes may be important for virus entry and that bound antibodies may interfere with virus interactions with other coreceptors or entry factors (33) or with the reorganization of E1E2 complex required for entry. Notably, the E2 MPER likely forms part of the AR4A epitope. It has been reported that the HCV E2 MPER is important for membrane fusion (37) and that the region 687–703 is predicted to form an amphipathic α-helix partially embedded in lipid membrane (38). If this prediction is correct, the helical register displaying D698 should be pointing away from the membrane for the binding of mAb AR4A. The HIV-1 MPER was reported to form a helix–hinge–helix structure on the viral membrane, and NAbs to this region inhibited the relative movement of the helices required for virus entry (39). It will be interesting to investigate whether NAbs to the E2 MPER also neutralize the viruses by a similar mechanism and if HCV shares a similar molecular feature in the viral envelope glycoprotein complex for membrane fusion and cell entry.
Sabo et al. recently reported that there are ostensibly cryptic epitopes on HCV similar to those found on other flaviviruses (40). The authors suggest that the HCV virions are not entirely rigid. Structural dynamics, or “breathing,” of the virions under different experimental conditions may expose partially occluded epitopes for some of their NAbs. It is currently unknown whether the HCV envelope glycoprotein complexes are packed in a regular and repetitive manner on the virus surface as in other flaviviruses for which structural dynamics of virion were observed or if they behave more like the virus envelope spikes of HIV-1, distributed sparsely on the virus surface with no evidence of virion breathing (41, 42). In any case, the AR3A, AR4A, and AR5A epitopes should be presented concurrently on the virus even if there is structural flexibility because the presence of one mAb did not adversely affect the activity of another mAb (Fig. S6).
Finally, the panel of mAbs and E1E2 mutants described here should provide new opportunities to study the HCV E1E2 glycoprotein complex. Soluble E2 can be expressed and purified independent of E1 for functional studies (43, 44), and E2 has been suggested to adopt a three-domain fold (44). It has been difficult to study HCV E1E2 complex because folded E1 and E1E2 cannot be produced as soluble proteins for biochemical analysis. The mAbs AR4A and AR5A can be useful tools for studying E1E2 complex formation because they recognize two distinct quaternary ARs on E1E2 and can neutralize HCV by blocking virus attachment and entry. The current study points to a region on the E1E2 complex involving the MPER of E2 as a target for broad neutralization of HCV. The mAb panel described here will also be useful for probing the antigenicity of E1E2-based HCV vaccine candidates and guide the design of immunogens to elicit cross-NAbs to HCV.
Materials and Methods
Human Anti-HCV Antibodies.
The panel of antibodies was generated from an HCV-immune antibody library by phage-display technology using the exhaustive-panning strategy (Fig. S1). The HCV-immune antibody library has been constructed previously with mRNA from the bone marrow mononuclear cells of an HCV-infected donor (genotype 1a), and some of the antibodies to AR1, AR2, and AR3 have been described previously (11). Antibody Fab fragments were expressed in Escherichia coli by using the pComb3H vector and purified with Protein G-conjugated agarose. Selected Fab fragments were converted into full-length IgG1 molecules by inserting the Fab heavy- and light-chain cDNAs into the pIgG1 vector and the antibodies produced in CHO-K1 cells.
Immunological Assays.
E1E2 and soluble E2 antigens were produced by transient transfection of 293T cells with expression plasmids encoding the corresponding HCV cDNA. E1E2 antigens were extracted from the cells by mild detergent, and soluble E2 was harvested as cell supernatant. The antigens were captured onto ELISA microwells precoated with Galanthus nivalis lectin for the detection of antigen-specific antibodies. Nonspecific background was detected with untransfected cell supernatant or lysate as antigen.
Virological Assays.
Antibody neutralization of HCV was determined by using the HCVcc (26–28, 32) and HCVpp (23, 24) virus systems. HCVcc was cultured with Huh-7 or Huh-7.5 cells (27), and the infectious foci were stained with the cross-reactive mAb AR3A or the NS5A-specific mAb 9E10 and the appropriate secondary antibody reagents. Neutralization of HCVcc was determined by the reduction of the number of infectious foci (11, 27). HCVpp was generated by cotransfection of 293T cells with the pNL4-3.lucR−E− and the corresponding expression plasmids encoding the E1E2 genes by polyethylenimine, and virus infection of Huh-7 cells was detected by using the firefly luciferase assay system (11). Neutralization of HCVpp was determined by the reduction of luciferase activity in Huh-7 cells infected with HCVpp displaying Env from different isolates (24, 25). Antibody synergism was calculated based on the median-effect equation using CalcuSyn software (Biosoft) (45, 46).
Antibody Protection Experiment.
The genetically humanized mouse model for HCV infection has been described previously (33). Briefly, Rosa26-Fluc mice containing the firefly luciferase gene, which is transcriptionally repressed by an upstream loxP-flanked STOP cassette, were injected i.v. with recombinant adenoviruses expressing the HCV entry factors human CD81, scavenger receptor class B type I (SR-BI), claudin 1, and occludin. Adenoviral gene delivery resulted in efficient transduction of murine hepatocytes in vivo. The animals were then injected i.v. with recombinant HCVcc expressing Cre recombinase, and infection was monitored by bioluminescence imaging of firefly luciferase activity at 3 d postchallenge. To study passive antibody protection against HCV, the animals were injected with the anti-HCV antibodies at 48 and 24 h before adenovirus infection.
Statistical Analysis.
Statistical analysis was performed with GraphPad Prism software. Statistical significance was calculated with one-way ANOVA, and P values below 0.05 were considered significant.
Full methods and the associated references can be found in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Travis Nieusma, Arthur Kim, Justin Robbins, Joshua Horwitz, and Marion Sourisseau for laboratory support; Christina Corbaci for graphics support; Frank Chisari for advice and encouragement; Takashi Wakita, Jonathan Ball, and François-Loïc Cosset for HCVcc and HCVpp reagents; and Harry Greenberg, Jean Dubuisson, Jane McKeating, Steven Foung, and Arvind Patel for antibody reagents. Support for this work was provided by National Institutes of Health Grants R21AI080916 and R01AI079031 (to M.L.), R01AI071084 (to D.R.B.), RC1DK087193 (to C.M.R. and A.P.), R01AI072613 (to C.M.R.), and R01AI079043 (to Frank Chisari that supported M. Dreux); The Starr Foundation (C.M.R.); the Greenberg Medical Institute (C.M.R.); a Center for Translational Science Award (to A.P., under parent Grant UL1 RR024143 to The Rockefeller University); and The Lundbeck Foundation (J.B.). M. Dorner was supported by a postdoctoral fellowship from the German Research Foundation (Deutsche Forschungsgesellschaft). This is The Scripps Research Institute (TSRI) manuscript number 21335.
Footnotes
Conflict of interest statement: The Scripps Research Institute has filed a provisional patent application on the mAb panel described in this paper.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1114927109/-/DCSupplemental.
References
- 1.Burton DR. Antibodies, viruses and vaccines. Nat Rev Immunol. 2002;2:706–713. doi: 10.1038/nri891. [DOI] [PubMed] [Google Scholar]
- 2.Dormitzer PR, Ulmer JB, Rappuoli R. Structure-based antigen design: A strategy for next generation vaccines. Trends Biotechnol. 2008;26:659–667. doi: 10.1016/j.tibtech.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dustin LB, Rice CM. Flying under the radar: The immunobiology of hepatitis C. Annu Rev Immunol. 2007;25:71–99. doi: 10.1146/annurev.immunol.25.022106.141602. [DOI] [PubMed] [Google Scholar]
- 4.Kuiken C, Simmonds P. Nomenclature and numbering of the hepatitis C virus. Methods Mol Biol. 2009;510:33–53. doi: 10.1007/978-1-59745-394-3_4. [DOI] [PubMed] [Google Scholar]
- 5.Miller FD, Abu-Raddad LJ. Evidence of intense ongoing endemic transmission of hepatitis C virus in Egypt. Proc Natl Acad Sci USA. 2010;107:14757–14762. doi: 10.1073/pnas.1008877107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neumann AU, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-α therapy. Science. 1998;282:103–107. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 7.Owsianka A, et al. Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glycoprotein. J Virol. 2005;79:11095–11104. doi: 10.1128/JVI.79.17.11095-11104.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broering TJ, et al. Identification and characterization of broadly neutralizing human monoclonal antibodies directed against the E2 envelope glycoprotein of hepatitis C virus. J Virol. 2009;83:12473–12482. doi: 10.1128/JVI.01138-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schofield DJ, et al. Human monoclonal antibodies that react with the E2 glycoprotein of hepatitis C virus and possess neutralizing activity. Hepatology. 2005;42:1055–1062. doi: 10.1002/hep.20906. [DOI] [PubMed] [Google Scholar]
- 10.Johansson DX, et al. Human combinatorial libraries yield rare antibodies that broadly neutralize hepatitis C virus. Proc Natl Acad Sci USA. 2007;104:16269–16274. doi: 10.1073/pnas.0705522104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Law M, et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med. 2008;14:25–27. doi: 10.1038/nm1698. [DOI] [PubMed] [Google Scholar]
- 12.Keck ZY, et al. Definition of a conserved immunodominant domain on hepatitis C virus E2 glycoprotein by neutralizing human monoclonal antibodies. J Virol. 2008;82:6061–6066. doi: 10.1128/JVI.02475-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabo MC, et al. Neutralizing monoclonal antibodies against hepatitis C virus E2 protein bind discontinuous epitopes and inhibit infection at a postattachment step. J Virol. 2011;85:7005–7019. doi: 10.1128/JVI.00586-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meunier JC, et al. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the e1 glycoprotein of hepatitis C virus. J Virol. 2008;82:966–973. doi: 10.1128/JVI.01872-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang P, et al. Depletion of interfering antibodies in chronic hepatitis C patients and vaccinated chimpanzees reveals broad cross-genotype neutralizing activity. Proc Natl Acad Sci USA. 2009;106:7537–7541. doi: 10.1073/pnas.0902749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–774. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- 17.Ditzel HJ. Rescue of a broader range of antibody specificities using an epitope-masking strategy. Methods Mol Biol. 2002;178:179–186. doi: 10.1385/1-59259-240-6:179. [DOI] [PubMed] [Google Scholar]
- 18.Tsui P, et al. Progressive epitope-blocked panning of a phage library for isolation of human RSV antibodies. J Immunol Methods. 2002;263:123–132. doi: 10.1016/s0022-1759(02)00032-7. [DOI] [PubMed] [Google Scholar]
- 19.Keck ZY, et al. Hepatitis C virus E2 has three immunogenic domains containing conformational epitopes with distinct properties and biological functions. J Virol. 2004;78:9224–9232. doi: 10.1128/JVI.78.17.9224-9232.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meunier JC, et al. Analysis of the glycosylation sites of hepatitis C virus (HCV) glycoprotein E1 and the influence of E1 glycans on the formation of the HCV glycoprotein complex. J Gen Virol. 1999;80:887–896. doi: 10.1099/0022-1317-80-4-887. [DOI] [PubMed] [Google Scholar]
- 21.Patel J, Patel AH, McLauchlan J. The transmembrane domain of the hepatitis C virus E2 glycoprotein is required for correct folding of the E1 glycoprotein and native complex formation. Virology. 2001;279:58–68. doi: 10.1006/viro.2000.0693. [DOI] [PubMed] [Google Scholar]
- 22.Owsianka AM, et al. Identification of conserved residues in the E2 envelope glycoprotein of the hepatitis C virus that are critical for CD81 binding. J Virol. 2006;80:8695–8704. doi: 10.1128/JVI.00271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu M, et al. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci USA. 2003;100:7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med. 2003;197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lavillette D, et al. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology. 2005;41:265–274. doi: 10.1002/hep.20542. [DOI] [PubMed] [Google Scholar]
- 26.Wakita T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong J, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci USA. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lindenbach BD, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 29.Gottwein JM, et al. Robust hepatitis C genotype 3a cell culture releasing adapted intergenotypic 3a/2a (S52/JFH1) viruses. Gastroenterology. 2007;133:1614–1626. doi: 10.1053/j.gastro.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 30.Scheel TK, et al. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc Natl Acad Sci USA. 2008;105:997–1002. doi: 10.1073/pnas.0711044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jensen TB, et al. Highly efficient JFH1-based cell-culture system for hepatitis C virus genotype 5a: Failure of homologous neutralizing-antibody treatment to control infection. J Infect Dis. 2008;198:1756–1765. doi: 10.1086/593021. [DOI] [PubMed] [Google Scholar]
- 32.Gottwein JM, et al. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: Role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology. 2009;49:364–377. doi: 10.1002/hep.22673. [DOI] [PubMed] [Google Scholar]
- 33.Dorner M, et al. A genetically humanized mouse model for hepatitis C virus infection. Nature. 2011;474:208–211. doi: 10.1038/nature10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walker LM, et al. Protocol G Principal Investigators Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326:285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cocquerel L, et al. Recognition of native hepatitis C virus E1E2 heterodimers by a human monoclonal antibody. J Virol. 2003;77:1604–1609. doi: 10.1128/JVI.77.2.1604-1609.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwarz AK, et al. Hepatoma cell density promotes claudin-1 and scavenger receptor BI expression and hepatitis C virus internalization. J Virol. 2009;83:12407–12414. doi: 10.1128/JVI.01552-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rychlowska M, et al. Comprehensive linker-scanning mutagenesis of the hepatitis C virus E1 and E2 envelope glycoproteins reveals new structure–function relationships. J Gen Virol. 2011;92:2249–2261. doi: 10.1099/vir.0.034314-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Albecka A, et al. Identification of new functional regions in hepatitis C virus envelope glycoprotein E2. J Virol. 2011;85:1777–1792. doi: 10.1128/JVI.02170-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song L, et al. Broadly neutralizing anti-HIV-1 antibodies disrupt a hinge-related function of gp41 at the membrane interface. Proc Natl Acad Sci USA. 2009;106:9057–9062. doi: 10.1073/pnas.0901474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sabo MC, et al. Hepatitis C virus epitope exposure and neutralization by antibodies is affected by time and temperature. Virology. 2012;422:174–184. doi: 10.1016/j.virol.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Law M, Hangartner L. Antibodies against viruses: Passive and active immunization. Curr Opin Immunol. 2008;20:486–492. doi: 10.1016/j.coi.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dowd KA, Pierson TC. Antibody-mediated neutralization of flaviviruses: A reductionist view. Virology. 2011;411:306–315. doi: 10.1016/j.virol.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whidby J, et al. Blocking hepatitis C virus infection with recombinant form of envelope protein 2 ectodomain. J Virol. 2009;83:11078–11089. doi: 10.1128/JVI.00800-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krey T, et al. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 2010;6:e1000762. doi: 10.1371/journal.ppat.1000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.ter Meulen J, et al. Human monoclonal antibody combination against SARS coronavirus: Synergy and coverage of escape mutants. PLoS Med. 2006;3:e237. doi: 10.1371/journal.pmed.0030237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58:621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 47.Dubuisson J, et al. Formation and intracellular localization of hepatitis C virus envelope glycoprotein complexes expressed by recombinant Vaccinia and Sindbis viruses. J Virol. 1994;68:6147–6160. doi: 10.1128/jvi.68.10.6147-6160.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Safran M, et al. Mouse reporter strain for noninvasive bioluminescent imaging of cells that have undergone Cre-mediated recombination. Mol Imaging. 2003;2:297–302. doi: 10.1162/15353500200303154. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.