

Abstract
Circulating tumor cells (CTC) in the peripheral blood could provide important information for diagnosis of cancer metastasis and monitoring treatment progress. However, CTC are extremely rare in the bloodstream, making their detection and characterization technically challenging. We report here the development of an aptamer-mediated, micropillar-based microfluidic device that is able to efficiently isolate tumor cells from unprocessed whole blood. High-affinity aptamers were used as an alternative to antibodies for cancer cell isolation. The microscope-slide-sized device consists of >59,000 micropillars, which enhanced the probability of the interactions between aptamers and target cancer cells. The device geometry and the flow rate were investigated and optimized by studying their effects on the isolation of target leukemia cells from a cell mixture. The device yielded a capture efficiency of >95% with purity of ~81% at the optimum flow rate of 600 nL/s. Further, we exploited the device for isolating colorectal tumor cells from non-processed whole blood; as few as 10 tumor cells were captured from 1 mL of whole blood. We also addressed the question of low throughput of a typical microfluidic device by processing 1 mL of blood within 28 minutes. In addition, we found that ~93% of the captured cells were viable, making them suitable for subsequent molecular and cellular studies.
Metastases from primary tumors are the leading causes of death for non-hematological cancers.1 During the progression of metastasis, cancer cells shed from solid tumors and enter the bloodstream, becoming circulating tumor cells (CTC),2,3 which has a potential to serve as important biomarkers for early diagnosis of cancer metastases.4,5 While most current methods for cancer diagnosis require invasive biopsy and following molecular analysis, CTC enumeration is less invasive and provides a means for cancer diagnosis and prognosis, as well as for monitoring the progress of treatment. However, CTC are extremely rare, comprising only a few out of >109 hematological cells in 1 mL of blood, making their isolation and characterization a significant technological challenge.6
A variety of techniques have been developed for CTC isolation and detection, ranging from the use of immunomagnetic beads7–9 (e.g., CellSearch from Veridex10), and size-based filtration systems,11 to microfluidic devices.12–16 Among these methods, microfluidic devices17 with high-affinity ligands, primarily antibodies, have provided unique opportunities for detecting CTC from patient blood.18–20 However, only a few antibodies have been identified for tumor cells and cancer cell lines. In contrast, a number of DNA aptamers with high affinity and excellent selectivity have been selected for numerous cancers.21,22 Aptamers are single-stranded oligonucleotides that can recognize and bind to their target cells by folding into unique secondary or tertiary structures. They can be easily generated using an in vitro selection process termed cell-SELEX (Systematic Evolution of Ligands by EXponential enrichment).23,24 Previously, we reported a flat channel polydimethylsiloxane (PDMS) device and utilized it to capture target cancer cells from a mixture of target and control cancer cells (1:1 ratio), with ~80% capture efficiency.25,26 To realize clinical utility, however, a device must be capable of isolating a few tumor cells in 1 mL of whole blood (>109 cells). As a result, the device must possess high capture efficiency (the percentage of tumor cells isolated relative to total tumor cells present), satisfactory cell purity (the percentage of target tumor cells in the cells isolated), and sufficient throughput (the amount of blood processed in a certain time period). In addition, the captured cells are preferred to remain viable so that they can be further analyzed at the cellular and molecular level (e.g., to study apoptosis of tumor cells).27
Herein, we report an aptamer-functionalized, micropillar-based microfluidic device that isolates cancer cells from unprocessed whole blood with the required metrics mentioned above. Aptamers with specific binding to cancer cells of interest are used as an alternative to antibodies that have been often used for CTC isolation. The micropillars in the microchannel enhanced the probability of the interactions between the cells and the aptamers coated on the channel/pillar surfaces, resulting in high capture efficiency. After optimizing the geometry of the micropillars, we have achieved efficient isolation of a few tumor cells from whole blood with sufficient throughput and high cell viability.
Experimental Section
Device Fabrication
The device was designed to be in the size of a microscope slide, consisting of eight parallel channels with an array of >59,000 isotropically-etched, elliptical micropillars as shown in Figure 1. The geometric design of the micropillar array was inspired by the deterministic-lateral-displacement-based particle separation,28,29 in which the flow streamlines are distorted to enhance cell-micropillar interactions. The dimension of the elliptical pillars is 30 μm (major axis) × 15 μm (minor axis) × 32 μm (height), with an interpillar distance of 80 μm (center to center) and an 80-μm shift after every 3 rows in the direction of the minor axis.
Figure 1.

(A) Picture of the device consisting of 1 inlet, 1 outlet, and 8 channels connected through bifurcation. The size of the device is 1” × 3”, the same size of a microscope slide. (B) Optical micrograph of a portion of micropillar array in a channel. (C) Scanning electron microscope (SEM) image of isotropically etched elliptical micropillars in the glass substrate. (D) Scheme of capturing cancer cells in the device. Avidin is immobilized on the surface of the microchannels/micropillars via physical adsorption, followed by immobilization of biotinylated aptamers through biotin-avidin chemistry. Target cancer cells are then captured via the interaction between the aptamer and the receptors on cell surfaces.
Glass devices were fabricated according to the procedures reported previously.30 In brief, the layout of the device was designed in AutoCAD and then sent to Photo Sciences (Torrance, CA) to produce a chrome photomask. Glass substrates coated with chromium and photoresist layers were purchased from TELIC (Valencia, CA). The pattern on the photomask was transferred to the glass substrate via photolithography. The glass substrate was then chemically etched to a channel depth of 24 to 44 μm using a mixture of HF, HNO3 and H2O. The channel depth was measured using a Dektak 150 profilometer, and the depth was controlled by the etching time. The glass substrate was then sealed with a 5 mm thick PDMS sheet, fabricated from Sylgard 184 reagents (Dow Corning, Midland, MI) according to the instructions of the manufacturer. Inlet and outlet wells were created at the channel ends by punching holes in the PDMS sheet.
The device was functionalized with aptamers through two-step surface modification (Figure 1D): physical adsorption of avidin (Invitrogen, Carlsbad, CA) onto the glass surface (1 minute incubation) and immobilization of biotinylated aptamers via biotin-avidin interaction (1 minute incubation). The attachment of aptamers onto the device surfaces was confirmed by fluorescence microscopy (Supporting Information). Target cancer cells were captured due to the specific binding between cell surface receptors and aptamers.
Cell Culture
CCRF-CEM cells (CCL-119, T cell line, human acute lymphoblastic leukemia), Ramos cells (CRL-1596, B-cell, human Burkitt’s lymphoma), DLD-1 cells (Dukes’ type C colorectal adenocarcinoma) and HCT 116 cells (colorectal carcinoma) were purchased from American Type Culture Collection (ATCC). CEM, Ramos and DLD-1 cells were cultured in RPMI medium 1640 (ATCC) supplemented with 10% FBS (heat-inactivated; GIBCO) and 100 units/mL penicillin-streptomycin (Cellgro, Manassas, VA); HCT 116 cells were cultured in McCoy’s 5A medium containing the same concentration of FBS and penicillin-streptomycin. All cultures were incubated at 37°C under 5% CO2 atmosphere. The colorectal cancer cell lines were grown as adherent monolayers in 100 mm × 20 mm culture dishes to 95% confluence. Cells were washed in the dish with Dulbecco’s Phosphate buffered saline (DPBS) (Sigma-Aldrich, St. Louis, MO), dissociated by 0.25% trypsin treatment (2 min) and seeded into culture dishes at a low concentration.
Aptamers and Buffers
DNA aptamers were synthesized in house. Aptamer sequences were as follows, sgc8, 5’-ATC TAA CTG CTG CGC CGC CGG GAA AAT ACT GTA CGG TTA GAT TTT TTT TTT- 3’-biotin; TD05, 5’-AAC ACC GTG GAG GAT AGT TCG GTG GCT GTT CAG GGT CTC CTC CCG GTG TTT TTT TTT T- 3’-biotin; KDED2a-3, 5’-TGC CCG CGA AAA CTG CTA TTA CGT GTG AGA GGA AAG ATC ACG CGG GTT CGT GGA CAC GGT TTT TTT TTT T-3’-biotin; KCHA10, 5’-ATC CAG AGT GAC GCA GCA GGG GAG GCG AGA GCG CAC AAT AAC GAT GGT TGG GAC CCA ACT GTT TGG ACA CGG TGG CTT AGT TTT TTT TTT T-3’-biotin. For flow cytometry and fluorescence microscopy, the 5’ end of an aptamer was labeled with fluorescein isothiocyanate (FITC) or carboxytetrame-thylrhodamine (TAMRA). All aptamers were synthesized using an ABI3400 DNA/RNA synthesizer (Applied Biosystems, Carlsbad, CA) with reagents purchased from Glen Research (Sterling, VA). DNA purification was performed with a ProStar HPLC (Varian, Walnut Creek, CA) using a C18 column (Econosil, 5U, 250 × 4.6 mm) from Alltech Associates (Deerfield, IL). DNA concentration was determined by UV-Vis measurements using a Cary Bio-300 UV spectrometer (Varian).
Several buffers were used in this work and each buffer contained the reagents specific for a particular task. A buffer called washing buffer contained 4.5 g/L glucose and 5 mM MgCl2 in Dulbecco’s phosphate buffered saline (DPBS) and it was used for rinsing cells. The binding buffer contained 0.1 mg/mL yeast tRNA (Sigma-Aldrich) and 1 mg/mL bovine serum albumin (BSA) (Fisher Scientific, Hampton, NH) in the washing buffer, and it served as the medium for cell-aptamer interactions. The BSA in the buffer can passivate the surfaces to reduce the nonspecific adsorption of cells in the channel. The capturing buffer was a mixture of 1:1 volume ratio of the binding buffer and Histopaque-1119 (Sigma-Aldrich), which was used to prevent cells from settling in the cell suspension while pumping. The density of the capturing buffer was approximately 1.06 g/ml, which was close to the density of blood/cells. Histopaque-1119 increased the viscosity of the buffer to the level of whole blood.31
The specific binding of aptamers and target cells was verified using confocal fluorescence microscopy and flow cytometry. For confocal fluorescence microscopy, cells were first incubated with 250 nM TAMRA-labeled aptamers in the binding buffer. After washing three times with the washing buffer, fluorescence microscope images were taken using a confocal microscope. Flow cytometry was performed with a FACScan cytometer (BD Immunocytometry Systems, San Jose, CA). Briefly, 200,000 cells were incubated with FITC-labeled DNA aptamers at 250 nM for 15 min in 200 μL of the binding buffer (or the capturing buffer as specified later), and 10,000 counts were measured in the flow cytometer.
Cell Capture in a Buffer
Immediately before experiments, cells were rinsed with the washing buffer and resuspended at 106 cells/mL. By following the manufacturer’s instructions, the cells were treated with Vybrant DiI or Vybrant DiD cell-labeling solutions (Invitrogen, Carlsbad, CA) for 5 min at 37 °C, then rinsed with the washing buffer, and resuspended at 106 cells/mL in the capturing buffer. Labeled cells were stored on ice and further diluted to the desired concentrations before experiments. Target CEM cells were spiked into the control Ramos cells to form a final concentration of 1,000 cells/mL for CEM cells and 106 cells/mL for Ramos cells.
To initiate cell capture experiments, one channel volume of 1 mg/mL avidin in phosphate buffered saline (PBS) was first introduced into the device, followed by incubation for 1 min and then three rinses with the binding buffer. Then, one channel volume of 30 μM biotinylated sgc8 aptamer with a poly-thymine (10-T) linker was introduced into the device and incubated for 1 min, followed by three times of rinsing with the binding buffer. Finally, 1 mL of a mixture of CEM cells (target) and Ramos cells (control) in the capturing buffer was pumped into the channel at a flow rate of 600 nL/s (or other flow rates specified in the text). At the end of the experiment, the microchannel was washed three times with the binding buffer, followed by taking fluorescent images for the determination of the cell concentrations. For the study of capturing Ramos cells using aptamer TD05, CEM cells were used as control cells. For the study of capturing DLD-1 cells and HCT 116 cells using aptamers KDED2a-3 and KCHA10, Ramos cells were again used as the control cells. The concentration of all target cells was at 1,000 cells/mL, and the concentration of all control cells was at 106 cells/mL for all cell capturing experiments in the buffer.
Tumor Cell Isolation from Whole Blood
The single donor human whole blood was obtained from Innovative Research (Novi, MI) and it contained anticoagulant of ethylenediaminetetraacetic acid (EDTA). The whole blood was used as received without any treatment, except for spiking colorectal cancer cells DLD-1 and HCT 116 into the blood sample. The protocol of avidin adsorption and aptamer immobilization was performed as described in the previous section. For capture studies, non-enzymatic cell dissociation reagent (1X) (MP Biomedicals, Solon, OH) was used (instead of trypsin) to detach DLD-1 and HCT 116 cells from the culture dishes to avoid possible damages by trypsin to the proteins on the cell surfaces. Immediately before the introduction into the device, these carcinoma cells were filtered using a 40 μm BD Falcon cell strainer (Becton, Dickinson and Company, Franklin Lakes, NJ) and then spiked into whole blood at a predetermined concentration of 10, 100, 1,000, 10,000 cells/mL.
Instrument Setup
The cell suspension (or whole blood) was introduced into the device by pumping. A Micro4 syringe pump (World Precision Instruments, Sarasota, FL) with a 1-mL syringe was connected to the inlet of the device via polymer tubing and a female luer-to-barb adapter (IDEX Health & Science, Oak Harbor, WA). The outlet of the device was connected to a waste collector. For tumor cell isolation from whole blood, no buffer additive (Histopaque-1119) was used to avoid the property change of blood. Instead, we used a tiny magnetic stirring bar inside the 1-mL syringe, with a stirring plate beneath the syringe, to avoid cell settling. The magnetic stirring bar kept cells in suspension while blood was being pumped through the device. The device was placed on the stage of an Olympus FV500-IX81 confocal microscope (Olympus America, Melville, NY) for detecting cell captures.
To determine cell concentrations, a set of three images corresponding to red fluorescent cells, blue fluorescent cells, and transmission images were acquired at eight positions in each channel. Images were then imported into ImageJ (NIH), and cell counts were obtained using the Analyze Particles function after setting an appropriate threshold. Cell numbers were further verified by comparing fluorescent images with transmission images; only those with appropriate cell morphology in the transmission images will be counted. Capture efficiency was calculated by dividing the number of the target cells captured by the number of total target cells introduced into the device. The purity of cells captured was determined by dividing the number of the target cells captured by the number of the total cells captured.
Cell Viability
To determine the viability of cancer cells captured in the device, we performed two assays on these cells: 1) propidium iodide (PI) and acridine orange (AO) staining (Invitrogen) and 2) MTS assay (CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay, Promega, Madison, WI). PI is a membrane-impermeant stain, thus it labels only the dead cells with red fluorescence by penetrating the membranes of dead cells and binding to their DNA. AO is a membrane-permeable dye that binds to nucleic acids of all cells, resulting in green fluorescence. By PI/AO staining, nonviable cells and viable cells can be differentiated by their difference in fluorescent images under a microscope. We followed the instructions of the manufacturer to carry out the assay. In brief, 200 μL of PI/AO working solution was prepared to contain 2 μM PI and 2 μM AO in PBS. After cell capture in the microfluidic device, one channel volume of the PI/AO solution was introduced into the device and incubated for 10 min. The cells were then examined under the confocal microscope and fluorescent images were taken to evaluate the viability of captured cells.
MTS assay is a colorimetric method for determining the number of viable cells through cell proliferation. To implement the assay, cells were first released from the device, followed by rinsing with the washing buffer. These cells were then collected and quantified. Typically, 20,000 captured cells in 100 μL of fresh cell culture medium were seeded in one well of a 96-well plate, and 3 repeats were simultaneously carried out. For comparison, cells without going through the capture experiment were seeded at the same concentration into other wells (3 repeats) of the same microplate. After the incubation of these cells at 37°C under 5% CO2 for 48 h, 20 μL of the MTS assay reagent was added to each well and incubated for another 3 h. A plate reader was used to measure the absorbance at 490 nm to evaluate the cell viability.
RESULTS AND DISCUSSION
Isolation of lymphocytes
The performance of the microfluidic device was demonstrated first by sorting leukemia cells: CCRF-CEM cells (human acute lymphoblastic leukemia) that function as target cancer cells and Ramos cells (human Burkitt’s lymphoma) that function as control cells. Biotinylated sgc8 aptamers have specific binding with CCRF-CEM cells, and they were immobilized onto the surfaces of the micropillars/microchannels. A cell mixture containing 1000 CEM cells and 106 Ramos cells (1:1000 ratio) in 1 mL of the capturing buffer was used as a sample. To differentiate these two types of cells during imaging, CEM and Ramos cells were pre-stained with Vybrant DiI (red) and DiD (blue), respectively.
Figure 2A shows an image of the cancer cell mixture prior to sorting in the device and it is essentially all Ramos cells in blue. Figure 2B shows an image of cells captured after processing 1 mL of the cell mixture, and the majority of cells are now target CEM cells in red. These images show qualitatively that significant enrichment of the cancer cells was obtained through the microfluidic device. Figure 2C shows a single cancer cell captured on a micropillar in the device, and the image was generated by overlapping a transparency image with a fluorescence image. To quantify the enrichment factor, we counted cells before and after sorting. Cell labeling and confocal fluorescence detection enabled the counting of the number of the target cells introduced into the device (Ti), the number of the target cells captured (Tc), and the number of the control cells captured (Cc). The capture efficiency (E) can be calculated by E = Tc/Ti and the cell purity (P) in the captured cells can be calculated by P = Tc/(Tc + Cc). As high as 98% capture efficiency was obtained from the cell mixture when the flow rate was at 300 nL/s as discussed in detail below. The high capture efficiency partially arose from the specific binding of sgc8 aptamers with CEM cells, with a dissociation constant of Kd = 0.8 ± 0.09 nM.21 In addition, a poly-T linker at the end of the biotinylated aptamer sequence minimized the steric effects of the device surface on the aptamers, preserving the aptamers’ binding affinity to the target cells.
Figure 2.

(A) Representative image of low-abundant target CEM cells (stained with a red fluorescent dye) among high-abundant control Ramos cells (blue) before sorting. (B) Representative image of CEM cells (red) among Ramos cells (blue) after sorting (1 mL of the cell mixture was enriched through the microfluidic device). (C) Image of a CEM cell captured on the wall of an elliptical micropillar in the device (scale bar = 20 μm). (D) Microscopy image of unstained CEM cell bound with fluorescently-labeled aptamers, with color only on the surface of target cells.
The specific and strong binding of sgc8 aptamer to CEM cells was verified by confocal fluorescence microscopy. Figure 2D shows the fluorescent micrograph of TAMRA-labeled sgc8 aptamer specifically bound to unstained CEM cells, but these aptamers did not bind to Ramos cells (a black image, not shown here). Fluorescence on cell surfaces not only demonstrated the binding of the aptamers to the target cells, but also showed a possible way to identify unstained CTC captured from a sample via a fluorescently-labeled aptamer. The specific binding of sgc8 aptamer to CEM cells was also verified by flow cytometry (Supporting Information).
Effects of Depth and Flow Rate
To optimize the performance of the microfluidic device, we investigated the effects of the channel depth and flow rate on capture efficiency and cell purity. By changing channel depth (or pillar height), the size of micropillars and the interpillar gap were altered simultaneously due to isotropic etching.32 As a result, the geometry of the micropillar device can be studied by varying the channel depth. Figure 3A shows that the capture efficiency reduced slightly with the increasing channel depth from 24 μm to 40 μm, whereas the cell purity in Figure 3B improved significantly with the same change in the channel depth. The decreased capture efficiency with the increasing channel depth is likely due to the reduction in the probability of cell encounters with the top and bottom surfaces in a deeper channel as well as cells interaction with aptamers on pillars with wider inter-pillar gap. Nevertheless, the results show that our design of the pillar-row-shift and the curved surfaces of micropillars enabled sufficient interaction opportunities of cells with the surfaces, essentially maintaining the capture efficiency when the channel depth was increased. However, the increased channel depth drastically reduced the non-specific binding, particularly geometric trapping of control cells, therefore significantly improving the cell purity. Based on the data in Figures 3A and 3B, we chose a depth of 40 μm as the best trade-off between the capture efficiency and cell purity.
Figure 3.

(A) The capture efficiency as a function of the channel depth. The capture efficiency was calculated by dividing the number of the target cells captured by the number of the target cells introduced into the device. (B) The purity of cells captured as a function of the channel depth. The purity was calculated by dividing the number of the target cells captured by the number of both target and control cells captured. The flow rate is 600 nL/s for both (A) and (B). (C) The capture efficiency as a function of the flow rate. (D) The purity of cells captured as a function of the flow rate. The channel depth is 40 μm for (C) and (D). In all experiments, CEM cells were used the target cells and Ramos Cells as the control. The error bars represent one standard deviation of 3 repeat experiments.
Figures 3C and 3D show the effects of the flow rate on the capture efficiency and the cell purity. The capture efficiency reduced with the increasing flow rate because of a larger shear force at a higher flow rate and the reduced interaction time between cells and surfaces. The cell purity improved with the increasing flow rate due to the fact that non-specifically-bound cells were washed away with a stronger shear force at a higher flow rate. Based on these results, we chose 600 nL/s as the best compromise between the capture efficiency and cell purity. In addition, this flow rate results in sufficient throughput.
Using the device with the optimal channel depth of 40 μm and at a flow rate of 600 nL/s, we obtained cell purity of (81 ± 3)% with capture efficiency of (95 ± 2)%. These experimental conditions were used for all subsequent experiments. To compare this device with the previous efforts using a flat channel device,25 we plotted both results in Figure 4A. They show that the capture efficiency of the device in this work is significantly better than the flat channel in the previous work. The results verified the device design, including (1) the increased surface area via micropillars enhanced the loading capacity of aptamers, and (2) the row shift of micropillars and channel geometry significantly increased the probability of cell encounters with aptamers on the surfaces.
Figure 4.

(A) Comparison of the capture efficiency between a flat channel device reported previously and the micropillar device in this work. (B) Capture efficiencies of 4 types of cancer cells in the microfluidic device with micropillars. A different aptamer with specific binding with cells of interest was used for each type of cancer cells. (C) Capture efficiencies of DLD-1 cells and HCT 116 cells in whole blood. (D) Regression analysis of the number of the cells captured by the microfluidic device versus the number of the cells spiked into 1 mL of samples. Both axes are in the logarithm scale. HCT 116 cells at different concentrations were spiked either into the capturing buffer with Ramos cells as the control or into whole blood. Two calibration curves overlap with each other, reflecting no significant difference between buffer and blood samples. The error bars represent one standard deviation of 6 repeats for 10-cell samples and 3 repeats for other cell numbers.
Using the optimal channel geometry and flow rate, we also studied 3 other types of cancer cells in the device as shown in Figure 4B. They were captured in a cell mixture using their respective aptamers. To study the isolation of Ramos cells, an aptamer called TD05 was used as it has specific binding with Ramos cells (with a dissociation constant of Kd = 74.7 ± 8.7 nM33). A binary cell mixture containing 1000 Ramos cells and 106 CEM cells in 1 mL was used and the capture efficiency was (93 ± 2)%. Two carcinoma cells, HCT 116 cells (Dukes’ type C colorectal adenocarcinoma) and DLD-1 cells (colorectal carcinoma) were also studied. HCT 116 cells have strong affinity with KCHA10 aptamer (Kd = 21.3 ± 1.7 nM) while DLD-1 cells show specific binding with KDED2a-3 aptamer (Kd = 29.2 ± 6.4 nM).34,35 Using a binary cell mixture containing 1000 of each type of carcinoma cell and 106 Ramos cells (as the control), we obtained capture efficiency of (97 ± 3)% for HCT 116 cells and (91 ± 1)% for DLD-1 cells, respectively.
Tumor Cell Isolation from Whole Blood
To mimic the isolation of CTC from patient blood, we spiked colorectal carcinoma cells, HCT 116 cells, into whole blood that was used as received. One mL of non-processed whole blood, spiked with 100 HCT 116 cells, was introduced into the microfluidic device at a flow rate of 600 nL/s. Using the procedures described in the Experimental Section, we obtained capture efficiency of (96 ± 8)% as shown in Figure 4C. A similar experiment using another carcinoma cells, DLD-1 cells, resulted in capture efficiency of (92 ± 6)%.
To illustrate the potential of the device for clinical applications, we evaluated the isolation of HCT 116 cells from whole blood at concentrations of 10,000, 1,000, 100 and 10 cells/mL. Capture efficiencies of >95% were achieved in all cases, and a calibration curve between the number of the cells spiked and the number of the cells captured is shown in Figure 4D. Comparable results were obtained for isolating HCT 116 cells from the buffer. The results show that the device has a potential to detect CTC in clinical samples since the number of CTC in 1 mL of peripheral blood of cancer patients is often in the range of 1–100.36
In addition, we addressed the question of low throughput of a typical microfluidic device by connecting 8 microchannels through bifurcation (Figure 1). The width of each channel is 2 mm. With the optimal flow rate of 600 nL/s, the time required to process 1 mL of whole blood in the device is 28 minutes, which is favorable compared with hours of operation required in the benchmark instruments.
Cell Viability
Isolation and enumeration of tumor cells in peripheral blood of cancer patients is important for medical diagnostics and prognosis. However keeping the cells viable during the isolation process is also important for subsequent molecular and cellular studies, which would help understand the metastasis mechanisms and lead to potential therapies.
The viability of the cancer cells captured was examined with PI/AO assay and MTS assay as described in the Experimental Section. Figure 5 shows fluorescent microscope image of all cells captured from the mixture of CEM and Ramos cells using PI/AO cell viability assay. The majority of cell colors are in green, indicating that the most of the captured cells are still viable. We obtained cell viability of (94 ± 2)% for CEM cells when they were processed through the device using the optimal flow rate and channel depth. In addition, we used cell proliferation (MTS assay) as an alternative method, and we found cell viability of (93 ± 3)%, which is statistically the same as in the PI/AO assay. These results indicate that the cells captured using our device are suitable for subsequent cell culture and molecular analysis.
Figure 5.
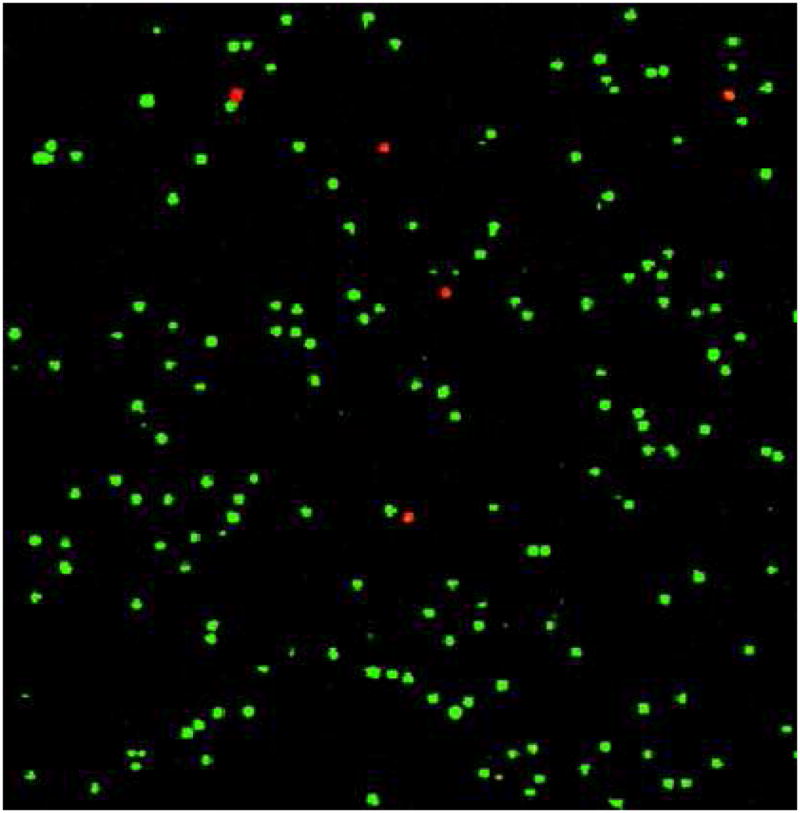
Fluorescent microscope image of CEM cells captured in the microfluidic device after PI/AO staining. The red color indicates nonviable cells (PI staining) while the green color indicates viable cells (AO staining).
CONCLUSION
We demonstrated using a DNA aptamer-enabled, micropillar-based microfluidic device to isolate cancer cells in non-processed perapheral blood. The unique geometry of the micropillar array in the device resulted in the high-performance cell isolation. This microfluidic device enabled the isolation of as few as 10 tumor cells from 1 mL of non-processed whole blood with >95% capture efficiency within 28 minutes. The advantages of such a device over the other methods include rapid analysis, no pre-treatment of blood samples, and low detection limit. As a result, the device has a potential to be used for clinical applications such as cancer diagnosis, prognosis, and monitoring the progress of therapeutic treatment.
Supplementary Material
Acknowledgments
This work was supported in part by National Cancer Institute of NIH (K25CA149080) and the University of Florida. The authors thank the Major Analytical Instrumentation Center (MAIC) at the University of Florida for the technical support in obtaining the SEM picture. We thank Meghan B. O’Donoghue and Pan Gu for their help.
Footnotes
Additional information as noted in text. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Fidler IJ. Nat Rev Cancer. 2003;3:453. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 2.Steeg PS. Nat Med. 2006;12:895. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 3.Gupta GP, Massagué J. Cell. 2006;127:679. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 4.Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LWMM, Hayes DF. N England J Med. 2004;351:781. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- 5.Danila DC, Fleisher M, Scher HI. Clin Cancer Res. 2011;17:3903. doi: 10.1158/1078-0432.CCR-10-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu M, Stott S, Toner M, Maheswaran S, Haber DA. J Cell Biol. 2011;192:373. doi: 10.1083/jcb.201010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Racila E, Euhus D, Weiss AJ, Rao C, McConnell J, Terstappen LWMM, Uhr JW. Procs Natl Acad Sci. 1998;95:4589. doi: 10.1073/pnas.95.8.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Talasaz AH, Powell AA, Huber DE, Berbee JG, Roh KH, Yu W, Xiao W, Davis MM, Pease RF, Mindrinos MN, Jeffrey SS, Davis RW. Proc Natl Acad Sci. 2009;106:3970. doi: 10.1073/pnas.0813188106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Danila DC, Heller G, Gignac GA, Gonzalez-Espinoza R, Anand A, Tanaka E, Lilja H, Schwartz L, Larson S, Fleisher M, Scher HI. Clin Cancer Res. 2007;13:7053. doi: 10.1158/1078-0432.CCR-07-1506. [DOI] [PubMed] [Google Scholar]
- 10.Riethdorf S, Fritsche H, Müller V, Rau T, Schindlbeck C, Rack B, Janni W, Coith C, Beck K, Jänicke F, Jackson S, Gornet T, Cristofanilli M, Pantel K. Clin Cancer Res. 2007;13:920. doi: 10.1158/1078-0432.CCR-06-1695. [DOI] [PubMed] [Google Scholar]
- 11.Vona G, Sabile A, Louha M, Sitruk V, Romana S, Schutze K, Capron F, Franco D, Pazzagli M, Vekemans M, Lacour B, Brechot C, Paterlini-Brechot P. Am J Pathol. 2000;156:57. doi: 10.1016/S0002-9440(10)64706-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Nature. 2007;450:1235. doi: 10.1038/nature06385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adams AA, Okagbare PI, Feng J, Hupert ML, Patterson D, GÖttert J, McCarley RL, Nikitopoulos D, Murphy MC, Soper SA. J Am Chem Soc. 2008;130:8633. doi: 10.1021/ja8015022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dharmasiri U, Balamurugan S, Adams AA, Okagbare PI, Obubuafo A, Soper SA. Electrophoresis. 2009;30:3289. doi: 10.1002/elps.200900141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang S, Wang H, Jiao J, Chen K-J, Owens GE, Kamei K-i, Sun J, Sherman DJ, Behrenbruch CP, Wu H, Tseng H-R. Angew Chem Int Ed. 2009;48:8970. doi: 10.1002/anie.200901668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhagat AAS, Hou HW, Li LD, Lim CT, Han J. Lab Chip. 2011;11:1870. doi: 10.1039/c0lc00633e. [DOI] [PubMed] [Google Scholar]
- 17.Whitesides GM. Nature. 2006;442:368. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 18.Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK, Floyd FP, Gilman AJ, Lord JB, Winokur D, Springer S, Irimia D, Nagrath S, Sequist LV, Lee RJ, Isselbacher KJ, Maheswaran S, Haber DA, Toner M. Proc Natl Acad Sci. 2010 doi: 10.1073/pnas.1012539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gleghorn JP, Pratt ED, Denning D, Liu H, Bander NH, Tagawa ST, Nanus DM, Giannakakou PA, Kirby BJ. Lab Chip. 2010;10:27. doi: 10.1039/b917959c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S, Liu K, Liu J, Yu ZTF, Xu X, Zhao L, Lee T, Lee EK, Reiss J, Lee YK, Chung LWK, Huang J, Rettig M, Seligson D, Duraiswamy KN, Shen CKF, Tseng HR. Angew Chem Int Ed. 2011;50:3084. doi: 10.1002/anie.201005853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shangguan D, Li Y, Tang Z, Cao ZC, Chen HW, Mallikaratchy P, Sefah K, Yang CJ, Tan W. Proc Natl Acad Sci. 2006;103:11838. doi: 10.1073/pnas.0602615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Famulok M, Hartig JS, Mayer G. Chem Rev. 2007;107:3715. doi: 10.1021/cr0306743. [DOI] [PubMed] [Google Scholar]
- 23.Tuerk C, Gold L. Science. 1990;249:505. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 24.Ellington AD, Szostak JW. Nature. 1990;346:818. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 25.Phillips JA, Xu Y, Xia Z, Fan ZH, Tan W. Anal Chem. 2009;81:1033. doi: 10.1021/ac802092j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu Y, Phillips JA, Yan J, Li Q, Fan ZH, Tan W. Anal Chem. 2009;81:7436. doi: 10.1021/ac9012072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dharmasiri U, Witek MA, Adams AA, Soper SA. Annu Rev Anal Chem. 2010;3:409. doi: 10.1146/annurev.anchem.111808.073610. [DOI] [PubMed] [Google Scholar]
- 28.Huang LR, Cox EC, Austin RH, Sturm JC. Science. 2004;304:987. doi: 10.1126/science.1094567. [DOI] [PubMed] [Google Scholar]
- 29.Inglis DW, Davis JA, Austin RH, Sturm JC. Lab Chip. 2006;6:655. doi: 10.1039/b515371a. [DOI] [PubMed] [Google Scholar]
- 30.Fan ZH, Harrison DJ. Anal Chem. 1994;66:177. [Google Scholar]
- 31.O'Brien S, Kent NJ, Lucitt M, Ricco AJ, McAtamney C, Kenny D, Meade G. IEEE Trans Biomed Eng. 2012;59:374. doi: 10.1109/TBME.2011.2172607. [DOI] [PubMed] [Google Scholar]
- 32.Madou MJ. Fundamentals of Microfabrication: The Science of Miniaturization. 2. CRC Press; New York: 2002. [Google Scholar]
- 33.Tang Z, Shangguan D, Wang K, Shi H, Sefah K, Mallikratchy P, Chen HW, Li Y, Tan W. Anal Chem. 2007;79:4900. doi: 10.1021/ac070189y. [DOI] [PubMed] [Google Scholar]
- 34.Martin JA, Phillips JA, Parekh P, Sefah K, Tan W. Molecular BioSystems. 2011;7:1720. doi: 10.1039/c0mb00311e. [DOI] [PubMed] [Google Scholar]
- 35.Sefah K, Meng L, Lopez-Colon D, Jimenez E, Liu C, Tan W. PLoS ONE. 2010;5:e14269. doi: 10.1371/journal.pone.0014269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zieglschmid V, Hollmann C, Böcher O. Crit Rev Clin Lab Sci. 2005;42:155. doi: 10.1080/10408360590913696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.