

Abstract
The majority of extracellular physiologic signaling molecules act by stimulating GTP-binding protein (G-protein)-coupled receptors (GPCRs). To monitor directly the formation of the active state of a prototypical GPCR, we devised a method to site specifically attach fluorescein to an endogenous cysteine (Cys-265) at the cytoplasmic end of transmembrane 6 (TM6) of the β2 adrenergic receptor (β2AR), adjacent to the G-protein-coupling domain. We demonstrate that this tag reports agonist-induced conformational changes in the receptor, with agonists causing a decline in the fluorescence intensity of fluorescein-β2AR that is proportional to the biological efficacy of the agonist. We also find that agonists alter the interaction between the fluorescein at Cys-265 and fluorescence-quenching reagents localized to different molecular environments of the receptor. These observations are consistent with a rotation and/or tilting of TM6 on agonist activation. Our studies, when compared with studies of activation in rhodopsin, indicate a general mechanism for GPCR activation; however, a notable difference is the relatively slow kinetics of the conformational changes in the β2AR, which may reflect the different energetics of activation by diffusible ligands.
Despite diverse physiologic roles, the majority of GTP-binding protein (G-protein)-coupled receptors (GPCRs) are thought to share a common activation mechanism. Briefly, agonists induce conformational changes in receptors, which then stimulate heterotrimeric G proteins. Activated G proteins influence cellular physiology by modulating specific effector enzymes and ion channels involved in cardiovascular, neural, endocrine, and sensory signaling systems (1). GPCRs share a common structural motif consisting of seven TM helices with an extracellular amino terminus. For all known GPCRs, the site of ligand binding is distant from the site of G-protein regulation (2). Therefore, the overall structural effects of agonists binding to extracellular sequences or TM domains must physically converge at the cytoplasmic interface between the receptor and its G protein.
We chose to characterize the conformational changes involved in G-protein activation by studying the human β2 adrenergic receptor (β2AR). The β2AR is an important pharmaceutical target for pulmonary and cardiovascular diseases, and a wide spectrum of pharmacologically well-characterized agonists and antagonists is readily available (3). Moreover, extensive mutagenesis studies have identified amino acids involved in ligand binding and G-protein coupling (Fig. 1A), thereby providing a basis for focusing our studies to domains likely to report conformational changes involved in receptor activation (4). We devised a means of labeling purified detergent-solubilized β2AR at Cys-265 in the carboxyl-terminal region of IC3 with the sulfhydryl-reactive fluorescent probe fluorescein maleimide (FM). Cys-265 is found in the native receptor, and labeling at this position did not require alteration of the receptor's amino acid sequence to introduce a labeling site. By homology with rhodopsin, for which a high-resolution structure is available (5), Cys-265 is predicted to be at the cytoplasmic end of the α helix of transmembrane (TM) 6 (Fig. 1A and B). Mutagenesis studies have demonstrated that sequences immediately carboxyl-terminal to Cys-265 are important for G-protein coupling (6–10), and mutation of the nearby Leu-272 leads to constitutive activation (11). In addition, synthetic peptides representing sequences from the carboxyl-terminal portion of IC3 are capable of directly stimulating G proteins (12–14). Therefore, a fluorophore covalently bound to Cys-265 is well positioned to detect agonist-induced conformational changes relevant to G-protein activation. Our results indicate that during agonist-induced activation, this portion of the receptor's G-protein-coupling domain rotates and/or tilts to a more hydrophobic environment, which is closer to both the surface of the micellar compartment and TM5. These observations provide insight into the real-time agonist-induced movement of the G-protein-coupling domain of a GPCR activated by diffusible ligands.
Figure 1.

Schematic diagram of the secondary structure of the β2AR illustrating the FM-labeling site at Cys-265. (A) There are 13 cysteines (yellow circles) in the β2AR, yet only Cys-265 is labeled with the relatively large polar fluorophore FM under the conditions described in Materials and Methods. Cys-106, Cys-184, Cys-190, and Cys-191 have been shown to be disulfide bonded (38, 39), and Cys-341 is palmitoylated (40). Cys-378 and Cys-406 in the carboxyl terminus form a disulfide bond during purification (data not shown). Labeling specificity was confirmed by peptide mapping and mutagenesis of potential reactive cysteines (data not shown). The sites of peptide cleavage by Factor Xa (line) and cyanogen bromide (black dots) are shown. (B) Schematic focusing on TM helices 5 and 6 and the connecting intracellular loop 3 (IC3). The location of the FM (F) site is highlighted. Fluorescence quenchers localized to either the aqueous milieu, the micellar environment, or the base of TM5 (oxyl-NHS bound to Lys-224, red square) were used to monitor conformational changes around Cys-265. (C) Cylinders representing the seven TM helices of the β2AR as viewed from the cytoplasmic side of the membrane, arranged according to the crystal structure of rhodopsin in the inactive state. In the inactive receptor, FM on Cys-265, is predicted to point toward the cytoplasmic extensions of TMs 3, 5, and 6. Also shown is the predicted position of the quencher oxyl-NHS on Lys-224 (red square).
Materials and Methods
Construction, Expression, and Purification of the β2AR.
Construction, expression, and purification of human β2AR were performed as described (15). The mutations Glu-224→Lys, Cys-378→Ala, and Cys-406→Ala were all generated on a background in which all of the lysines in the receptor had been mutated to arginine (16). A sequence coding for the cleavage site for the tobacco etch virus protease (GIBCO/BRL) was added to the 5′ end of the receptor construct via the linker-adapter method. All mutations were confirmed by restriction enzyme analysis and sequenced. The mutant receptor demonstrated only minor alterations in the general pharmacological properties of the receptor, as assessed by the affinity of the mutant receptor for (−)-isoproterenol (ISO) and (−)-alprenolol (ALP) [KI for ISO = 150 ± 40 μM for the mutant receptor vs. 210 ± 21 μM for the wild-type receptor (17); KD for ALP = 4.3 ± 0.6 nM for the mutant receptor vs. 1.7 ± 0.9 nM for wild-type receptor (18)].
Fluorescent Labeling of Purified β2AR.
Purified detergent-soluble receptor was diluted to 1 μM in HS buffer [20 mM Tris, pH 7.5/500 mM NaCl/0.1% N-dodecyl maltoside (NDM)] and reacted with 1 μM FM (Molecular Probes) for 2 h on ice in the dark. The reaction was quenched with the addition of 1 mM cysteine. The receptor was bound to a 250 μl Ni-chelating Sepharose column, and the column was washed alternately with 250 μl high salt (HS) buffer and 250 μl no salt buffer (20 mM Tris, pH 7.5/0.1% NDM) for a total of 10 cycles to remove free FM. The labeled protein (FM-β2AR) was eluted with HS buffer with 200 mM imidazole, pH 8.0. FM-β2AR was diluted ≈1:100 in HS buffer for fluorescence measurements. Fluorescence in control samples without receptor was negligible. The labeling procedure resulted in incorporation of 0.6 mol of FM per mol of receptor, on the basis of an extinction coefficient of 83,000 M−1 cm−1 for FM and a molecular mass of 50 kDa for β2AR. For labeling the Q224K site on the mutant receptor, the sample was split after labeling with FM (1 h) and dialyzed for 1 h at room temperature into a Hepes HS buffer. Half of the sample was treated with 1 mM oxyl-N-hydroxysuccinimide (NHS) for 1 h on ice. Both the FM alone and the FM + oxyl-NHS samples were then treated with tobacco etch virus (TEV) protease (GIBCO/BRL), according to the manufacturer's instructions, and then washed on a Ni-chelating Sepharose column, as above. Equivalent amounts of FM- and FM + oxyl-NHS-labeled receptor, as confirmed by protein assay (Bio-Rad DC Kit), were thus prepared for comparison. The TEV protease site at the amino terminus of the receptor allowed us to remove any probe located at the amino terminus after labeling the receptor with an amine-reactive tag. The location of the FM-labeling site at Cys-265 in both the wild-type and mutant receptors was verified by peptide mapping with protease factor Xa and cyanogen bromide (the cleavage sites are indicated in Fig. 1A).
Fluorescence Spectroscopy.
Experiments were performed on a Spex (Spex Industries, Metuchen, NJ) Fluoromax spectrofluorometer with photon counting mode by using an excitation and emission bandpass of 4.2 nm. Approximately 25 pmol of FM-labeled β2AR was used in 500 μl of HS buffer. Excitation was at 490 nm, and emission was measured from 500 to 599 nm with an integration time of 0.3 s/nm for emission scan experiments. For time course experiments, excitation was at 490 nm, and emission was monitored at 517 nm. For studies measuring ligand effects, no difference was observed when using polarizers in magic angle conditions. Unless otherwise indicated, all experiments were performed at 25°C, and the sample underwent constant stirring. Fluorescence intensity was corrected for dilution by ligands in all experiments and normalized to the initial value. All of the compounds tested had an absorbance of less than 0.01 at 490 and 517 nm in the concentrations used, excluding any inner filter effect in the fluorescence experiments.
Fluorescence Lifetime Determination.
Fluorescence lifetime measurement of the FM-labeled β2AR was carried out by using a PTI (South Brunswick, NJ) Laserstrobe fluorescence lifetime instrument. Measurements were taken at 25°C by using 490-nm excitation pulses (full-width half-maximum ≈1.4 ns) to excite the samples, and emission was monitored through a combination of three >550-nm long-pass filters. Measurements used 225 μl of a 5 μM sample placed in a 4 × 4-mm cuvette and represent 3 average shots of 5 shots per point collected in 150 channels. The fluorescence decays were fit to a single exponential by using the commercial pti program (TimeMaster Software, Photon Technologies, South Brunswick, NJ).
Quenching of Fluorescence.
FM was diluted to 1 μM in HS buffer. The dye was diluted into 375 μl of a buffer containing 20 mM Hepes, pH 7.5/0.1% N-dodecyl maltoside. Experiments were performed at the indicated concentration of potassium iodide, freshly made in 10 mM Na2S2O3, whereas the total salt concentration was maintained at 250 mM with potassium chloride in all experiments. Potassium iodide and potassium chloride at concentrations up to 250 mM do not alter the ligand-binding properties of the β2AR (18). For nitroxide quenching, receptor was diluted into HS buffer. Experiments were performed at the indicated concentration of nitroxide fatty acids (Molecular Probes), while maintaining total fatty acid concentration at 100 μM with stearic acid. After each addition of quencher, samples were thoroughly mixed, incubated for 10 min (KI) or 5 min (nitroxides), and fluorescence was recorded by exciting at 490 nm and performing an emission scan from 500 to 599 nm. Data were plotted according to the Stern–Volmer equation, Fo/F = 1 + Ksv[KI], where Fo/F is the ratio of fluorescence intensity in the absence and presence of KI, and Ksv is the Stern–Volmer quenching constant. The Ksv values thus obtained were then used with the measured fluorescence lifetimes (τo) to determine the bimolecular quenching constant, kq (Ksv = kq ⋅ τo) (19). For quenchers, a time scan was initiated after the emission scan and 100 μM (−)-ISO was added after 2 min. At 10 min, 20 μM ALP was added and the extent of reversal determined. The quenchers used did not alter the ability of ISO or ALP to compete with [3H]DHA.
Results and Discussion
The Effect of Full and Partial Agonists on the Fluorescence of FM-β2AR Correlates with the Biological Properties of the Agonists.
Only Cys-265 is labeled when purified detergent-solubilized β2AR (1 μM) is reacted with FM at a 1:1 stoichiometry. This polar fluorophore does not label TM cysteines, and the two other potentially accessible cysteines in the carboxyl terminus (Fig. 1A) form a disulfide bond during purification (data not shown). The specificity of labeling was confirmed by peptide mapping studies with factor Xa (which cleaves only in the third intracellular loop) and cyanogen bromide (which cleavage at methionines, shown in Fig. 1A). When FM-β2AR is cleaved with factor Xa, fluorescence labeling is observed only on the carboxyl-terminal half of the protein. After cleavage of FM-β2AR with cyanogen bromide, labeling is localized to a 7-kDa peptide representing a portion of the third intracellular loop containing Cys-265 (data not shown). Labeling of the β2AR with fluorescein did not alter ligand binding or G protein coupling in a reconstitution assay (data not shown).
The fluorescence properties of FM-β2AR were examined by monitoring fluorescence as a function of time. The addition of the full-agonist ISO induces a conformational change that results in a decrease in fluorescence intensity of FM-β2AR (Fig. 2A). This agonist effect is readily reversed by the addition of the high-affinity neutral antagonist ALP. The partial agonists epinephrine, salbutamol, and dobutamine produce progressively smaller changes in receptor fluorescence. The magnitude of the effect of agonists on the fluorescence intensity of FM-β2AR correlates with the biological efficacy of these drugs in β2AR-mediated activation of Gs in membranes (Fig. 2B). These experiments verify that fluorescence intensity changes in FM-β2AR reflect biologically relevant ligand-induced conformational changes.
Figure 2.

Effect of agonists and partial agonists on fluorescence intensity of FM-β2AR. (A) The change in intensity of FM-β2AR in response to the addition of the full agonist ISO and the strong partial agonist epinephrine was reversed by the neutral antagonist ALP. In most experiments, we use the ALP reversal to quantitate the magnitude of the agonist-induced change. We found the ALP reversal to be the most consistent measure for comparison of agonist-induced conformational changes, because ALP reversal occurs over a shorter period relative to agonist responses and therefore is less subject to nonspecific effects on fluorescence intensity (e.g., photobleaching, receptor denaturation) that affect the baseline. ALP alone did not induce any changes in fluorescence, and treatment with ligands did not cause a change in the wavelength of maximum emission (data not shown). (B) Agonist and partial agonist effects on the intensity of FM-β2AR are compared with an assay of biological efficacy (GTPγS binding). FM-β2AR was treated with different agonists, and the change in fluorescence was measured at a time equal to 5 times the calculated t1/2 for each drug. All agonists were used at 100 μM to ensure saturation of the receptors and eliminate the effect of variations in agonist affinities. The ability of these ligands to stimulate GTPγS binding in a β2AR-Gαs fusion protein was determined, as previously described (41). All data represent experiments performed in triplicate.
Kinetics of Agonist-Induced Conformational Change.
Rhodopsin has long been used as a model system for direct biophysical analyses of GPCR activation because of its natural abundance, inherent stability, and spectroscopically defined activation scheme (20). The recent crystal structure of bovine rhodopsin (5) provides the first high-resolution picture of the inactive state of this highly specialized GPCR. Although the general features of this structure presumably apply across the broad family of GPCRs, the mechanism of rhodopsin activation is unique among GPCRs because of the presence of a covalent linkage between the receptor and its ligand, retinal. Thus, the dynamic processes of agonist association and dissociation common to the GPCRs for hormones, neurotransmitters, and other sensory stimuli are not part of the activation mechanism of rhodopsin. In contrast to rhodopsin, the β2AR is activated by a functionally broad spectrum of diffusible ligands.
This difference between rhodopsin and the β2AR is reflected in the rate of agonist-induced structural changes. Conformational changes induced in detergent-solubilized preparations of rhodopsin by light activation are very rapid, occurring with a t1/2 of milliseconds (21, 22). In contrast, as shown in Fig. 2, agonist activation of the β2AR is slow, despite the rapid on-rate of agonist binding (t1/2 ≈ 20 sec) [calculated from the agonist affinity, the off-rate estimated from the ALP reversal of the agonist effect (Fig. 2A) and the concentration of agonists used in these experiments (100 μM)]. Under these conditions, the on-rate of agonist is comparable to the more rapid rate of reversal of the agonist effect by the antagonist ALP (t1/2 at 25°C = 22.8 ± 3.6 sec, mean ± SEM, n = 3). We have observed the same slow rate of agonist-induced conformational change with a different fluorescent reporter on Cys-125 in TM3 and on Cys-285 in TM6 of the β2AR (Fig. 1A) (23), and Salamon et al. observed a similar rate of agonist-induced conformational changes in the δ-opioid receptor analyzed by surface plasmon resonance spectroscopy (24). Thus, agonist binding precedes the conformational change. The rate of conformational change is temperature dependent, with the rate at 37°C ≈3 times that at 25°C (data not shown). The slow temperature-dependent rate of conformational change and the rapid reversal suggest that the active state is a relatively high energy state that may be reached through one or more intermediate states (Eq. 1).
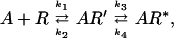 |
1 |
where R is the inactive receptor, R′ is the agonist-bound inactive receptor, and R* is the active receptor.
Our data would predict that k3 is slow relative to k1, k2, and k4. Moreover, the agonist-binding site in R′ may not be identical to the binding site in R*. The ligand-binding site for the β2AR has been well characterized by mutagenesis studies and lies relatively deep in the TM domains (Fig. 1A) (2, 4). A possible explanation for the sequence of binding followed by conformational change is that agonist binding is a stepwise process in which some receptor–ligand interactions form rapidly, whereas other interactions cannot form until stochastic conformational fluctuations make the complete binding pocket accessible (25). In the case of the β2AR, the energy to stabilize this conformational change comes from the energy of binding, which is calculated to be 6–10 Kcal/mol [on the basis of the binding affinity of ISO to detergent-solubilized β2AR (18) or to β2AR in membranes (26)]. In contrast, the energy released by the photoisomerization of retinal is ≈33 Kcal/mol (27). Thus, the difference in the rate of conformational change between rhodopsin and the β2AR can be attributed to the need for the ligand to diffuse into the binding pocket and the smaller energy associated with agonist binding. It should be emphasized that our studies are done in the absence of G protein. Precoupling of the β2AR to its cognate G protein in cell membranes may accelerate agonist-induced conformational changes.
Agonist-Induced Movement of FM Bound to Cys-265 Relative to Molecular Landmarks.
To characterize the agonist-induced structural changes in the G-protein-coupling domain containing Cys-265, we examined agonist-induced changes in the interaction of FM-β2AR with a variety of fluorescence quenchers. The results of these experiments were interpreted in the context of a three-dimensional model of the β2AR generated from the recent crystal structure of rhodopsin in the inactive state (5). Viewed from the cytoplasmic surface of the receptor, we would predict that in the absence of agonist, fluorescein bound to Cys-265 would be surrounded by the cytoplasmic extensions of TM3, TM5, and TM6 (Fig. 1C).
We first determined the accessibility of the water-soluble quencher potassium iodide to the fluorescein bound to Cys-265 (Fig. 3). As represented in the Stern–Volmer plot, steady-state fluorescence quenching by KI is much lower for fluorescein bound to the receptor when compared with FM bound to free cysteine in solution. This observation indicates that the fluorescein site on the receptor is relatively inaccessible to the water-soluble quencher KI, as expected on the basis of the predicted position of the fluorescein bound to Cys-265 (Fig. 1C). To determine the effect of agonist on KI quenching, we measured the fluorescence lifetimes of FM-β2AR in the presence ISO and ALP, which permitted us to calculate the bimolecular quenching constant (kq = Ksv/τo) by using the average value of the lifetime of FM-β2AR in the presence of either ISO (kq = 0.45 ± 0.01 × 10−9 M−1 s−1) or ALP (kq = 0.51 ± 0.01 × 10−9 M−1 s−1) (28). There was no difference between the extent of KI quenching in the ligand-free or ALP-bound receptor (data not shown). However, the lower kq in the ISO-bound state clearly shows that the fluorescein label on the β2AR is less accessible to the water-soluble quenching reagent KI in the presence of the agonist ISO (29). As a result, the fractional change in fluorescence intensity induced by ISO in the presence of 250 mM KI is smaller than in the presence of 250 mM KCl (Fig. 3B). Thus, ISO induces a conformational change that enhances the intrareceptor quenching of FM bound to Cys-265 but reduces access of Cys-265 to exogenous aqueous quencher KI. The burial of Cys-265 away from the aqueous milieu could be accomplished by a movement of TM6 toward the plasma membrane (Fig. 1B) and/or by a movement of TM6 that would bring Cys-265 closer to either TM3 or TM5 (Fig. 1C).
Figure 3.

Response of FM-β2R to agonist in the presence of KI or oxyl-NHS. (A) Stern–Volmer plots of KI quenching of FM-labeled β2AR. KI was added to FM reacted with cysteine, to labeled receptor incubated with 20 μM ALP, and to labeled receptor incubated with 100 μM ISO. Fluorescence was measured and plotted as described in Materials and Methods. The quenching constant Ksv was 7.9 ± 0.4 M−1 for fluorescein alone, 2.19 ± 0.06 M−1 for labeled receptor incubated with ALP, and 1.66 ± 0.06 M−1 for labeled receptor incubated with ISO. The difference between ISO and ALP was significant (P < 0.05, unpaired t test). There was no difference in Ksv between buffer alone and ALP treatments. All values are mean ± SEM, n = 3. (B) The effect of quenchers KI and oxyl-NHS on the magnitude of the ISO-induced decrease in fluorescence was determined. Percentage of control ISO response was calculated by using the formula [100 (ISO induced change in fluorescence in the presence of quencher)/(ISO induced change in fluorescence in the absence of quencher)]. For the aqueous quencher KI, the ISO-induced change in fluorescence in the presence of 250 mM KI was less than that in the presence of 250 mM KCl (55.4 ± 8.3% of control ISO response). In contrast, covalent binding of the spin-labeled quencher oxyl-NHS to K224 in TM5 increased the magnitude of the ISO response relative to the control (158 ± 8% control ISO response). In these experiments, the magnitude of the ALP reversal of the ISO-induced change in fluorescence was used as a measure of the magnitude of the ISO response for reasons given in the legend for Fig. 2. All values are mean ± SEM, n = 3.
Agonist-Induced Movement of Cys-265 Relative to Lys-224.
To distinguish between the movement of Cys-265 toward either TM3 or TM5, we generated a modified β2AR that permits site-specific attachment of an amine-reactive spin-labeled quencher at the cytoplasmic border of TM5 (Fig. 1C). To position the quencher at the base of TM5, we began with a template β2AR in which all of the lysines have been replaced by arginine (16) and changed Glu-224 to lysine. We purified this mutant and studied the interaction between FM at Cys-265 and oxyl-NHS at Lys-224. Although the baseline quenching of FM on Cys-265 with oxyl-NHS bound to Lys-224 was less that 10%, we observed that the effect of ISO on the FM fluorescence intensity (as reflected in the magnitude of the ALP reversal) was enhanced by more than 50% with the quencher bound to Lys-224 (Fig. 3B). Because the effect of this quencher is distance dependent, the increase in the extent of quenching reflects an agonist-induced conformational change that brings these regions of TM6 and TM5 closer together.
Agonist Induces Movement of FM Bound to Cys-265 Relative to a Lipophilic Quencher in the Detergent Micelle.
Because of the location of the fluorophore close to the predicted protein-lipid interface (Fig. 1B) of TM6, we next studied the interaction between the fluorophore and nitroxide spin-labeled fatty acids that partition into the detergent micelle to observe relative motion between the Cys-265 and the micelle (Fig. 4A). Because of their ability to quench the excited state of a variety of fluorophores in a distance-dependent manner, these spin-labeled fatty acid derivatives have been used extensively to study the distribution, location, and dynamics of fluorescently tagged proteins and lipids (30). We examined fatty acid derivatives with spin labels at two different locations along the carbon chain (Fig. 4A) and observed the best quenching of fluorescein by 4-(N,N-dimethyl-N-hexadecyl)ammonium-2,2,6,6-tetramethylpiperidine-1-oxyl iodide (CAT-16), which has a charged spin label on the head group of the fatty acid (Fig. 4B). The magnitude of the change in fluorescence intensity of FM-β2AR in response to the agonist ISO is dramatically increased in the presence of CAT-16 compared with the control fatty acid stearate (Fig. 4C). This effect was not observed with 5-doxyl stearate (5-DOX) (Fig. 4C). For example, 100 μM 5-DOX quenched baseline fluorescence by 12% (Fig. 4B) but had no significant effect on the magnitude of the agonist-induced change in fluorescence (Fig. 4C). In contrast, 50 μM CAT-16 produced a similar (≈12%) quenching in baseline fluorescence (Fig. 4B) but increased the magnitude of the agonist-induced fluorescence change by more than 2-fold (Fig. 4C). This indicates that ISO induces a conformational change at Cys-265, which brings the fluorophore closer to the nitroxide spin label of CAT-16 in the detergent micelle border, but not significantly closer to nitroxide spin label in 5-DOX, which would be buried within the hydrophobic core of the micelle. According to the models shown in Figs. 4A and 5, a piston-like movement of TM6 into the detergent micelle would bring fluorescein closer to the quenchers on both 5-DOX and CAT-16, but a clockwise rotation of TM6 and/or a tilting of TM6 would bring fluorescein closer to CAT-16 without significantly changing its position relative to 5-DOX.
Figure 4.

Comparison of effects of quenchers localized to the micelle on the response of FM-β2R to ISO. (A) Schematic depicting the structure of CAT-16 and 5-DOX, as well as the putative location of these quenching groups in the micelle. The quenching group on CAT-16 is localized on the polar surface of the micelle. The quenching group on 5-DOX is located within the hydrophobic core of the micelle. (B) Stern–Volmer plots depicting the extent of quenching of FM-β2AR by increasing concentrations of CAT-16 or 5-DOX. Quenchers were added to labeled receptor, and fluorescence was measured and plotted as in Fig. 3 and Materials and Methods. The total lipid concentration was kept constant at 100 μM with stearic acid. The quenching constant Ksv was 2.4 ± 0.1 mM−1 in the presence of CAT-16 and 1.4 ± 0.2 mM−1 in the presence of 5-DOX. (C) Differing effects of CAT-16 and 5-DOX on agonist-induced fluorescence change of FM-β2AR. The extent of response to ISO is presented as a percentage of control ISO response, calculated as in Fig. 3. (D) An example of the experiments used to generate the ratios in C. In this example, FM-β2AR was incubated with either 100 μM CAT-16 or 100 μM stearic acid. The response to agonist was monitored as described in Fig. 2. In the presence of the quencher CAT-16, ISO induced a 24.2 ± 0.3% decrease in fluorescence vs. 4.1 ± 0.6% in the presence of the stearic acid. All values are mean ± SEM, n = 3.
Figure 5.

A schematic indicating agonist-induced conformational changes in around Cys-265. The model represents TM domains 3, 5, and 6, as viewed from the cytoplasmic surface of the receptor, arranged according to the crystal structure of rhodopsin. In the inactive receptor, FM (green circle) on Cys-265 is predicted to point toward the cytoplasmic extensions of TMs 3, 5, and 6. Also shown is the predicted position of the quencher oxyl-NHS on Lys-224 (red square). The results from quenching experiments can best be explained by either a clockwise rotation of TM6 (A) and/or a tilting of TM6 (B) toward TM5 during agonist-induced activation of the receptor, as described in the text.
Conclusions
Electron paramagnetic resonance spectroscopic studies provide evidence that photoactivation of rhodopsin involves a rotation and tilting of TM6 relative to TM3 (31). Further support for motion of TM6 during rhodopsin activation was provided by chemical reactivity measurements and fluorescence spectroscopy (29), as well as by UV absorbance spectroscopy (32) and zinc crosslinking of histidines (33).
In earlier biophysical studies with β2AR labeled at multiple TM cysteines, we obtained evidence for movement of TM3 and TM6 on agonist activation (23). Recent fluorescence spectroscopic analysis of mutant β2ARs labeled on the cytoplasmic side of TM6 suggests that a rigid body motion occurs in this region during agonist-induced activation (34), whereas additional support for movement of TM3 and TM6 in the β2AR comes from zinc crosslinking studies (35) and chemical reactivity measurements in constitutively active β2AR mutants (36, 37). Our present experiments provide a detailed picture of the real-time movement of the G-protein-coupling domain of this receptor. By site-specific labeling with a single fluorophore on the cytoplasmic extension of TM6 and with a single quencher on the cytoplasmic extension of TM5 (Fig. 1B), we obtained evidence for movement of these two labeling sites toward each other (Fig. 3B). This observation, and the results of studies by using an aqueous quencher (Fig. 3A) and quenchers that partition into the detergent micelle (Fig. 4), allow us to deduce the agonist-induced movement of Cys-265 and the adjacent G-protein-coupling domain (Fig. 5). The results are most consistent with a clockwise rotation of TM6 and/or a tilting of the cytoplasmic end of TM6 toward TM5 (Fig. 5 A and B). These movements would further bury FM on Cys-265 in the interface between TM5 and TM6, rendering it less accessible to the aqueous quencher KI. Also, these conformational changes would bring FM on Cys-265 closer to both a nitroxide spin quencher covalently bound to Lys-224 and to a nitroxide quencher (CAT-16) on the polar surface of the detergent micelle surrounding the TM domains. Our data are not consistent with a large piston-like movement of TM6 into the membrane, although they do not exclude a small screw-like movement of the fluorophore toward the membrane that might accompany a clockwise rotation of TM6. Thus, our findings suggest that the conformational changes associated with β2AR activation are similar to those in rhodopsin and indicate a shared mechanism of GPCR activation.
Acknowledgments
P.G. and B.K.K. thank Joseph Lakowicz, Harel Weinstein and Irache Visiers for helpful discussions, and Roland Seifert for providing baculovirus for the β2AR-Gαs fusion protein. P.G. is a member of the Medical Scientist Training Program and is supported in part by National Institutes of Health Training Grant 5T32GM07365. This work was supported in part by National Institutes of Health 5 RO1 NS28471 and by the Mathers Charitable Foundation.
Abbreviations
- G protein
GTP-binding protein
- β2AR
β2 adrenergic receptor
- TM
transmembrane
- ISO
(−)-isoproterenol
- ALP
(−)-alprenolol
- NHS
N-hydroxysuccinimide
- 5-DOX
5-doxyl stearate
- CAT-16
4-(N,N-dimethyl-N-hexadecyl)ammonium-2,2,6,6-tetramethylpiperidine-1-oxyl iodide
- GPCR
G-protein-coupled receptor
References
- 1.Strader C D, Fong T M, Tota M R, Underwood D, Dixon R A. Annu Rev Biochem. 1994;63:101–132. doi: 10.1146/annurev.bi.63.070194.000533. [DOI] [PubMed] [Google Scholar]
- 2.Ji T H, Grossmann M, Ji I. J Biol Chem. 1998;273:17299–17302. doi: 10.1074/jbc.273.28.17299. [DOI] [PubMed] [Google Scholar]
- 3.Kobilka B. Annu Rev Neurosci. 1992;15:87–114. doi: 10.1146/annurev.ne.15.030192.000511. [DOI] [PubMed] [Google Scholar]
- 4.Gether U. Endocr Rev. 2000;21:90–113. doi: 10.1210/edrv.21.1.0390. , in press. [DOI] [PubMed] [Google Scholar]
- 5.Palczewski K, Kumasaka T, Hori T, Behnke C A, Motoshima H, Fox B A, Le Trong I, Teller D C, Okada T, Stenkamp R E, et al. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 6.Dixon R A, Sigal I S, Rands E, Register R B, Candelore M R, Blake A D, Strader C D. Nature (London) 1987;326:73–77. doi: 10.1038/326073a0. [DOI] [PubMed] [Google Scholar]
- 7.Strader C D, Dixon R A, Cheung A H, Candelore M R, Blake A D, Sigal I S. J Biol Chem. 1987;262:16439–16443. [PubMed] [Google Scholar]
- 8.O'Dowd B F, Hnatowich M, Regan J W, Leader W M, Caron M G, Lefkowitz R J. J Biol Chem. 1988;263:15985–15992. [PubMed] [Google Scholar]
- 9.Liggett S B, Caron M G, Lefkowitz R J, Hnatowich M. J Biol Chem. 1991;266:4816–4821. [PubMed] [Google Scholar]
- 10.Kobilka B K, Kobilka T S, Daniel K, Regan J W, Caron M G, Lefkowitz R J. Science. 1988;240:1310–1316. doi: 10.1126/science.2836950. [DOI] [PubMed] [Google Scholar]
- 11.Samama P, Cotecchia S, Costa T, Lefkowitz R J. J Biol Chem. 1993;268:4625–4636. [PubMed] [Google Scholar]
- 12.Okamoto T, Murayama Y, Hayashi Y, Inagaki M, Ogata E, Nishimoto I. Cell. 1991;67:723–730. doi: 10.1016/0092-8674(91)90067-9. [DOI] [PubMed] [Google Scholar]
- 13.Palm D, Munch G, Dees C, Hekman M. FEBS Lett. 1989;254:89–93. doi: 10.1016/0014-5793(89)81015-4. [DOI] [PubMed] [Google Scholar]
- 14.Cheung A H, Huang R R, Graziano M P, Strader C D. FEBS Lett. 1991;279:277–280. doi: 10.1016/0014-5793(91)80167-2. [DOI] [PubMed] [Google Scholar]
- 15.Ghanouni P, Schambye H, Seifert R, Lee T W, Rasmussen S G, Gether U, Kobilka B K. J Biol Chem. 2000;275:3121–3127. doi: 10.1074/jbc.275.5.3121. [DOI] [PubMed] [Google Scholar]
- 16.Parola A L, Lin S, Kobilka B K. Anal Biochem. 1997;254:88–95. doi: 10.1006/abio.1997.2361. [DOI] [PubMed] [Google Scholar]
- 17.Seifert R, Wenzel-Seifert K, Lee T W, Gether U, Sanders-Bush E, Kobilka B K. J Biol Chem. 1998;273:5109–5116. [PubMed] [Google Scholar]
- 18.Gether U, Lin S, Kobilka B K. J Biol Chem. 1995;270:28268–28275. doi: 10.1074/jbc.270.47.28268. [DOI] [PubMed] [Google Scholar]
- 19.Lakowicz J R. Principles of Fluorescence Spectroscopy. New York: Plenum; 1983. [Google Scholar]
- 20.Sakmar T P. Prog Nucleic Acid Res Mol Biol. 1998;59:1–34. doi: 10.1016/s0079-6603(08)61027-2. [DOI] [PubMed] [Google Scholar]
- 21.Arnis S, Fahmy K, Hofmann K P, Sakmar T P. J Biol Chem. 1994;269:23879–23881. [PubMed] [Google Scholar]
- 22.Farahbakhsh Z T, Hideg K, Hubbell W L. Science. 1993;262:1416–1419. doi: 10.1126/science.8248781. [DOI] [PubMed] [Google Scholar]
- 23.Gether U, Lin S, Ghanouni P, Ballesteros J A, Weinstein H, Kobilka B K. EMBO J. 1997;16:6737–6747. doi: 10.1093/emboj/16.22.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salamon Z, Cowell S, Varga E, Yamamura H I, Hruby V J, Tollin G. Biophys J. 2000;79:2463–2474. doi: 10.1016/S0006-3495(00)76489-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gether U, Kobilka B K. J Biol Chem. 1998;273:17979–17982. doi: 10.1074/jbc.273.29.17979. [DOI] [PubMed] [Google Scholar]
- 26.Weiland G A, Minneman K P, Molinoff P B. Nature (London) 1979;281:114–117. doi: 10.1038/281114a0. [DOI] [PubMed] [Google Scholar]
- 27.Shieh T, Han M, Sakmar T P, Smith S O. J Mol Biol. 1997;269:373–384. doi: 10.1006/jmbi.1997.1035. [DOI] [PubMed] [Google Scholar]
- 28.Dewey T G. Biophysical and Biochemical Aspects of Fluorescence Spectroscopy. New York: Plenum; 1991. [Google Scholar]
- 29.Dunham T D, Farrens D L. J Biol Chem. 1999;274:1683–1690. doi: 10.1074/jbc.274.3.1683. [DOI] [PubMed] [Google Scholar]
- 30.Matko J, Ohki K, Edidin M. Biochemistry. 1992;31:703–711. doi: 10.1021/bi00118a010. [DOI] [PubMed] [Google Scholar]
- 31.Farrens D L, Altenbach C, Yang K, Hubbell W L, Khorana H G. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 32.Lin S W, Sakmar T P. Biochemistry. 1996;35:11149–11159. doi: 10.1021/bi960858u. [DOI] [PubMed] [Google Scholar]
- 33.Sheikh S P, Zvyaga T A, Lichtarge O, Sakmar T P, Bourne H R. Nature (London) 1996;383:347–350. doi: 10.1038/383347a0. [DOI] [PubMed] [Google Scholar]
- 34.Jensen A D, Guarnieri F, Rasmussen S G, Asmar F, Ballesteros J A, Gether U. J Biol Chem. 2000;276:9279–9290. doi: 10.1074/jbc.M004871200. [DOI] [PubMed] [Google Scholar]
- 35.Sheikh S P, Vilardarga J P, Baranski T J, Lichtarge O, Iiri T, Meng E C, Nissenson R A, Bourne H R. J Biol Chem. 1999;274:17033–17041. doi: 10.1074/jbc.274.24.17033. [DOI] [PubMed] [Google Scholar]
- 36.Javitch J A, Fu D, Liapakis G, Chen J. J Biol Chem. 1997;272:18546–18549. doi: 10.1074/jbc.272.30.18546. [DOI] [PubMed] [Google Scholar]
- 37.Rasmussen S G, Jensen A D, Liapakis G, Ghanouni P, Javitch J A, Gether U. Mol Pharmacol. 1999;56:175–184. doi: 10.1124/mol.56.1.175. [DOI] [PubMed] [Google Scholar]
- 38.Dixon R A, Sigal I S, Candelore M R, Register R B, Scattergood W, Rands E, Strader C D. EMBO J. 1987;6:3269–3275. doi: 10.1002/j.1460-2075.1987.tb02645.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dohlman H G, Caron M G, DeBlasi A, Frielle T, Lefkowitz R J. Biochemistry. 1990;29:2335–2342. doi: 10.1021/bi00461a018. [DOI] [PubMed] [Google Scholar]
- 40.O'Dowd B F, Hnatowich M, Caron M G, Lefkowitz R J, Bouvier M. J Biol Chem. 1989;264:7564–7569. [PubMed] [Google Scholar]
- 41.Lee T W, Seifert R, Guan X, Kobilka B K. Biochemistry. 1999;38:13801–13809. doi: 10.1021/bi9908282. [DOI] [PubMed] [Google Scholar]