

Abstract
Glutathione peroxidase 4 (Gpx4) is an antioxidant defense enzyme important in reducing hydroperoxides in membrane lipids and lipoproteins. Gpx4 is essential for survival of embryos and neonatal mice; however, whether Gpx4 is required for adult animals remains unclear. In this study, we generated a floxed Gpx4 mouse (Gpx4(f/f)), in which exons 2–4 of Gpx4 gene are flanked by loxP sites. We then cross-bred the Gpx4(f/f) mice with a tamoxifen (tam)-inducible Cre transgenic mouse (R26CreER mice) to obtain mice in which the Gpx4 gene could be ablated by tam administration (Gpx4(f/f)/Cre mice). After treatment with tam, adult Gpx4(f/f)/Cre mice (6–9 months of age) showed a significant reduction of Gpx4 levels (a 75–85 % decrease) in tissues such as brain, liver, lung and kidney. Tam-treated Gpx4(f/f)/Cre mice lost body weight and died within 2 weeks, indicating that Gpx4 is essential for survival of adult animals. Tam-treated Gpx4(f/f)/Cre mice exhibited increased mitochondrial damage, as evidenced by the elevated 4-hydroxylnonenal (4-HNE) level, decreased activities of electron transport chain complex I and IV, and reduced ATP production in liver. Tam treatment also significantly elevated apoptosis in Gpx4(f/f)/Cre mice. Moreover, tam-treated Gpx4(f/f)/Cre mice showed neuronal loss in hippocampus region and had increased astrogliosis. These data indicate that Gpx4 is essential for mitochondria integrity and survival of neurons in adult animals.
Keywords: Gpx4, knockout mice, lipid peroxidation, mitochondria, neurodegeneration, apoptosis, oxidative stress
Introduction
Reactive oxygen species (ROS), such as superoxide anions (O2˙−) and hydrogen peroxide (H2O2), are generated in aerobic organisms by a variety of pathways [1]. Although ROs may be essential for cell signaling [2], high concentrations of ROS is deleterious by inducing oxidative damage to macromolecules such as lipids, proteins, and DNA [3]. Polyunsaturated fatty acids in membranes are especially vulnerable to ROS, and the resultant lipid peroxidation is a primary mechanism in the injury and death of cells, as lipid peroxidation not only impairs membrane fluidity and the function of membrane proteins [4], but also generates reactive aldehydes such as 4-hydroxylnonenal (4-HNE) and malondialdehyde (MDA) that are highly toxic to cells [5].
Glutathione peroxidase 4 (Gpx4) is a member of selenoproteins glutathione peroxidase family that catalyzes the reduction of peroxides using glutathione as electron donor [6]. However, Gpx4 is unique in that it can reduce hydroperoxides in membrane lipids (e.g., phospholipids, cholesterol, and cholestryl ester) because of its small size, large hydrophobic surface, and high affinity for lipid hydroperoxides [6, 7]. Mitochondria are the major source of cellular ROS. ROS can damage mitochondria membrane lipids, leading to impaired mitochondrial functions [8] and elevated apoptosis [9]. Because of its ability to repair oxidation damage to cardiolipin, a mitochondria specific phospholipid [10, 11], Gpx4 plays an important role in protecting mitochondrial function and suppressing apoptosis.
The importance of Gpx4 in vivo is shown by the phenotypes of Gpx4 knockout mice. For example, Gpx4 homozygous knockout (Gpx4−/−) embryos were shown to die in utero by midgestation (E7.5) [12, 13]. And Seiler et al showed that neuron-specific Gpx4 knockout mice were neonatally lethal [14]. These studies demonstrate that Gpx4 is essential for the survival of embryos and neonatal animals that are undergoing rapid development; however, the importance of Gpx4 in adult animals remains unclear. To determine the importance of Gpx4 in adult animals, we generated adult mice that were deficient in Gpx4 using an inducible knockout approach. Our results indicated that ablation of Gpx4 in adult mice was lethal. Moreover, adult mice with deficiency in Gpx4 showed mitochondrial dysfunction, increased apoptosis and neurodegeneration in brain.
Materials and methods
1. Animals
To generate a mouse model with inducible knockout of Gpx4, we used the targeting vector (shown in Figure 1) containing a floxed allele of Gpx4 gene (the exon 2, 3 and 4 of Gpx4 gene were flanked by two LoxP sites in the same direction) as well as a neo cassette flanked by two Frt sites. The targeting vector was electroporated into mouse ES cells at the Transgenic Core of University of Michigan, and the targeted allele was screened by Southern blots (shown in Figure 1B and 1C). ES cell clones that carried the targeted allele were identified, expanded and injected into C57/B6J blastocysts. Chimera mice were bred with female C57/B6J mice to obtain mice with a targeted allele of Gpx4 gene. Mice with the targeted allele of Gpx4 gene were then bred with transgenic mice expressing FLPe recombinase (stock#005703, The Jackson Laboratory, Bar Harbor, ME 04609) to remove the neo cassette to obtain mice with the floxed allele of Gpx4 gene [Gpx4(f/f)]. A PCR based genotyping protocol was developed to genotype the Gpx4(f/f) mice (Forward, 5'-TAC TGC AAC AGC TCC GAG TTC-3'; Reverse, 5'-GGT GCC AAA GAA AGA AAG TCC-3') (Figure 1D).
Figure 1.

Generation of a floxed Gpx4 mouse.
A. Illustrations of the wild-type Gpx4 (WT) gene allele, the targeting vector, the targeted allele, and the floxed allele of Gpx4 gene. The black boxes indicate exons: E1, E1a, and E2–E6 (left to right). The targeting vector was electroporated into ES cells. After standard selection and screening procedures, ES cells were used to produce mice with the targeted allele of Gpx4 gene. Mice with the targeted allele of Gpx4 gene were then cross-bred with Flpe mice to generate mice with a floxed allele of Gpx4 gene, in which E2, E3 and E4 are flanked by two loxP sites. B&C. Southern blot results showing the presence of the targeted allele in ES cells (the left lane). B. Genomic DNA from two ES cell clones was digested with HindIII and hybridized with Probe 2. The 15.5 kb fragment and the 11.0 kb fragment were indicative of the WT allele and the targeted allele, respectively. C. Genomic DNA from two ES cell clones was digested with EcoRI and hybridized with Probe 1. The 5.3 kb fragment and the 7.6 kb fragment were indicative of the WT allele and the targeted allele, respectively. The graph was rearranged. D. Identification of the floxed allele of Gpx4 gene by PCR. Using primers described in the Materials and Methods, a 200bp fragment and a 600bp fragment were amplified from the WT allele and the floxed allele of tail DNA, respectively. The genotypes are shown above.
Gpx4(f/f) mice were cross-bred with R26CreER transgenic mice (The Jackson Laboratory, Bar Harbor, ME 04609), which express a tamoxifen (tam)-inducible Cre recombinase driven by the Gt(ROSA)26Sor promoter [15], to generate the Gpx4(f/f)/Cre mice.
Tamoxifen (T5648, Sigma Chemical Co., St. Louis, MO) was dissolved in corn oil at a concentration of 30 mg/ml. Tam was administered to mice (6–9 months of age, male and female) intraperitoneally at a dose of 60mg/kg for a total of four injections (once every other day)
2. Western blots
Measurement of Gpx4 expression
Tissues were homogenized in RIPA buffer [20 mM Tris, pH 7.4, 0.25 M NaCl, 1 mM EDTA, 0.5% NP-40, and 50 mM sodium fluoride] supplemented with protease inhibitors (539134, EMD Biosciences Inc., San Diego, CA). Gpx4 protein levels in tissues and other antioxidant defense enzymes were determined by Western blots as described previously [12]. The protein level of β-actin was used to adjust for loading.
Measurement of mitochondrial 4-HNE level
Mitochondria from liver were isolated by homogenization followed by two-step centrifugation: 10 min at 1000 g to remove unbroken cells/tissue and at 10,000 g for 10 min to pellet the mitochondria. Mitochondria were homogenized in RIPA buffer supplemented with protease inhibitors (539134, EMD Biosciences Inc., San Diego, CA). Protein concentration was measured with the Bradford method. The mitochondrial proteins were separated by 4–20% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membrane was blocked for 1 hour in 10% non-fat dry milk and incubated with an anti-4-HNE antibody (R&D Systems, Minneapolis, MN) overnight at 4°C. The bands were visualized using the ECL Kit (RPN2132, GE Healthcare, Piscataway, NJ). 4-HNE adducts bands were quantified using ImageQuant 5.0 software and normalized to β-actin. The mean level of 4-HNE adducts (the ratio of 4HNE to β-actin determined by densitometry) in Gpx4(f/f) mice was assigned as 1 arbitrarily, and relative data are expressed as mean ± SEM.
3. Measurement of mitochondrial function
Liver tissues were homogenized in buffer 1 (250 mM mannitol, 75 mM sucrose, 500 μM EGTA, 100 μM EDTA, and 10 mM Hepes, pH 7.4) supplemented with protease inhibitor cocktail (539134, EMD Biosciences Inc., San Diego, CA).To obtain mitochondria, the homogenates were centrifuged at 600 g for 10 min at 4°C, the supernatants were then centrifuged at 10,000 g for 20 min at 4°C. The mitochondrial pellets were washed twice and used to measure ATP production and mitochondrial respiratory chain complex activities. The supernatants were further centrifuged at 100, 000 g for 60 min at 4°C to obtain the cytosolic fraction. Mitochondrial pellets were resuspended in ACA/BT buffer (750 mM 6-aminocaproic acid and 50 mM bistris, pH 7.0) plus n-dodecyl-maltoside (1%) and protease inhibitors and incubated at 4°C for 45 min. After centrifugation at 100,000 g at 4°C for 15 min, the supernatants were removed to measure the activities of the mitochondrial respiratory chain complexes. Complex I activity was measured by monitoring the oxidation of NADH at 340nm using ubiquinone-2 as electron acceptor in the presence of DCPIP (dichlorophenol indophenol). Complex II activity was measured by monitoring succinate-dependent reduction of DCPIP at 600nm using ubiquinone-2 as an electron acceptor as described by Hatefi [16]. Complex III activity was measured by monitoring the reduction of cyt. c (III) at 550 nm using D-ubiquinol-2 as an electron donor as described by Camacho et al [17]. The activity of Complex IV was measured by monitoring the oxidation of cyt. c+2 at 550 nm. All data are expressed as μmol/min/ mg protein.
ATP production was measured by ATP luminescence Assay Kit CLS II (Roche, Germany). In brief, 1 μg of isolated mitochondrial proteins was added to 200 μl of reaction mixture with 2.5 mM glutamate and 2.5 mM malate or with 10 mM succinate plus 1 μM rotenone. ADP was added to a final concentration of 6.25 mM. The luminescence was chased at 560 nm for 5 min. The linear portion of the curve was used to extrapolate the variation in luminescence per unit of time. The variation in luminescence was then converted into ATP concentration based on the values obtained from an ATP standard curve, and the data were expressed as nmol of ATP/min/mg proteins.
4. Apoptosis measurements
The number of apoptotic nuclei in liver was determined using the In Situ Oligo Ligation (ISOL) Kit with OligoB (S7200, Chemicon/Millipore, Billerica, MA), as described previously [11]. The slides were visualized using a light microscopy using a 40× objective. Ten random fields from each slide were selected for analysis in a blinded manner. The percentage of apoptotic cells in total cell population was calculated.
To measure cyt. c release, liver cytosolic fractions were obtained as described above. Cytosolic proteins were separated by 4–20% SDS-polyacrylamide gel, transferred to nitrocellulose membranes, and subjected to Western blot analysis with an anti-cyt. c antibody (4272S, Cell Signaling Technology, Beverly, MA). The intensities of cyt. c bands were quantified by ImageQuant 5.0 and normalized to that of IκB-a, a cytosolic protein loading control. Cleaved caspase-3 protein levels in liver tissues were determined by Western blots using an anti-caspase-3 antibody (sc-7148, Santa Cruz Biotechnology, Inc., Santa Cruz, CA)
5. Measurement of neurodegeneration
Mice were anesthetized and perfused transcardially, first with saline, then with 4% para-formaldehyde. Perfused brains were collected, post-fixed in 4% paraformaldehyde at 4°C overnight, equilibrated in 30% sucrose in PBS for 1–2 days at 4°C. The brains were then frozen by submersion in 2-metylbutane chilled in dry ice. The brain sections at a thickness of 30 μm were made using a frozen microtome.
For immunofluorescence staining, brain sections were blocked with blocking buffer (PBS supplemented with 0.05% Triton-X100 and 10% goat serum) for 1 hour and then incubated with primary antibody anti-NeuN (MAB377, Millipore, Billerica, Massachusetts) or anti-GFAP (3670, Cell Signaling Technology, Beverly, MA) in PBS supplemented with 0.05% Triton-X100 at 4°C overnight. The sections then washed 3 times with PBS and incubated with the fluorphore-conjugated secondary antibody (Alexa Fluor 488, goat anti-mouse IgG, Invitrogen, Carlsbad, CA) in PBS for 1 hour at room temperature. After washing 3 times, slides were mounted with ProLong Gold Antifade Reagent (P36930, Invitrogen, Carlsbad, CA). Brain slides were examined under a confocal microscope (Nikon TE-2000U).
Hippocampus tissue was homogenized in PBS buffer supplemented with 0.5% Triton X-100 and protease inhibitor (539134, EMD Biosciences Inc., San Diego, CA). NeuN, synapthophysin and GFAP levels were determined by Western blots using respective antibodies (anti-NeuN, Millipore, Billerica, MA; anti-Synaptophysin and anti-GFAP, Cell Signaling Technology, Beverly, MA). The intensities of the bands were quantified with ImageQuant 5.0 software and normalized to that of β-actin.
6. Statistics
Data are expressed as mean ± SEM. Results were statistically analyzed using two-way ANOVA or Student's t-test when appropriate. Statistical significance was set to a minimum of p < 0.05.
Results
1. Ablation of Gpx4 in adult mice was lethal
As shown in Figure 1, we generated a floxed Gpx4 mouse model (Gpx4(f/f) mouse), in which exons 2, 3 and 4 of the Gpx4 gene are flanked by loxP sites. We then cross-bred Gpx4(f/f) mice with R26CreER transgenic mice to generate Gpx4(f/f) mice with the R26CreER transgene: Gpx4(f/f)/Cre mice. Because the R26CreER transgene encodes a tamoxifen (tam)-activatable Cre recombinase, administration of tam will lead to knockout of Gpx4 expression in Gpx4(f/f)/Cre mice.
To determine the role of Gpx4 in adult mice, we administrated tam to Gpx4(f/f)/Cre mice and control Gpx4(f/f) mice (which lacked the tam-activatable Cre recombinase), at 6–9 months of age. Mice received four injections (i.p.) of tam over a period of eight days. The body weights of tam-treated Gpx4(f/f)/Cre mice and control Gpx4(f/f) mice were monitored. As shown in Figure 2A, the body weights of tam-treated Gpx4(f/f)/Cre mice and Gpx4(f/f) mice were similar until after the 3rd injection of tam. After the 3rd injection, a decrease in body weight was observed in tam-treated Gpx4(f/f)/Cre mice, and the difference in body weight between Gpx4(f/f)/Cre mice and control Gpx4(f/f) mice became significant one week after the last injection. As shown in Figure 2B, all tam-treated Gpx4(f/f)/Cre mice died within two weeks after the tam treatment. Tam-treated Gpx4(f/f)/Cre mice were lethargic and cold to the touch with virtually no movement before they died. No gender difference in body weight loss and time of death was observed. In contrast, all tam-treated Gpx4(f/f) mice survived and showed no abnormally behaviors. In subsequent experiments, we selected to study Gpx4(f/f)/Cre mice at one week after the 4th injection of tam, because at this point Gpx4(f/f)/Cre mice showed abnormalities but the majority of mice were still alive.
Figure 2.

Lethal phenotype of adult Gpx4(f/f)/Cre mice treated with tam.
A. Body weights of Gpx4(f/f)/Cre mice and Gpx4(f/f) mice (6–9 months of age) during and after tam treatment (4 injections of tam) are shown (n=19 mice per group). *: p <0.05 B. Survival of tam-treated Gpx4(f/f)/Cre mice (n = 21) and tam-treated Gpx4(f/f) mice (n = 21). The curves were found to be significantly different by Log-Rank test.
To determine the effect of tam administration on Gpx4 expression, we compared Gpx4 protein levels in Gpx4(f/f)/Cre mice and Gpx4(f/f) mice by Western blots. Figure 3 shows the levels of Gpx4 protein in liver, lung, kidney, and brain (cortex and hippocampus) tissues of tam-treated Gpx4(f/f)/Cre mice and tam-treated Gpx4(f/f) mice at one week post tam injection. Compared to Gpx4(f/f) mice, Gpx4(f/f)/Cre mice showed a 75–85% reduction of Gpx4 protein levels in tissues at this point. This result indicates that tam administration resulted in knockout of Gpx4 expression in Gpx4(f/f)/Cre mice. The presence of small amount of Gpx4 protein in Gpx4(f/f)/Cre mice is expected, as residual Gpx4 from cells that did not express the tam-activatable Cre recombinase or from Gpx4 that had not turned over could still be detected.
Figure 3.
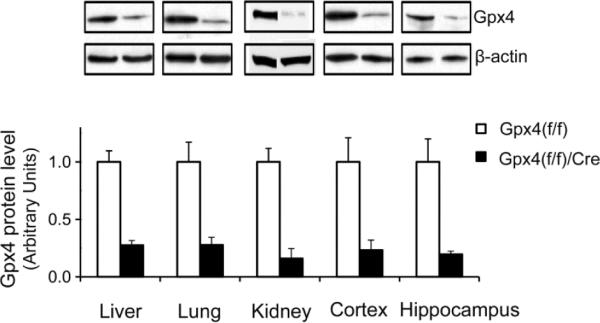
Reduced Gpx4 levels in tissues from tam-treated adult Gpx4(f/f)/Cre mice.
Gpx4 levels were measured by Western blots (Representative blots are shown. Some graphs were rearranged.) in liver, lung, kidney, and brain (cortex and hippocampus) from tam-treated Gpx4(f/f)/Cre mice and tam-treated Gpx4(f/f) mice. The mean levels of Gpx4 protein (the ratio of Gpx4 to β-actin determined by densitometry) in Gpx4(f/f) mice were assigned as 1 arbitrarily, and the relative data representing mean ± SEM are shown. *: p <0.05. n=3
2. Mitochondrial dysfunction in tam-treated Gpx4(f/f)/Cre mice
We previously showed that a deficiency of Gpx4 increased lipid peroxidation in mitochondria [18]. 4-HNE is a production of oxidation of lipids such as cardiolipin in mitochondria [19]; to determine whether ablation of Gpx4 increased lipid peroxidation in adult mice, we compared 4-HNE protein adducts levels in whole homogenates and mitochondrial proteins from livers of tam-treated Gpx4(f/f)/Cre mice and Gpx4(f/f) mice by Western blots. 4-HNE protein adducts level was significantly increased (a 50% increase) in mitochondrial proteins from liver of tam-treated Gpx4(f/f)/Cre mice (Figure 4A); however, no difference in 4-HNE levels in liver whole homogenates of tam-treated Gpx4(f/f)/Cre mice and tam-treated Gpx4(f/f) mice was observed (Figure 4A), indicating that Gpx4 ablation specifically elevated mitochondrial lipid peroxidation in adult mice.
Figure 4.

Effect of Gpx4 ablation on mitochondrial function.
A. The levels of 4-HNE protein adducts in liver whole homogenates and mitochondrial fractions of tam-treated Gpx4(f/f)/Cre mice and tam-treated Gpx4(f/f) mice. The activities of electron transport chain complex I (B), II (C), III (D), IV (E), and ATP generation (F) of isolated mitochondria from liver were measured in tam-treated Gpx4(f/f)/Cre mice and tam-treated Gpx4(f/f) mice. The data representing mean ± SEM are shown. (*: p <0.05, n=3). G/M: glutamate plus malate; Suc/Rot: succinate plus rotenone.
To determine the effect of Gpx4 ablation on mitochondrial function in adult mice, we measured electron transport chain (ETC) complex activities and ATP production in mitochondria isolated from tam-treated Gpx4(f/f)/Cre mice and tam-treated Gpx4(f/f) mice. As shown in Figure 4B–4E, tam-treated Gpx4(f/f)/Cre mice had significantly reduced activities of complex I (a 37% reduction) and complex IV (a 30% reduction) compared to tam-treated control Gpx4(f/f) mice. Mitochondria from the livers of tam-treated Gpx4(f/f)/Cre mice also had a significant decrease (40% reduction) in ATP production (Figure 4F). Thus, Gpx4 ablation significantly decreased mitochondrial electron transport chain complex I and complex IV activities and reduced ATP generation in adult mice.
3. Increased apoptosis in tam-treated Gpx4(f/f)/Cre mice
Our previous results indicated that Gpx4 levels regulate mitochondrial apoptosis [11]; therefore, we also determined whether apoptosis was higher in tam-treated Gpx4(f/f)/Cre mice. We measured apoptosis by three independent methods. First, we used the ISOL assay to detect the presence of double-strand DNA breaks, which are indicative of end-stage apoptosis [20]. Figure 5A and 5B are image of liver sections from tam-treated Gpx4(f/f)/Cre mice and tam-treated Gpx4(f/f) mice that show the presence of apoptotic cells in tam-treated Gpx4(f/f)/Cre mice. The percentages of apoptotic cells were quantified and shown in Figure 5C. A significant increase (11 folds) in numbers of apoptotic cells was observed in tam-treated Gpx4(f/f)/Cre mice. Second, we measured caspase-3 activation. The levels of activated caspase-3 (i.e., cleaved caspase-3) were determined by Western blots (Figure 5D), and the data in Figure 5E show that tam-treated Gpx4(f/f)/Cre mice had a significant increase (3.3 folds) in levels of activated caspase-3. Third, we measured cyt. c release into cytosol. As shown in Figure 5F, tam-treated Gpx4(f/f)/Cre mice had significantly higher (an increase of 50%) levels of cyt. c in cytosolic subcellular fractions. Therefore, these data demonstrate that Gpx4 ablation significantly elevated apoptosis in livers of adult mice.
Figure 5.

Elevated apoptosis in livers from tam-treated Gpx4(f/f)/Cre mice.
A&B. Graphs of liver sections stained for the presence of double-strand DNA breaks from tam-treated Gpx4(f/f) mice (A) and tam-treated Gpx4(f/f)/Cre mice (B). Arrows indicate cells positive for double-strand DNA breaks. C. The levels (expressed as percentages) of apoptotic cells in livers of tam-treated Gpx4(f/f) mice and tam-treated Gpx4(f/f)/Cre mice. The values are expressed as mean ± SEM. *: p<0.05. n=3. D. A graph of Western blots showing levels of caspase-3 and cleaved caspase-3 in tam-treated Gpx4(f/f)/Cre mice and tam-treated Gpx4(f/f) mice. E. The levels of cleaved caspase-3 in tam-treated Gpx4(f/f) mice and tam-treated Gpx4(f/f)/Cre mice. F. The levels of cytosolic cyt. c in tam-treated Gpx4(f/f) mice and tam-treated Gpx4(f/f)/Cre mice. The mean level of cleaved caspase-3 or cytosolic cyt. c in Gpx4(f/f) mice was assigned as 1 arbitrarily, and the relative data representing mean ± SEM are shown. *: p <0.05, n=3.
4. Neurodegeneration and increased glia activation in tam-treated adult Gpx4(f/f)/Cre mice
Neuronal loss was reported in the hippocampus region of neuron-specific Gpx4 knockout mice [14], and we had previously shown that Gpx4 was important in protecting neurons against oxidative insults and Aβ toxicity [21]. To determine whether ablation of Gpx4 affected neuron survival in adult mice, we performed histological analysis of brain sections stained with an antibody to NeuN, a neuron-specific marker. Figure 6A and 6B show images of NeuN-stained brain sections from tam-treated Gpx4(f/f)/Cre mice and tam-treated Gpx4(f/f) mice, respectively. Compared to tam-treated Gpx4(f/f) mice, tam-treated Gpx4(f/f)/Cre mice showed noticeable neuronal loss in the CA1 region of hippocampus. To confirm the histological observation, we compared levels of two neural specific proteins, NeuN and synaptophysin, in hippocampus of tam-treated Gpx4(f/f)/Cre mice and tam-treated Gpx4(f/f) mice by Western blots (Figure 6C). The data in Figure 6D and 6E show that, compared to tam-treated Gpx4(f/f) mice, tam-treated Gpx4(f/f)/Cre mice had significantly reduced levels of both NeuN and synaptophysin in hippocampus. Thus, these data demonstrate that Gpx4 ablation led to neurodegeneration in hippocampus region of adult Gpx4(f/f)/Cre mice.
Figure 6.

Neurodegeneration in tam-treated Gpx4(f/f)/Cre mice.
A&B. Images of brain sections of hippocampus regions stained with an anti-NeuN antibody from tam-treated Gpx4(f/f)mice (A) and tam-treated Gpx4(f/f)/Cre mice (B). Arrows indicate areas of neuronal loss. C. A graph of Western blots showing NeuN and Synaptophysin (syn) levels in hippocampus of tam-treated Gpx4(f/f) mice and tam-treated Gpx4(f/f) /Cre mice. D&E. Quantified results of NeuN levels (D) and Syn levels (E). The mean level of NeuN or Syn in Gpx4(f/f) mice was assigned as 1 arbitrarily, and the relative data representing mean ± SEM are shown. *: p <0.05, n=3.
Activation of glia cells such as astrocytes could contribute to neurodegeneration, so we also determined the effect of Gpx4 ablation on glia activation by staining brain sections with an antibody against glial fibrillary acidic protein (GAFP), a marker of activated astrocytes. Figure 7A and 7B show images of brain sections of hippocampus region from tam-treated Gpx4(f/f)/Cre mice and tam-treated Gpx4(f/f) mice, respectively. Compared to tam-treated Gpx4(f/f) mice, tam-treated Gpx4(f/f)/Cre mice showed increased GFAP staining in hippocampus. We further compared GFAP protein levels in these two groups of mice by Western blots (Figure 7C). As shown in Figure7D, tam-treated Gpx4(f/f)/Cre mice had significantly increased levels of GFAP protein, indicating elevated astrocyte activation.
Figure 7.
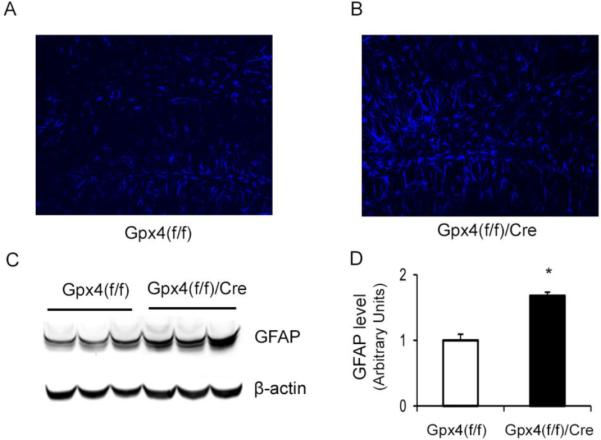
Astrocyte activation in hippocampus of tam-treated Gpx4(f/f)/Cre mice.
A&B. Images of brain sections of hippocampus regions stained with an anti-GFAP antibody from tam-treated Gpx4(f/f) mice (A) and tam-treated Gpx4(f/f)/Cre mice (B). C. A graph of Western blots showing GFAP protein levels in hippocampus of tam-treated Gpx4(f/f) mice and tam-treated Gpx4(f/f) /Cre mice. D. Quantified results of GFAP protein levels. The mean level of GFAP in Gpx4(f/f) mice was assigned as 1 arbitrarily, and the relative data representing mean ± SEM are shown. *: p <0.05, n=3.
Discussion
Gpx4, a member of the selenoprotein glutathione peroxidase family [6], is present at a relatively low level in mitochondria, cytosol, and nucleus of somatic tissues [22, 23]. While all selenoprotein glutathione peroxidases reduce hydrogen peroxide and alkyl hydroperoxides (e.g., cumene hydroperoxide, tert-butylhydroperoxide, and hydroperoxy fatty acids) using glutathione as hydrogen donor, Gpx4 is unique in that it also reduces hydroperoxides in complex lipids such as phospholipids, cholesterol and cholesterol esters [24]. Moreover, Gpx4 has the ability to reduce hydroperoxides integrated in membranes. It is believed that the small size and unique structure allow Gpx4 to react directly with lipid hydroperoxides in membranes [25]. Gpx4 is an essential for development, as whole body Gpx4 knockout mice were embryonically lethal [12, 13], and neuron-specific Gpx4 knockout mice were neonatally lethal [14]. However, whether Gpx4 is essential for adult animals was not clear. In this study, we generated a floxed-Gpx4 mouse model and used it to investigate the effect of Gpx4 ablation in adult animals. In our experiments, we ablated Gpx4 in adult Gpx4(f/f)/Cre mice at 6 to 9 months of age by administration of tam. We showed that Gpx4(f/f)/Cre mice treated with tam had a decline in body weight and died within two weeks. At present, we are not sure about the cause of death in Gpx4(f/f)/Cre mice treated with tam. However, because those mice looked sick and died rather rapidly with significant loss of body weight, we suspect that cachexia due to reduced food intake or loss of appetite may be a contributing factor. Although the exact cause of death for tam-treated Gpx4(f/f)/Cre mice remains to be determined, our data nonetheless demonstrate that Gpx4 is essential for the survival of mice at adult stage.
Gpx4 plays an important role in regulating mitochondrial functions because of its role in repairing oxidative damage to cardiolipin (CL). CL is a phospholipid found almost exclusively in mitochondrial inner membrane. CL plays a pivotal role in maintaining functions of mitochondrial inner membrane proteins [10] and exerts a major impact on apoptosis because cyt. c, one of the important apoptogenic proteins in mitochondria, is tightly associated with CL [26]. Oxidation of CL results in the dissociation of cyt. c from the mitochondrial inner membrane [26] and the opening of the mitochondrial PT pore, which lead to the release of cyt. c and other apoptogenic proteins to cytosol to induce apoptosis [10]. Using oxidative lipidomics approach, Kagan et al identified CL oxidation as an early event in apoptosis [27]. Gpx4 associates with the mitochondrial inner membranes and reduces CL hydroperoxides efficiently [28]. The reduction of CL hydroperoxides by Gpx4 prevents cyt. c dissociation from mitochondrial inner membrane and its subsequent release from mitochondria [10]. Liu et al showed recently that oxidation of CL results in 4-HNE formation [19]. In our study, we observed increased levels of 4-HNE protein adducts in mitochondria from tam-treated Gpx4(f/f)/Cre mice. The increase in mitochondrial 4-HNE protein adducts in Gpx4(f/f)/Cre mice was correlated with decreased activities of mitochondrial electron transport chain complex I and IV and reduced level of ATP production. Moreover, the numbers of apoptotic cells and the levels of activated caspase-3 and cytosolic cyt. c were increased in livers of tam-treated Gpx4(f/f)/Cre mice. Therefore, consistent with previous reports, our results demonstrate that Gpx4 plays a key role in protecting the integrity of mitochondria.
Gpx4 appears to be very important for the survival of neurons. We previously showed that overexpression of Gpx4 protected neurons against Aβ toxicity [21] and that Gpx4 deficiency facilitated Aβ accumulation in an Alzheimer's mouse model [29]. In this study, we found that Gpx4 ablation resulted in loss of neurons in hippocampal CA1 region of adult Gpx4(f/f)/Cre mice. The CA1 region of the hippocampus is a site of major neuron loss in Alzheimer's brain [30, 31], and it was reported that hippocampal pyramidal neurons in this region are particularly sensitive to oxidative stress [32–34]. Thus, our data indicate that Gpx4 plays an important in the survival of CA1 neurons. Our results also showed that tam-treated Gpx4(f/f)/Cre mice had increased activation of astrocytes, indicating that Gpx4 may protect neurons by suppressing glia activation. An early report by Seiler et al showed that mice with neuron specific knockout of Gpx4 was neonatally lethal and had abnormal behaviors such as seizure [14]. This study further showed that mice with neuron specific knockout of Gpx4 had neuronal loss at CA3 region of hippocampus. The different patterns of neuronal loss between mice with neuron specific knockout of Gpx4 and tam-treated Gpx4(f/f)/Cre mice are likely due to differences in cell-type specific expression of the Cre and/or ages of animals. Despite the difference in regions of neuronal loss observed in these two models, the data obtained from these two models clearly indicate that Gpx4 is important for neuron survival.
In conclusion, our findings from this study indicate that Gpx4 is essential for survival of adult mice. Moreover, our results indicate that Gpx4 is a key neural protective enzyme in adult mice.
Highlights
A conditional Gpx4 knockout mouse model was generated.
Gpx4 ablation in adult mice was lethal.
Adult Gpx4 knockout mice showed elevated mitochondrial damage.
Adult Gpx4 knockout mice had neurodegeneration in hippocampus.
Acknowledgements
This work was supported by NIH grants P01 AG19316 and P01AG020591, the San Antonio Nathan Shock Aging Center (1P30-AG13319), and a Merit Review grant (Q.R.) from the Department of Veteran Affairs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- [1].Fridovich I. The biology of oxygen radicals. Science. 1978;201:875–80. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- [2].Stone JR, Yang S. Hydrogen peroxide: a signaling messenger. Antioxid Redox Signal. 2006;8:243–70. doi: 10.1089/ars.2006.8.243. [DOI] [PubMed] [Google Scholar]
- [3].Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- [4].Richter C. Biophysical consequences of lipid peroxidation in membranes. Chem Phys Lipids. 1987;44:175–89. doi: 10.1016/0009-3084(87)90049-1. [DOI] [PubMed] [Google Scholar]
- [5].Yu BP, Yang R. Critical evaluation of the free radical theory of aging. A proposal for the oxidative stress hypothesis. Ann N Y Acad Sci. 1996;786:1–11. doi: 10.1111/j.1749-6632.1996.tb39047.x. [DOI] [PubMed] [Google Scholar]
- [6].Brigelius-Flohe R. Tissue-specific functions of individual glutathione peroxidases. Free Radic Biol Med. 1999;27:951–65. doi: 10.1016/s0891-5849(99)00173-2. [DOI] [PubMed] [Google Scholar]
- [7].Ursini F, Bindoli A. The role of selenium peroxidases in the protection against oxidative damage of membranes. Chem Phys Lipids. 1987;44:255–76. doi: 10.1016/0009-3084(87)90053-3. [DOI] [PubMed] [Google Scholar]
- [8].Paradies G, Petrosillo G, Pistolese M, Ruggiero FM. Reactive oxygen species affect mitochondrial electron transport complex I activity through oxidative cardiolipin damage. Gene. 2002;286:135–41. doi: 10.1016/s0378-1119(01)00814-9. [DOI] [PubMed] [Google Scholar]
- [9].Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci U S A. 2002;99:1259–63. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Imai H, Nakagawa Y. Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med. 2003;34:145–69. doi: 10.1016/s0891-5849(02)01197-8. [DOI] [PubMed] [Google Scholar]
- [11].Ran Q, Liang H, Gu M, Qi W, Walter CA, Roberts LJ, et al. Transgenic mice overexpressing glutathione peroxidase 4 are protected against oxidative stress-induced apoptosis. J Biol Chem. 2004;279:55137–46. doi: 10.1074/jbc.M410387200. [DOI] [PubMed] [Google Scholar]
- [12].Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34:496–502. doi: 10.1016/s0891-5849(02)01360-6. [DOI] [PubMed] [Google Scholar]
- [13].Imai H, Hirao F, Sakamoto T, Sekine K, Mizukura Y, Saito M, et al. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun. 2003;305:278–86. doi: 10.1016/s0006-291x(03)00734-4. [DOI] [PubMed] [Google Scholar]
- [14].Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–48. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- [15].Badea TC, Wang Y, Nathans J. A noninvasive genetic/pharmacologic strategy for visualizing cell morphology and clonal relationships in the mouse. J Neurosci. 2003;23:2314–22. doi: 10.1523/JNEUROSCI.23-06-02314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hatefi Y. Introduction--preparation and properties of the enzymes and enzymes complexes of the mitochondrial oxidative phosphorylation system. Methods Enzymol. 1978;53:3–4. doi: 10.1016/s0076-6879(78)53004-8. [DOI] [PubMed] [Google Scholar]
- [17].Camacho A, Moreno-Sanchez R, Bernal-Lugo I. Control of superoxide production in mitochondria from maize mesocotyls. FEBS Lett. 2004;570:52–6. doi: 10.1016/j.febslet.2004.06.024. [DOI] [PubMed] [Google Scholar]
- [18].Ran Q, Liang H, Ikeno Y, Qi W, Prolla TA, Roberts LJ, et al. Reduction in glutathione peroxidase 4 increases life span through increased sensitivity to apoptosis. J Gerontol A Biol Sci Med Sci. 2007;62:932–42. doi: 10.1093/gerona/62.9.932. [DOI] [PubMed] [Google Scholar]
- [19].Liu W, Porter NA, Schneider C, Brash AR, Yin H. Formation of 4-hydroxynonenal from cardiolipin oxidation: Intramolecular peroxyl radical addition and decomposition. Free Radic Biol Med. 2011;50:166–78. doi: 10.1016/j.freeradbiomed.2010.10.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tanaka M, Nakae S, Terry RD, Mokhtari GK, Gunawan F, Balsam LB, et al. Cardiomyocyte-specific Bcl-2 overexpression attenuates ischemia-reperfusion injury, immune response during acute rejection, and graft coronary artery disease. Blood. 2004;104:3789–96. doi: 10.1182/blood-2004-02-0666. [DOI] [PubMed] [Google Scholar]
- [21].Ran Q, Gu M, Van Remmen H, Strong R, Roberts JL, Richardson A. Glutathione peroxidase 4 protects cortical neurons from oxidative injury and amyloid toxicity. J Neurosci Res. 2006;84:202–8. doi: 10.1002/jnr.20868. [DOI] [PubMed] [Google Scholar]
- [22].Knopp EA, Arndt TL, Eng KL, Caldwell M, LeBoeuf RC, Deeb SS, et al. Murine phospholipid hydroperoxide glutathione peroxidase: cDNA sequence, tissue expression, and mapping. Mamm Genome. 1999;10:601–5. doi: 10.1007/s003359901053. [DOI] [PubMed] [Google Scholar]
- [23].Nam S, Nakamuta N, Kurohmaru M, Hayashi Y. Cloning and sequencing of the mouse cDNA encoding a phospholipid hydroperoxide glutathione peroxidase. Gene. 1997;198:245–9. doi: 10.1016/s0378-1119(97)00321-1. [DOI] [PubMed] [Google Scholar]
- [24].Brigelius-Flohe R, Friedrichs B, Maurer S, Schultz M, Streicher R. Interleukin-1-induced nuclear factor kappa B activation is inhibited by overexpression of phospholipid hydroperoxide glutathione peroxidase in a human endothelial cell line. Biochem J. 1997;328(Pt 1):199–203. doi: 10.1042/bj3280199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Scheerer P, Borchert A, Krauss N, Wessner H, Gerth C, Hohne W, et al. Structural basis for catalytic activity and enzyme polymerization of phospholipid hydroperoxide glutathione peroxidase-4 (GPx4) Biochemistry. 2007;46:9041–9. doi: 10.1021/bi700840d. [DOI] [PubMed] [Google Scholar]
- [26].Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J. 2000;351:183–93. doi: 10.1042/0264-6021:3510183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–32. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- [28].Liang H, Ran Q, Jang YC, Holstein D, Lechleiter J, McDonald-Marsh T, et al. Glutathione peroxidase 4 differentially regulates the release of apoptogenic proteins from mitochondria. Free Radic Biol Med. 2009;47:312–20. doi: 10.1016/j.freeradbiomed.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chen L, Na R, Gu M, Richardson A, Ran Q. Lipid peroxidation up-regulates BACE1 expression in vivo: a possible early event of amyloidogenesis in Alzheimer's disease. J Neurochem. 2008;107:197–207. doi: 10.1111/j.1471-4159.2008.05603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kril JJ, Patel S, Harding AJ, Halliday GM. Neuron loss from the hippocampus of Alzheimer's disease exceeds extracellular neurofibrillary tangle formation. Acta Neuropathol. 2002;103:370–6. doi: 10.1007/s00401-001-0477-5. [DOI] [PubMed] [Google Scholar]
- [31].Simic G, Kostovic I, Winblad B, Bogdanovic N. Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer's disease. J Comp Neurol. 1997;379:482–94. doi: 10.1002/(sici)1096-9861(19970324)379:4<482::aid-cne2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- [32].Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–95. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- [33].Patel M, Day BJ, Crapo JD, Fridovich I, McNamara JO. Requirement for superoxide in excitotoxic cell death. Neuron. 1996;16:345–55. doi: 10.1016/s0896-6273(00)80052-5. [DOI] [PubMed] [Google Scholar]
- [34].Guo Q, Sebastian L, Sopher BL, Miller MW, Ware CB, Martin GM, et al. Increased vulnerability of hippocampal neurons from presenilin-1 mutant knock-in mice to amyloid beta-peptide toxicity: central roles of superoxide production and caspase activation. J Neurochem. 1999;72:1019–29. doi: 10.1046/j.1471-4159.1999.0721019.x. [DOI] [PubMed] [Google Scholar]