

Abstract
Background
Carotid intima-media thickness (CIMT) is a subclinical measure for atherosclerosis. Previously, we have mapped quantitative trait loci (QTLs) for CIMT to chromosomes 7p (MLOD=3.1) and to 14q (MLOD=2.3). We sought to identify the underlying genetic variants within those QTLs,
Methods and Results
Using the 100 extended Dominican Republican (DR) families (N=1312) used in the original linkage study, we fine mapped the QTLs with 2031 tagging single nucleotide polymorphisms (SNPs). Promising SNPs in the family dataset were examined in an independent population-based subcohort comprised of DR individuals (N=553) from the Northern Manhattan Study. Among the families, evidence for association (P<0.001) was found in multiple genes (ANLN, AOAH, FOXN3, CCDC88C, PRiMA1, and an intergenic SNP rs1667498), with the strongest association at PRiMA1 (P=0.00007, corrected P=0.047). Additional analyses revealed that the association at these loci, except PRiMA1, was highly significant (P= 0.00004~0.00092) in families with evidence for linkage but not in the rest of families (P=0.13~0.80) and the population-based cohort, suggesting the genetic effects at these SNPs are limited to a subgroup of families. In contrast, the association at PRiMA1 was significant in both families with and without evidence for linkage (P=0.002 and 0.019, respectively), and the population-based subcohort (P=0.047), supporting a robust association.
Conclusions
We identified several candidate genes for CIMT in DR families. Some of the genes manifest genetic effects within a specific subgroup and others were generalized to all groups. Future studies are needed to further evaluate the contribution of these genes to atherosclerosis.
Introduction
As with other common complex diseases, the pathogenesis of stroke and myocardial infarction (MI) is heterogeneous, involving multiple genes, environmental exposures, and their interactions. Carotid intima-media thickness (IMT) is a highly reproducible and quantitative phenotype that is widely accepted as a subclinical marker of vascular disease1–3, with slightly greater relative risk for stroke than for MI4. Given the complexity of the pathogenesis of stroke and MI, evaluation of carotid IMT rather than the ultimate manifestation of diseases may reduce etiologic heterogeneity and increase statistical power in genetic studies.
We and several groups reported the heritability of carotid IMT to range from 0.30 to 0.60 in various populations, supporting a strong genetic contribution to carotid IMT5–8. In an attempt to map the genetic determinants of carotid IMT, we have completed a genome-wide linkage analysis in our Family Study of Stroke Risk and Carotid Atherosclerosis. Using 100 extended Dominican Republican (DR) families with an average of 13 individuals with genotype and phenotype data per family, we have previously reported strong evidence for linkage on chromosomes 7p for carotid bifurcation IMT (MLOD=3.1) and chromosome 14q for total carotid IMT (MLOD=2.3)9. Two additional genome-wide linkage studies on carotid IMT have been reported10, 11. In the Framingham Heart Study Offspring cohort10, suggestive evidence for linkage (two point LOD=1.75) has also been reported at the same peak marker D14S606 as in our study. The replication of evidence for linkage greatly strengthens our linkage findings.
There have been other attempts to elucidate the genetic basis for carotid IMT using candidate gene and genome-wide association study (GWAS) designs12. While there is some consensus on the associations with apolipoprotein E (APOE), angiotensin converting enzyme (ACE), and interleukin-6 (IL-6) genes, other findings have been conflicting13. In the GWAS on carotid IMT in the Framingham Heart Study14, no SNP met criteria for genome-wide significance. In a recent GWAS meta-analysis organized by the CHARGE consortium, only one SNP rs11781551 near ZHX2 gene reached genome-wide significance in the discovery stage. After conducting a second meta-analysis, two additional SNPs reached genome-wide significance in the combined meta-analysis15. These studies suggest that the association of carotid IMT and any specific common genetic variant is likely to be small and highlight the challenges to elucidate the genetic architecture of this important precursor phenotype for cardiovascular diseases. Indeed, although GWAS has led to interesting findings in a number of complex traits, the associated SNPs only account for a small fraction of the phenotypic variations, which has been referred as the “missing heritability”. Rare variants that are underrepresented in the GWAS are believed to account for some of the “missing heritability”. Family-based studies offer unique advantages in identifying rare variants using a linkage and association combined approach. Herein, we sought to fine map the quantitative trait loci (QTLs) identified in our original linkage study using a two-stage design. First, we surveyed the QTLs using high-density tagSNPs in the 100 extended DR families. Then, we evaluated the associations detected in the family data set in an independent population-based DR sample.
Materials and Methods
Subjects
The design of the family study has been described in detail previously16. In brief, the family study was derived from the Northern Manhattan Study (NOMAS)17. Probands were selected from Caribbean Hispanic members of NOMAS with high risk for cardiovascular disease. Families were enrolled if the proband was able to obtain the family members’ permission for the research staff to contact them, and had at least 3 first-degree relatives able to participate. The probands were initially identified in Northern Manhattan and family members were enrolled in New York at Columbia University as well as in the Dominican Republic at the Clinicas Corazones Unidos in Santo Domingo. Demographic, socioeconomic and risk factor data were collected through direct interview18. All subjects provided informed consent and the study was approved by the Institutional Review Boards of Columbia University, University of Miami, the National Bioethics Committee, and the Independent Ethics Committee of Instituto Oncologico Regional del Cibao in the DR. The subset of DR subjects in the NOMAS cohort who had carotid measurements and were not members of the Family Study served as an independent validation sample.
Carotid Measurements
All family members and NOMAS subjects had high resolution B-mode ultrasound measurement of carotid IMT. Carotid ultrasound was performed according to the standard scanning and reading protocols by a trained and certified sonologist as detailed previously19. In brief, the carotid IMT protocols yield measurements of the distance between lumen-intima and media-adventitia ultrasound echoes, from which the IMT and arterial diameter are derived for the 3 carotid segments. The carotid segments were defined as follows: (1) the near and the far wall of the segment extending from 10 to 20 mm proximal to the tip of the flow divider into the common carotid artery (CCA); (2) the near and the far wall of the carotid bifurcation beginning at the tip of the flow divider and extending 10 mm proximal to the flow divider tip (BIF); and (3) the near and the far wall of the proximal 10 mm of the internal carotid artery (ICA).
IMT measurements were performed outside the areas of plaque as recommended by consensus documents20. IMT was measured using an automated computerized edge tracking software M’Ath (Intelligence in Medical Technologies, Inc., Paris, France) from the recorded ultrasound clips which improves precision and reduces variance of the measurements.17 Total IMT is calculated as a mean composite measure of the means of the near and the far wall IMT of all carotid sites (IMTx), and of the maximum of the near and the far wall IMT of all carotid sites (IMTm). We also examined carotid segment-specific IMT phenotypes in the carotid bifurcation (BIF), the common carotid artery (CCA) and the internal carotid artery (ICA) (BIFx, BIFm, CCAx, CCAm, ICAx, ICAm). Our carotid IMT reliability statistics demonstrated excellent results.17 Among 88 subjects, inter-reader reliability between 2 readers was demonstrated with a mean absolute difference in IMT of 0.11±0.09 mm, variation coefficient 5.5%, correlation coefficient 0.87, and the percent error 6.7%. Intra-reader mean absolute IMT difference was 0.07±0.04 mm, variation coefficient 5.4%, correlation coefficient 0.94, and the percent error 5.6%.
SNP Selection and Genotyping
We have completed a GWAS in a subcohort of NOMAS to study subclinical brain phenotypes using the Human SNP Array 6.0 chip (AffyMetrix). Among the genotyped individuals, about 400 samples were DR subjects, thus providing a unique database on SNP allele frequency and linkage disequilibrium (LD) structure in the DR population. Using this database, we selected SNPs with minor allele frequency greater than 0.05 and pairwise LD (r2) less than 0.8 to cover the one-LOD-unit down regions on chromosomes 7p and 14q. Assays for selected SNPs were manufactured on Infinium iSelectHD Custom BeadChips (Illumina Inc). Genotyping results were analyzed using the Illumina GenomeStudio® Genotyping module. To ensure high genotyping quality, samples with a GenTrain score less than 0.15 were regarded as poorly performing samples and dropped from further analysis. Then the genotype calling algorithm was re-run based on high quality DNA samples only. Genotypes on SNPs with a GenCall score greater than 0.8 were automatically called by the GenomeStudio® Genotyping module. SNPs with lower GenCall scores were manually called. Upon completion of the association analysis in the family data set, SNPs that were nominally significant (P<0.05) in more than one statistical model (see statistics section) or were nominally significant in at least one statistical model and reside in an exon were prioritized for follow-up in the independent population-based NOMAS subcohort of DR individuals. This set of SNPs were genotyped on OpenArray (Applied Biosystems) using Taqman® Allelic Discrimination assays. Genotyping results were analyzed using Taqman Genotyper software (ver.1.0.1).
All SNP genotyping was performed in the Genotyping Core of the Hussman Institute for Human Genomics following manufacturer’s instruction. SNPs with less than 95% call rate and DNA samples with genotyping efficiency <95% were removed from the statistical analysis in both the family data set and population-based NOMAS subcohort. No SNP was found to be severely deviated from Hardy-Weinberg equilibrium (p >10−3).
Statistics
In the family data set, SNP genotypes were used to verify and adjust family structure using the program PREST21 and GRR22. Mendelian error checking was performed on the final family structure using PLATO23.
In SOLAR24, an initial polygenic model for IMT at each carotid segment was used to identify significant covariates as described before9. Covariates that were tested included age, sex, waist hip ratio (WHR), body mass index (BMI), hypertension, hypercholesterolemia, diabetes, and smoking (pack years). Only significant covariates (P<0.10) were adjusted in all final analyses. The IMT measurements were nature-log transformed to ensure a normal distribution in the overall data set as well as in the subsets of families (Kurtosis =-0.39 ~0.17; Skewness=0.10~0.31).
For the association analysis in the family data set, the Quantitative Transmission-Disequilibrium test (QTDT) was used as the primary analytical tool because it is designed to evaluate association in the presence of linkage without inflating type I error. In addition, QTDT is robust against population stratification and uses data from all available relatives in a pedigree, which makes it more powerful in analyzing data sets with missing parental data, a common problem in the study of complex traits such as carotid IMT. Three additional analytical approaches, Measured Genotype (MG), Quantitative Trait Linkage-Disequilibrium (QTLD), and family-based association (PBAT), were included to assess consistency for the top associated SNPs identified by QTDT. Since MG, QTLD, and PBAT are not used to determine significance in the peak-wide association mapping, we did not adjust for multiple statistical methods used. In SOLAR, the QTDT, MG, and QTLD tests were simultaneously generated using the qtld command. PBAT is a stand-alone software package for the testing of family-based association (http://www.biostat.harvard.edu/~clange/default.htm). For the association analysis in the NOMAS DR subcohort, linear regression analysis was carried out using an additive genetic model adjusting for the same covariates used in the family data set.
Stratified association analysis was performed based on family-specific LOD scores from the original linkage analysis25. Families with a positive LOD score on the trait of interest were used to construct the subset with evidence for linkage, while families with a negative LOD score on the trait of interest were used to construct the subset without evidence for linkage. Association tests were then carried out in each subset of families separately. To evaluate the contribution of associated SNPs to linakge results, linkage analysis were re-run in SOLAR including the genotype of SNPs as a covariate. We used a likelihood ratio test to determine if there was a significant decrease in the LOD score after conditioning on the genotype. This was done separately for each genotype as well as jointly for all associated SNPs on each chromosome.
Although we have selected tagSNPs to perform the peak-wide association mapping, there is still considerable LD between tested SNPs. To correct for the multiple testing in the presence of correlation between these SNPs, we used the “SNP effnum” command implemented in the SOLAR to estimate the effective number of independent tests based on the pairwise genotypic correlations between tested SNPs in our dataset. The peak-wide significance threshold was defined as 0.05/effective number of tests under each linkage peak.
Quanto was used estimate statistical power for the NOMAS cohort. We assumed independence of individuals, minor allele frequency of 0.20, an additive genetic effect, a two-sided alpha of 0.05, and a population mean (SD) of −0.096 (0.091) and −0.052 (0.106) for the natural logarithm of total IMT and BIF IMT, respectively (estimated in our data set). With our NOMAS cohort, we had over 80% power to detect an effect size of 0.0191 for IMT and 0.0222 for BIF (corresponding to an approximate change of 0.0175 and 0.0213 mm from the mean of IMT and BIF, respectively).
Results
Association mapping in the overall family data set
The one-LOD-unit-down region extends from 29 megabase (Mb) to 43 Mb on chromosome 7, and 77 Mb to 94 Mb on chromosome 14. To survey these linkage regions, 2211 highly informative tagSNPs were selected for genotyping. 174 SNPs failed manufacturing and 6 SNPs did not pass genotyping quality controls. As a result, 2031 SNPs (846 on chromosome 7 and 1185 on chromosome 14) were available for the family-based association analysis (Figure). The number of effective independent tests is 426 and 542 for chromosomes 7 and 14, respectively. Overall, 1312 subjects from the 1o0 DR families had complete genotype and phenotype data for analysis.
Figure 1.
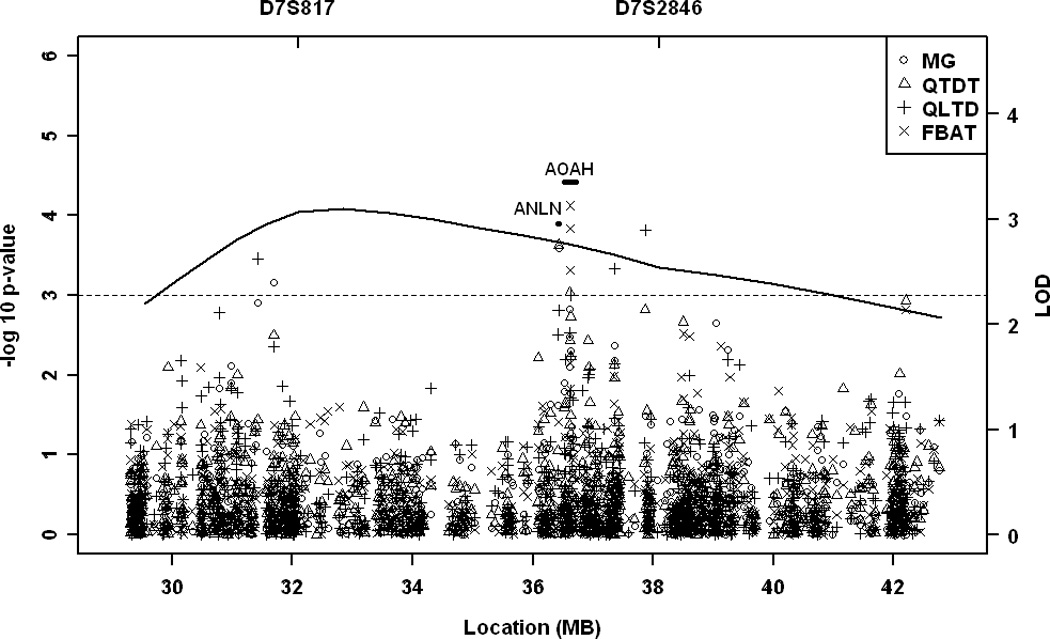
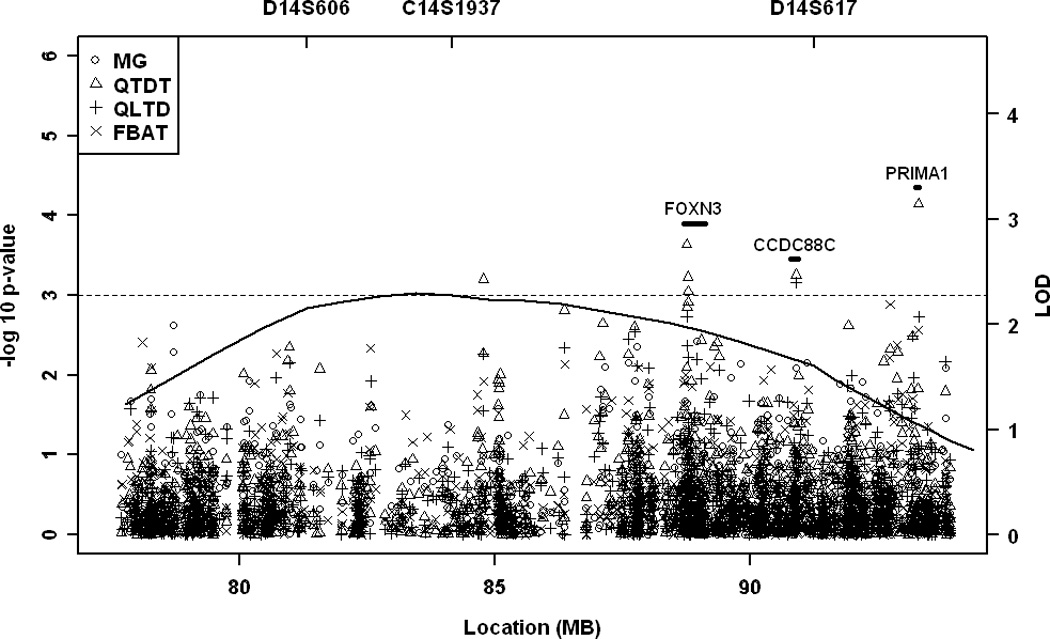
Maximum multipoint linkage and fine mapping association results for maximum BIF IMT on chromosome 7p (A) and for maximum total IMT on chromosome 14q (B). Multipoint LOD score curve over the one-LOD unit down region is depicted as a solid line. Each symbol represents an association test the Measured Genotype (MG), Quantitative Transmission-Disequilibrium (QTDT), and Quantitative Trait Linkage-Disequilibrium (QTLD), and Family-based association test (FBAT).
Since genetic influences on carotid IMT may vary at different anatomical sites, we examined maximum and mean IMT at CCA, ICA, BIF, as well as the total IMT (a composite measure of IMT at all three sites) in our previous linkage study. The strongest evidence for linkage was found on chromosome 7p for the maximum BIF IMT, and on chromosome 14q for the maximum total IMT25. To fine map these QTLs, we first evaluated associations within each QTL with the corresponding carotid IMT measurement that provided the strongest evidence for linkage (Figure). Among the clinically relevant covariants, age, sex, age2, BMI, WHR, and pack years were significantly associated with both BIF IMT and total IMT. In addition, an age-sex interaction significantly contributed to BIF IMT variations in our data set. All the significant covariants were adjusted in the family-based association tests.
Table 1 displays the top associated SNPs (P<0.001, QTDT) in the fine mapping study. On chromosome 7, SNPs in anillin (ANLN) and acyloxyacyl hydrolase (AOAH) are associated with maximum BIF IMT; on chromosome 14, SNPs in forkhead box N3 (FOXN3), coiled-coil domain containing 88C (CCDC88C), proline-rich membrane anchor (PRiMA1), and an inter-genic region are associated with maximum total IMT. The strongest association was found at rs7152362 in PRiMA1 (P=0.00007), which remained significant after correcting for the multiple tests performed on chromosome 14 (corrected P=0.047). Three additional analytical approaches, QTLD, MG, and FBAT, supported associations at those genes (Table 1).
Table 1.
Top SNP associations with BIF IMT on chromosome 7p and total IMT on 14q.
| SNP | Gene | SNP Function |
P values | Minor Allele |
Allele Frequency |
|||
|---|---|---|---|---|---|---|---|---|
| QTDT | QLTD | MG | FBAT | |||||
| Association with BIF IMT on chromosome 7p | ||||||||
| rs3815483 | ANLN | intronic | 0.00023 | 0.00155 | 0.00026 | 0.03865 | G | 0.069 |
| rs2001600 | AOAH | intronic | 0.00092 | 0.00291 | 0.00149 | 0.00008 | G | 0.410 |
| Association with total IMT on chromosome 14q | ||||||||
| rs1667498 | intergenic | 0.00062 | 0.00550 | 0.10298 | 0.09966 | T | 0.081 | |
| rs10144627 | FOXN3 | intronic | 0.00023 | 0.00183 | 0.02274 | 0.04671 | T | 0.442 |
| rs10151997 | FOXN3 | intronic | 0.00059 | 0.04717 | 0.03241 | 0.45169 | T | 0.053 |
| rs10151430 | FOXN3 | intronic | 0.00089 | 0.06167 | 0.06992 | 0.22593 | A | 0.038 |
| rs17803546 | CCDC88C | intronic | 0.00056 | 0.00069 | 0.00811 | 0.08898 | A | 0.218 |
| rs7152362 | PRIMA1 | intronic | 0.00007 | 0.00183 | 0.09479 | 0.00273 | T | 0.441 |
Family-based association tests were performed in the 100 DR families. SNPs with a p value (generated by the QTDT test) less than 0.001 are displayed. The chromosome 7 SNPs were tested for association with IMT at bifurcation and the chromosome 14 SNPs were tested for association with total IMT. P values from the QTDT, QLTD, MG and FBAT are displayed in the table.
Association tests with IMT in other carotid segments
To explore whether these top SNPs display a site-specific association with carotid IMT, we then evaluated the associations between these top SNPs and other carotid IMT measurements (i.e. total IMT, CCA, and ICA for chromosome 7 SNPs, and BIF, CCA, and ICA for chromosome 14 SNPs). Since the maximum and mean IMT at each site are highly correlated with each other (Pearson correlation coefficient >0.90), we focused on the maximum IMT in our analysis. On chromosome 7p, the top SNPs for BIF IMT (P=0.0002 for ANLN and 0.0009 for AOAH) were poorly associated with IMT in CCA and ICA (P=0.03~0. 22, Table 2). These SNPs were modestly associated with total IMT (P=0.003 for ANLN and 0.004 for AOAH), which is probably driven by the BIF IMT component. On chromosome 14q, the top SNPs for total IMT (P=0.0007~0.0009) were modestly associated with the carotid IMT measurement at all three carotid segments that make up the composite measure of total IMT: BIF (P=0.001~0.03), CCA (P=0.002~0.04), ICA (P=0.002~0.1).
Table 2.
Association between SNPs and carotid IMT at different anatomical sites
| SNP | Gene | P values | |||
|---|---|---|---|---|---|
| BIF | CCA | ICA | Total | ||
| Chromosome 7 | |||||
| rs3815483 | ANLN | 0.00023 | 0.08852 | 0.11102 | 0.00360 |
| rs2001600 | AOAH | 0.00092 | 0.02931 | 0.22562 | 0.00422 |
| Chromosome 14 | |||||
| rs1667498 | 0 | 0.00679 | 0.04423 | 0.00627 | 0.00062 |
| rs10144627 | FOXN3 | 0.00131 | 0.00864 | 0.00088 | 0.00023 |
| rs10151997 | FOXN3 | 0.00160 | 0.00638 | 0.03823 | 0.00059 |
| rs10151430 | FOXN3 | 0.00547 | 0.02747 | 0.00764 | 0.00089 |
| rs17803546 | CCDC88C | 0.02980 | 0.00369 | 0.00216 | 0.00056 |
| rs7152362 | PRIMA1 | 0.00262 | 0.00187 | 0.09807 | 0.00007 |
Each SNP was tested for association with maximum IMT at bifurcation (BIF), common carotid artery (CCA), internal carotid artery (ICA), and a mean composite measure of the IMT of all three carotid sites (Total). P values from the QTDT test in the 100 DR families are reported in the table.
Association tests in families with and without evidence for linkage
As an intermediate phenotype, carotid IMT is expected to have less genetic heterogeneity in comparison to the ultimate manifestation of the disease such as stroke or myocardial infarction. There is, however, still substantial genetic heterogeneity in carotid IMT. To further reduce genetic heterogeneity, we performed a subset analysis using families with and without evidence for linkage based on family-specific LOD scores at each QTL. For the BIF IMT QTL on chromosome 7p, there were 733 individuals from 53 families with positive LOD scores, and 579 individuals from 47 families with negative LOD scores. For the total IMT QTL on chromosom 14q, there were 739 individuals from 51 families with positive LOD scores, and 570 individuals from 49 families with negative LOD scores. In the subset of families with evidence for linkage, despite a smaller sample size (about half of the overall analysis), the association to the top SNPs (except for the PRiMA1/rs7152362 and FOXN3/rs10144627) became more significant (P=0.00004 ~0.0009) than in the overall analysis. In the subset of families without evidence for linkage, the association to these SNPs were not significant (P=0.13~0.79) anymore (Table 3). Therefore, the evidence for association at these SNPs was mainly driven by the families with evidence for linkage, suggesting that these SNPs could account for, at least partially, the studied QTLs. In contrast, PRiMA1/rs7152362 and FOXN3/rs10144627 were significant in both subsets (P<0.05), suggesting that variants at or linked to these two SNPs may contribute to the carotid IMT variations in broader populations.
Table 3.
Subset analysis in families with and without evidence for linkage
| SNP | Gene | p-values | |
|---|---|---|---|
| LOD>0 | LOD<0 | ||
| Association with BIF IMT on chromosome 7p | |||
| rs3815483 | ANLN | 0.00024 | 0.13153 |
| rs2001600 | AOAH | 0.00017 | 0.41570 |
| Association with total IMT on chromosome 14q | |||
| rs1667498 | 0 | 0.00034 | 0.34681 |
| rs10144627 | FOXN3 | 0.01615 | 0.00266 |
| rs10151997 | FOXN3 | 0.00010 | 0.43651 |
| rs10151430 | FOXN3 | 0.00092 | 0.17991 |
| rs17803546 | CCDC88C | 0.00004 | 0.79038 |
| rs7152362 | PRIMA1 | 0.00169 | 0.01878 |
Each SNP was tested for association with the corresponding trait in a subset of DR families with (LOD>0) or without (LOD<0) evidence for linkage. P values from the QTDT test are reported in the table.
To determine if these SNPs contribute to the linkage signal, we performed a linkage analysis including each significant SNP listed in Table 1 as covariant. The SNPs on chr 7 indeed contribute to the linkage results significantly as evidenced by a significant decrease of LOD score from 3.34 to 2.35, 2.92 and 1.89, for rs3815483, rs2001600, and the two SNPs together, respectively (P<0.01 for each SNP individually, P<0.0005 for the two SNPs together, Supplementary Table). The chr 14 SNPs have a smaller contribution to the linkage signal (LOD decreased from 2.00 to 1.09, P=0.017, after adjusting for all chr 14 SNPs, Supplementary Table).
Validation in the NOMAS DR subcohort
Based on the strength of association in the family data set and the locations of SNPs, we prioritized 50 SNPs for further evaluation in the NOMAS DR subcohort (Supplementary Table). To construct an independent validation data set, probands of the family study were excluded from the NOMAS DR subcohort. As a result, 553 NOMAS DR subjects (213 males and 340 females) with carotid IMT measurements were included in the validation data set. Supplementary Table displays results of the 50 SNPs in both the family data set and the NOMAS DR cohort, including minor allele frequencies, regression coefficients for effect size and p values. Among the top associated SNPs listed in Table 1, only PRiMA1/rs7152362 was significant in the validation data set (P=0.047, Supplementary Table). Furthermore, the effect size was in the same trend in both the discovery family data set and the validation cohort data set (β= −0.0197 and −0.0098, respectively). Among those SNPs with a nominal p value of less than 0.05 in the family data set, only GLi3/rs3801216 was significant in the validation data set (P=0.019, Supplementary Table). The effect size, however, was opposite between the two data sets (β=0.0237 and-0.0146, respectively).
Discussion
In previous studies, we have established high heritability of carotid IMT. Using our collection of extended Dominican families, we have also mapped QTLs for carotid IMT measurements. In the current study, we have extended these observations to the identification of susceptibility genes within the prominent QTLs. Our study design includes a battery of family-based association tests and an independent DR subset from a population-based cohort for follow up. Using the two-stage design and high-density tagSNPs, we identified several potential novel candidate genes for carotid IMT in Dominicans, an underrepresented population in genetic studies of cardiovascular diseases.
On chromosome 7, strong evidence for association with BIF IMT was found to SNPs in ANLN and AOAH in the family data set, although they did not meet the peak-wide significance. The two genes are located next to each other with no LD between the significant SNPs in these two genes (Figure and Supplementary Figure), suggesting independent associations. In AOAH, another SNP that is not in high LD with the top SNP (r2 <0.01) was also associated with BIF IMT (P=0.006, QTDT) (Supplementary Table and Supplementary Figure), further supporting a role of this gene in carotid atherogenesis. AOAH encodes acyloxyacyl hydroxylase, a lipase that deactivates lipopolysaccharides (LPS) on cell walls of gram-negative bacteria. Upon exposure to LPS, B-cells are stimulated to proliferate and produce antibody through Toll-like receptor signaling. It has been shown that AOAH modulates this Toll-like receptor mediated response to LPS in vivo26. Given that inflammation is a major cellular process underlying atherogenesis, AOAH is a biologically plausible susceptibility gene for carotid IMT. Interestingly, the QTL on chromosome 7p has been linked to asthma, serum total IgE, osteoarthritis, and Hereditary hemorrhagic telangiectasia (HHT)27–29. One molecular mechanism shared between these traits and carotid IMT is inflammation and AOAH could be an underlying susceptibility gene for these traits as well. Indeed, SNPs in AOAH have been reported to be associated with serum total IgE30. Recently, higher serum IgE levels were observed in patients with MI in comparison to patients without MI or with stable angina. Subsequent animal and cell culture studies supported a role of IgE in atherogenesis, at least partially, through inducing cytokine expression and apoptosis in marcrophage31. Our stratification and validation analyses suggest that the association between AOAH variants and BIF IMT is restricted to a specific subset of subjects. Further studies are needed to elucidate the environmental exposure and/or genetic makeup required for the AOAH variants to manifest their effects. Within the chromosome 7 QTL, we also found evidence for association at the GLi3 gene in both the family and the population-based data sets but with different directions for association, which could suggest a false positive finding.
The most interesting finding is within our chromosome 14 QTL with the peak marker of D14S606. Suggestive evidence for linkage (two point LOD=1.75) with carotid IMT at D14S606 has been reported in the Framingham Heart Study Offspring cohort10. This marker has also been associated with arterial stiffness in African Americans in the Hypertension Genetic Epidemiology Network study32. The replication of linkage evidence at D14S606 in different ethnic groups not only strengthens the finding but also suggests that the genetic variants within the chromosome 14 QTL have a more generalizable effect on atherosclerosis. Within the QTL, the strongest association was found to PRiMA1/rs7152362, which remained significant after adjusting for multiple testing. Furthermore, the association was significant in all the family subsets, across all carotid IMT segments, and confirmed in the population-based NOMAS DR subcohort. The consistent association in multiple data sets supports a robust effect of PRiMA1 variants on carotid IMT. The molecular mechanisms whereby PRiMA1 confers genetic influence on carotid IMT remain elusive. Since the gene was initially identified as the anchor for the acetylcholinesterase33, an enzyme hydrolyzing the neurotransmitter acetylcholine, most of the studies on PRIMA1 and its protein product have been conducted in the nervous system34–36. The expression of PRIMA1 has been found in both neurons and muscle cells surrounding the basal lamina of the neuromuscular junction where acetylcholinesterase is anchored. It is possible that PRIMA1 contributes to carotid IMT by regulating the cellular activities of smooth muscle cells in vessels but this hypothesis needs further investigation.
Genetic heterogeneity represents a daunting challenge in the study of complex traits as it could wash out a true signal that may only manifest in a subset of subjects. In our original linkage analysis, not all families showed evidence for linkage within the QTLs on chromosomes 7p and 14q. This indicates genetic heterogeneity among families; some families may carry genetic variants outside these QTLs that contribute to the inter-individual IMT variations. Consistent with this notion, our subset analysis revealed that some SNPs, such as the ones in AOAH, ANLN, and CCDC88C, are only significant in the families with evidence for linage but not in the families without evidence for linkage, supporting that other variants are yet to be identified for the families that are not linked to the QTLs. These “private associations" could be related to the differential presence pattern of these variants in different families, or the specific environmental exposures of a subgroup of families that are required for the variants to display an association with IMT, or the combination of both. Our data provided an example that subset analysis is useful in overcoming genetic heterogeneity and facilitating discovery of private variants.
In our previous candidate gene study, we showed that different sets of genes influence IMT indifferent carotid segments, e.g., the common carotid artery (CCA), the internal carotid artery (ICA), and the carotid bifurcation (BIF)37. Consistent with that observation, in the current study genetic variants within the chromosome 7p QTL are strongly associated with IMT in BIF but to a lesser degree in CCA or ICA. Furthermore, the association of IMT with vascular risk factors varies by carotid segment. In the NOMAS, we have reported that sex is a strong risk factor for BIF and ICA IMT (β=2.8 and 2.3 respectively, P<0.01 for being male) but not for CCA IMT (β=0.7, not significant). In contrast, hypertension is strongly associated with IMT in CCA (β=2.9, P<0.01) but not BIF or ICA (β=0.4 and −0.7, respectively, not significant)37. Therefore, separate analyses of IMT in different carotid segments may increase the homogeneity of the IMT phenotype and enhance analytical power. Other genetic studies on carotid IMT have mainly focused on CCA while ours provides a comprehensive analysis on IMT in all carotid segments.
We acknowledge several limitations in the current study. First, we used a tagSNP approach to conduct the fine mapping study. Such approach is cost-efficient for screening large numbers of genes and individuals by capturing the majority of common variants in the population. Additional follow up studies, however, are often required to identify the functional variants responsible for the detected association. In addition, it is likely that the inter-individual variations of carotid IMT result from many rare variants collectively, which are not covered by the tagSNP approach. The recent advance of next-generation sequencing technologies will exhaustively catalogue sequence variants and facilitate the evaluation of rare variants in future studies. Second, to reduce heterogeneity and increase our power to map the QTLs, we restricted our fine mapping and validation efforts to Dominican subjects only. As a result, our findings may not be directly generalized to other populations. However, our study provides the much needed knowledge on the rapidly growing Hispanic population given that the majority of the genetic studies on carotid IMT have primarily focused on non-Hispanic white populations. Finally, despite the successful validation of the association at PRiMA1, the statistical power of our validation data set is not optimal given that the effect size of SNP for carotid IMT is less than 0.02 in general, as seen in our data set (Supplementary Table) and in the CHARGE consortium15. Therefore, we cannot exclude the possibility of other loci under chromosome 7p and 14q QTL contributing to carotid IMT.
In summary, using a unique collection of extended Dominican families, we have mapped novel candidate genes for carotid IMT. Some of the genes manifest genetic effects within a specific subset of families, such as the AOAH gene, and others demonstrate generalized association, such as the PRiMA1 gene. Further studies are required to validate these associations and functionally characterize these genes to examine their role in carotid atherosclerosis.
Supplementary Material
Acknowledgements
The authors are grateful to all the families and research staff who participated in the study. We thank Drs. Katihurca Almonte and Carlos Garcia Ligthgow for their support in the Dominican Republic. We also thank Dr. Luis Cuello Mainardi, Director of the Clinicas Corazones Unidos, where subjects were enrolled in the DR, and Drs. Rafael Lantigua and Andres Peralta for their guidance with regulatory environment in the DR.
This research was supported by grants from the National Institute of Neurological Disorders and Stroke R01 NS40807, R37 NS29993 and RO1 NS047655.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: The authors have no conflicts of interest to disclose regarding this work.
References
- 1.Chambless LE, Folsom AR, Clegg LX, Sharrett AR, Shahar E, Nieto FJ, Rosamond WD, Evans G. Carotid wall thickness is predictive of incident clinical stroke: the Atherosclerosis Risk in Communities (ARIC) study. Am J Epidemiol. 2000;151:478–487. doi: 10.1093/oxfordjournals.aje.a010233. [DOI] [PubMed] [Google Scholar]
- 2.Touboul PJ, Labreuche J, Vicaut E, Amarenco P. GENIC Investigators. Carotid intima-media thickness, plaques, and Framingham risk score as independent determinants of stroke risk. Stroke. 2005;36:1741–1745. doi: 10.1161/01.STR.0000174490.23495.57. [DOI] [PubMed] [Google Scholar]
- 3.O'Leary DH, Polak JF, Kronmal RA, Manolio TA, Burke GL, Wolfson SK., Jr Carotid-artery intima and media thickness as a risk factor for myocardial infarction and stroke in older adults. Cardiovascular Health Study Collaborative Research Group. N Engl J Med. 1999;340:14–22. doi: 10.1056/NEJM199901073400103. [DOI] [PubMed] [Google Scholar]
- 4.Lorenz MW, Markus HS, Bots ML, Rosvall M, Sitzer M. Prediction of clinical cardiovascular events with carotid intima-media thickness: a systematic review and meta-analysis. Circulation. 2007;115:459–467. doi: 10.1161/CIRCULATIONAHA.106.628875. [DOI] [PubMed] [Google Scholar]
- 5.Lange LA, Bowden DW, Langefeld CD, Wagenknecht LE, Carr JJ, Rich SS, Riley WA, Freedman BI. Heritability of carotid artery intima-medial thickness in type 2 diabetes. Stroke. 2002;33:1876–1881. doi: 10.1161/01.str.0000019909.71547.aa. [DOI] [PubMed] [Google Scholar]
- 6.Xiang AH, Azen SP, Buchanan TA, Raffel LJ, Tan S, Cheng LS, Diaz J, Toscano E, Quinonnes M, Liu CR, Liu CH, Castellani LW, Hsueh WA, Rotter JI, Hodis HN. Heritability of subclinical atherosclerosis in Latino families ascertained through a hypertensive parent. Arterioscler Thromb Vasc Biol. 2002;22:843–848. doi: 10.1161/01.atv.0000015329.15481.e8. [DOI] [PubMed] [Google Scholar]
- 7.Fox CS, Polak JF, Chazaro I, Cupples A, Wolf PA, D'Agostino RA, O'Donnell CJ Framingham Heart Study. Genetic and environmental contributions to atherosclerosis phenotypes in men and women: heritability of carotid intima-media thickness in the Framingham Heart Study. Stroke. 2003;34:397–401. doi: 10.1161/01.str.0000048214.56981.6f. [DOI] [PubMed] [Google Scholar]
- 8.Juo SH, Lin HF, Rundek T, Sabala EA, Boden-Albala B, Park N, Lan MY, Sacco RL. Genetic and environmental contributions to carotid intima-media thickness and obesity phenotypes in the Northern Manhattan Family Study. Stroke. 2004;35:2243–2247. doi: 10.1161/01.STR.0000142132.20442.d8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sacco RL, Blanton SH, Slifer S, Beecham A, Glover K, Gardener H, Wang L, Sabala E, Juo SH, Rundek T. Heritability and linkage analysis for carotid intima-media thickness: the family study of stroke risk and carotid atherosclerosis. Stroke. 2009;40:2307–2312. doi: 10.1161/STROKEAHA.109.554121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fox CS, Cupples LA, Chazaro I, Polak JF, Wolf PA, D'Agostino RB, Ordovas JM, O'Donnell CJ. Genomewide Linkage Analysis for Internal Carotid Artery Intimal Medial Thickness: Evidence for Linkage to Chromosome 12. Am J Hum Genet. 2004;74:253–261. doi: 10.1086/381559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang D, Yang H, Quinones MJ, Bulnes-Enriquez I, Jimenez X, De La RR, Modilevsky T, Yu K, Li Y, Taylor KD, Hsueh WA, Hodis HN, Rotter JI. A genome-wide scan for carotid artery intima-media thickness: the Mexican-American Coronary Artery Disease family study. Stroke. 2005;36:540–545. doi: 10.1161/01.STR.0000155746.65185.4e. [DOI] [PubMed] [Google Scholar]
- 12.Mayosi BM, Avery PJ, Baker M, Gaukrodger N, Imrie H, Green FR, Farrall M, Watkins H, Keavney B. Genotype at the −174G/C polymorphism of the interleukin-6 gene is associated with common carotid artery intimal-medial thickness: family study and meta-analysis. Stroke. 2005;36:2215–2219. doi: 10.1161/01.STR.0000182254.47941.96. [DOI] [PubMed] [Google Scholar]
- 13.Juo SH. Genetics of carotid atherosclerosis. Front Biosci. 2009;14:4525–4534. doi: 10.2741/3545. [DOI] [PubMed] [Google Scholar]
- 14.O'Donnell CJ, Cupples LA, D'Agostino RB, Fox CS, Hoffmann U, Hwang SJ, Ingellson E, Liu C, Murabito JM, Polak JF, Wolf PA, Demissie S. Genome-wide association study for subclinical atherosclerosis in major arterial territories in the NHLBI's Framingham Heart Study. BMC Med Genet. 2007;8(Suppl 1):S4. doi: 10.1186/1471-2350-8-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bis JC, Kavousi M, Franceschini N, Isaacs A, Abecasis GR, Schminke U, Post WS, Smith AV, Cupples LA, Markus HS, Schmidt R, Huffman JE, Lehtimaki T, Baumert J, Munzel T, Heckbert SR, Dehghan A, North K, Oostra B, Bevan S, Stoegerer EM, Hayward C, Raitakari O, Meisinger C, Schillert A, Sanna S, Volzke H, Cheng YC, Thorsson B, Fox CS, Rice K, Rivadeneira F, Nambi V, Halperin E, Petrovic KE, Peltonen L, Wichmann HE, Schnabel RB, Dorr M, Parsa A, Aspelund T, Demissie S, Kathiresan S, Reilly MP, Taylor K, Uitterlinden A, Couper DJ, Sitzer M, Kahonen M, Illig T, Wild PS, Orru M, Ludemann J, Shuldiner AR, Eiriksdottir G, White CC, Rotter JI, Hofman A, Seissler J, Zeller T, Usala G, Ernst F, Launer LJ, D'Agostino RBS, O'Leary DH, Ballantyne C, Thiery J, Ziegler A, Lakatta EG, Chilukoti RK, Harris TB, Wolf PA, Psaty BM, Polak JF, Li X, Rathmann W, Uda M, Boerwinkle E, Klopp N, Schmidt H, Wilson JF, Viikari J, Koenig W, Blankenberg S, Newman AB, Witteman J, Heiss G, Duijn CV, Scuteri A, Homuth G, Mitchell BD, Gudnason V, O'Donnell CJ the CARDIoGRAM Consortium. Meta-analysis of genome-wide association studies from the CHARGE consortium identifies common variants associated with carotid intima media thickness and plaque. Nat Genet. 2011 doi: 10.1038/ng.920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sacco RL, Sabala EA, Rundek T, Juo SH, Huang JS, DiTullio M, Homma S, Almonte K, Lithgow CG, Boden-Albala B. Design of a family study among high-risk Caribbean Hispanics: the Northern Manhattan Family Study. Ethn Dis. 2007;17:351–357. [PMC free article] [PubMed] [Google Scholar]
- 17.Sacco RL, Anand K, Lee HS, Boden-Albala B, Stabler S, Allen R, Paik MC. Homocysteine and the risk of ischemic stroke in a triethnic cohort: the NOrthern MAnhattan Study. Stroke. 2004;35:2263–2269. doi: 10.1161/01.STR.0000142374.33919.92. [DOI] [PubMed] [Google Scholar]
- 18.Elkind MS, Sciacca R, Boden-Albala B, Rundek T, Paik MC, Sacco RL. Moderate alcohol consumption reduces risk of ischemic stroke: the Northern Manhattan Study. Stroke. 2006;37:13–19. doi: 10.1161/01.STR.0000195048.86810.5b. [DOI] [PubMed] [Google Scholar]
- 19.Rundek T, Elkind MS, Pittman J, Boden-Albala B, Martin S, Humphries SE, Juo SH, Sacco RL. Carotid intima-media thickness is associated with allelic variants of stromelysin-1, interleukin-6, and hepatic lipase genes: the Northern Manhattan Prospective Cohort Study. Stroke. 2002;33:1420–1423. doi: 10.1161/01.STR.0000015558.63492.B6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Touboul PJ, Hennerici MG, Meairs S, Adams H, Amarenco P, Bornstein N, Csiba L, Desvarieux M, Ebrahim S, Fatar M, Hernandez Hernandez R, Jaff M, Kownator S, Prati P, Rundek T, Sitzer M, Schminke U, Tardif JC, Taylor A, Vicaut E, Woo KS, Zannad F, Zureik M. Mannheim carotid intima-media thickness consensus (2004–2006). An update on behalf of the Advisory Board of the 3rd and 4th Watching the Risk Symposium, 13th and 15th European Stroke Conferences, Mannheim, Germany, 2004, and Brussels, Belgium, 2006. Cerebrovasc Dis. 2007;23:75–80. doi: 10.1159/000097034. [DOI] [PubMed] [Google Scholar]
- 21.Sun L, Wilder K, McPeek MS. Enhanced pedigree error detection. Hum Hered. 2002;54:99–110. doi: 10.1159/000067666. [DOI] [PubMed] [Google Scholar]
- 22.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 23.Grady BJ, Torstenson E, Dudek SM, Giles J, Sexton D, Ritchie MD. Finding unique filter sets in plato: a precursor to efficient interaction analysis in gwas data. Pac Symp Biocomput. 2010:315–326. [PMC free article] [PubMed] [Google Scholar]
- 24.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sacco RL, Blanton SH, Slifer S, Beecheam A, Glover K, Garderner H, Wang L, Sabala E, Juo SH, Rundek T. Heritability and Linkage Analysis of Carotid Intima-Media Thickness: The Family Study of STroke Risk and Carotid Atherosclerosis. Stroke. 2009;40 doi: 10.1161/STROKEAHA.109.554121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu M, Zhang M, Takashima A, Weiss J, Apicella MA, Li XH, Yuan D, Munford RS. Lipopolysaccharide deacylation by an endogenous lipase controls innate antibody responses to Gram-negative bacteria. Nat Immunol. 2005;6:989–994. doi: 10.1038/ni1246. [DOI] [PubMed] [Google Scholar]
- 27.Bayrak-Toydemir P, McDonald J, Akarsu N, Toydemir RM, Calderon F, Tuncali T, Tang W, Miller F, Mao R. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet A. 2006;140:2155–2162. doi: 10.1002/ajmg.a.31450. [DOI] [PubMed] [Google Scholar]
- 28.Laitinen T, Polvi A, Rydman P, Vendelin J, Pulkkinen V, Salmikangas P, Makela S, Rehn M, Pirskanen A, Rautanen A, Zucchelli M, Gullsten H, Leino M, Alenius H, Petays T, Haahtela T, Laitinen A, Laprise C, Hudson TJ, Laitinen LA, Kere J. Characterization of a common susceptibility locus for asthma-related traits. Science. 2004;304:300–304. doi: 10.1126/science.1090010. [DOI] [PubMed] [Google Scholar]
- 29.Hunter DJ, Demissie S, Cupples LA, Aliabadi P, Felson DT. A genome scan for joint-specific hand osteoarthritis susceptibility: The Framingham Study. Arthritis Rheum. 2004;50:2489–2496. doi: 10.1002/art.20445. [DOI] [PubMed] [Google Scholar]
- 30.Barnes KC, Grant A, Gao P, Baltadjieva D, Berg T, Chi P, Zhang S, Zambelli-Weiner A, Ehrlich E, Zardkoohi O, Brummet ME, Stockton M, Watkins T, Gao L, Gittens M, Wills-Karp M, Cheadle C, Beck LA, Beaty TH, Becker KG, Garcia JG, Mathias RA. Polymorphisms in the novel gene acyloxyacyl hydroxylase (AOAH) are associated with asthma and associated phenotypes. J Allergy Clin Immunol. 2006;118:70–77. doi: 10.1016/j.jaci.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 31.Wang J, Cheng X, Xiang MX, Alanne-Kinnunen M, Wang JA, Chen H, He A, Sun X, Lin Y, Tang TT, Tu X, Sjoberg S, Sukhova GK, Liao YH, Conrad DH, Yu L, Kawakami T, Kovanen PT, Libby P, Shi GP. IgE stimulates human and mouse arterial cell apoptosis and cytokine expression and promotes atherogenesis in Apoe−/− mice. J Clin Invest. 2011;121:3564–3577. doi: 10.1172/JCI46028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sherva R, Miller MB, Lynch AI, Devereux RB, Rao DC, Oberman A, Hopkins PN, Kitzman DW, Atwood LD, Arnett DK. A whole genome scan for pulse pressure/stroke volume ratio in African Americans: the HyperGEN study. Am J Hypertens. 2007;20:398–402. doi: 10.1016/j.amjhyper.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perrier AL, Massoulie J, Krejci E. PRiMA: the membrane anchor of acetylcholinesterase in the brain. Neuron. 2002;33:275–285. doi: 10.1016/s0896-6273(01)00584-0. [DOI] [PubMed] [Google Scholar]
- 34.Xie HQ, Choi RC, Leung KW, Siow NL, Kong LW, Lau FT, Peng HB, Tsim KW. Regulation of a transcript encoding the proline-rich membrane anchor of globular muscle acetylcholinesterase. The suppressive roles of myogenesis and innervating nerves. J Biol Chem. 2007;282:11765–11775. doi: 10.1074/jbc.M608265200. [DOI] [PubMed] [Google Scholar]
- 35.Noureddine H, Carvalho S, Schmitt C, Massoulie J, Bon S. Acetylcholinesterase associates differently with its anchoring proteins ColQ and PRiMA. J Biol Chem. 2008;283:20722–20732. doi: 10.1074/jbc.M801364200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dobbertin A, Hrabovska A, Dembele K, Camp S, Taylor P, Krejci E, Bernard V. Targeting of acetylcholinesterase in neurons in vivo: a dual processing function for the proline-rich membrane anchor subunit and the attachment domain on the catalytic subunit. J Neurosci. 2009;29:4519–4530. doi: 10.1523/JNEUROSCI.3863-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liao YC, Lin HF, Rundek T, Cheng R, Guo YC, Sacco RL, Juo SH. Segment-specific genetic effects on carotid intima-media thickness: the Northern Manhattan study. Stroke. 2008;39:3159–3165. doi: 10.1161/STROKEAHA.108.522789. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.