

Summary
The roles of nitric oxide (NO) in physiology and pathophysiology merit the use of NO as a therapeutic for certain biomedical applications. Unfortunately, limited NO payloads, too rapid NO release, and the lack of targeted NO delivery have hindered the clinical utility of NO gas and low molecular weight NO donor compounds. A wide-variety of NO-releasing macromolecular scaffolds has thus been developed to improve NO’s pharmacological potential. In this tutorial review, we provide an overview of the most promising NO release scaffolds including protein, organic, inorganic, and hybrid organic-inorganic systems. The NO release vehicles selected for discussion were chosen based on their enhanced NO storage, tunable NO release characteristics, and potential as therapeutics.
1. Introduction
Ignarro, Furchgott, and Murad’s Nobel Prize-winning discovery that the endothelial derived relaxation factor (EDRF) was in fact nitric oxide (NO) inaugurated an extensive swell of research into the pivotal role of NO in numerous other physiological systems.1 The diverse list of biological processes that NO is associated with includes vasodilation2, platelet aggregation and adhesion,2 the immune response to infection,3 wound repair,4 as well as cancer biology and pathology.5
Nitric oxide is produced endogenously from L-arginine via the enzyme nitric oxide synthase (NOS).2 Due to its integral role in human physiology, deficiencies in NO biosynthesis have been linked to a number of disease states.6 Strategies for delivering exogenous NO thus hold promise for a number of biomedical applications ranging from cardiovascular regulation to antimicrobial and tumoricidal therapies. However, the reactivity of NO gas has hindered the development of effective NO-based therapies.7
In response to the need for controlled NO delivery, much work has focused on the synthesis of NO donors.8 Many classes of NO donors exist, including organic nitrates, nitrites, metal – NO complexes, nitrosamines, N-diazeniumdiolates (NONOates), and S-nitrosothiols (RSNOs). Based on their ability to spontaneously release NO in physiological media, NONOates and RSNOs represent the most widely used NO donor systems.
N-Diazeniumdiolates are arguably the most extensively studied and used class of NO donors. Stable NONOates are formed via the reaction of secondary amines with high pressures (i.e., 5 atm) of NO (Scheme 1).9 Efficient diazeniumdiolate formation requires the presence of a second basic residue, either an unreacted amine substrate or an added metal alkoxide base, to deprotonate the backbone amine and promote its nucelophilic attack on NO. The cation (e.g., protonated additional amine or metal from alkoxide base) stabilizes the charge of the resulting anionic NONOate. N-Diazeniumdiolates are attractive as NO donors because they decompose to NO upon protonation of the amine bearing the NONOate moiety. Indeed, NONOates regenerate the parent amine compound and two moles of NO per mole of donor in physiological solution (i.e., 37°C, pH 7.4).9 The structure of the amine precursor directly affects the NO release kinetics. Certain N-diazeniumdiolate polyamines exhibit lengthy durations of NO release due to hydrogen bonding stabilization from additional amines. For example, diethylenetriamine (DETA/NO) features an NO release half-life (t1/2) of 20 h.9 In contrast, the NO release from metal cation-stabilized N-diazeniumdiolate adduct of the amino acid proline (PROLI/NO) is rapid with a t1/2 of only 1.8 s.9
Scheme 1.

N-Diazeniumdiolate formation and decomposition of representative secondary amine-bearing compounds, illustrating the difference between metal cation and protonated amine formation and stabilization.
In contrast to the strictly exogenous NONOates, RSNOs are an endogenous class of NO donor touted as the physiological transporters of NO in vivo.10 In the lab, RSNOs are prepared upon reaction of a thiol with a nitrosating agent (e.g., alkyl nitrite, dinitrogen trioxide, or nitrous acid) (Scheme 2).8 Confirmation of RSNO formation is easily made by their color as they absorb light in both the UV (225–261 and 330–350 nm) and the visible (550–600 nm) regions.8
Scheme 2.

S-Nitrosothiol formation and decomposition.
Whereas NONOates decompose and release NO as a function of pH, RSNOs may decompose via multiple triggers.8 For example, thermal and photoinitiated decomposition of RSNOs results in homolytic cleavage of the S–N bond to generate a thiyl radical and NO.10 The thiyl radical further reacts with another RSNO to yield a disulfide and additional NO. Trace copper ions also affect RSNO decomposition.10 Reduction of Cu(II), usually by trace thiolate, forms Cu(I) that subsequently reacts with RSNOs to produce NO, the thiolate and Cu(II). The thiolate may then regenerate Cu(I) from Cu(II), propagating the catalytic decomposition of RSNOs to NO. Transnitrosation or the direct transfer of the nitroso functionality to a free thiol (R’SH) also impacts RSNO stability, resulting in the formation of R’SNO and RSH.10 The newly generated R’SNO then decomposes by one of the aforementioned triggers releasing NO.
Low molecular weight (LMW) NO donors have been used to study the influence of exogenous NO on a number of cardiovascular, central nervous system, and immunological-related disorders.8 These compounds have demonstrated remarkable antiplatelet, antimicrobial, antitumor, and vasodilatory activities with great potential as therapeutics in the biomedical arena. However, the inability to target NO to a specific site of action, the rapid systemic clearance associated with LMW species, toxicity concerns, and the undesirable low doses of NO have hindered their clinical development. As such, much recent research has focused on the synthesis of macromolecular NO-releasing vehicles.
An ideal NO-releasing macromolecular vehicle would be multifunctional and consist of multiple NO donors to achieve a desirable payload. Multifunctionalization would allow for advanced tailoring of the scaffold, including chemistry for targeting delivery, tuning biodistribution, and controlling toxicity. In this tutorial review, we provide an overview of the most promising NO release macromolecular scaffolds including NO donor functionalization of proteins, organic, inorganic, and hybrid polymeric materials. Our goal is to provide a glimpse into this emerging discipline by introducing representative examples of each category. For further breadth, we guide the reader to prior reviews on NO-releasing macromolecular materials.11–15
2. Proteins
The innate specificity associated with biological function makes proteins attractive as macromolecular NO donor scaffolds capable of targeting NO to a site of physiological interest. Seeking the ability to target NO, Hrabie et al. modified the lysine residues within human and bovine serum albumin (BSA) with a methoxymethyl-protected (MOM) diazeniumdiolated piperazine.16 These NO donor conjugates were characterized as having extremely long NO release half-lives (on the order of 20 d) due to the stability of the MOM protecting group that required hydrolysis prior to diazeniumdiolate decomposition to NO. The multiple (59) lysine residues on the albumins enabled enhanced NO donor loading, although mass spectroscopic analysis indicated that only ~20 of the 59 lysine residues were converted to NO donor form. Nevertheless, the authors reported ~40 mol NO per mol of protein with NO release levels approaching ~1 pmol NO mg−1 s−1.
Due to their physiological ubiquity, more work has been done with S-nitrosothiol-modified proteins and biomolecules with respect to NO donor modification. Although the single free cysteine residue on serum albumin restricts storage to 1 mole of RSNO per mole of protein, Stamler and coworkers reduced the 17 disulfide bonds that maintain the tertiary structure of BSA to increase the number of NO reactive thiol sites available for nitrosation, resulting in 19.2 ± 3.1 mol of NO storage (as RSNO) per mol of protein.17 The resulting protein denaturation to enable enhanced NO storage capability was an obvious drawback to this approach. Thus, others have developed more mild chemical modification strategies to store NO onto serum albumin. For example, Ewing et al. polythiolated BSA with N-acetylhomocysteine thiolactone to store 12 ± 3 mol of NO per mol of BSA after nitrosation.18 Unfortunately, the thiolation induced protein oligomerization and a heterogeneous molecular mass. This change in protein structure also affected protein activity. Otagiri and coworkers nitrosated a variant of human serum albumin (HSA), albumin Liprizzi (R410C), that inherently contained an additional cysteine residue at position 410.19 The separation between this cysteine residue and (normal) Cys-34 was hypothesized not to alter the protein structure upon nitrosation. Indeed, the resulting nitrosated macromolecule had a homogenous molecular mass of 67 kDA and could theoretically store 1 more mol of NO per mol of protein than native HSA. Although such modification did not influence protein activity, the total NO loading was improved only slightly (1.3 mol per mol of protein).
To improve the thermal stability of RSNO moieties by decreasing homolytic cleavage of the S–N bond via a cage effect, Katsumi et al. modified BSA with polyethylene glycol (PEG).20 The PEG-modified BSA was then reacted with N-succinimidyl S-acetylthiolate (SATA). Deacetylation of the thiol groups on SATA and subsequent nitrosation yielded the PEG-poly SNO-BSA macromolecule. This NO-releasing vehicle was estimated to have an average molecular mass of 250 kDA and with ~37 mol of PEG and ~10 mol of NO per mol of protein. As expected, the PEG modification both reduced protein aggregation and extended the NO release half-life (147 ± 9 h), roughly 11 times greater than that of nitrosated BSA and 108 times greater than that of the LMW S-nitroso-N-acetyl penicillamine (SNAP). While NO-releasing proteins represent an advancement over LMW analogues, their limited NO payloads ultimately restrict their potential utility.
3. Encapsulation of NO and low molecular weight NO donors
To enhance NO loading, macromolecular vesicles have been used to encapsulate large quantities of NO active species (e.g., gaseous NO or LMW NO donors). As shown in Figure 1A, NO gas encapsulation offers the distinct advantage of utilizing the active therapeutic directly versus only delivering a prodrug requiring an activation trigger to produce NO. The amount of active species encapsulated within such a macromolecular scaffold would be enhanced because space would not be required for the inactive portion of the prodrug. Cavalieri et al. developed poly(vinyl alcohol) (PVA) microbubbles loaded with gaseous NO.21 Briefly, PVA scaffolds were synthesized at pH 5 and at room temperature to yield microbubbles 4.6 ± 0.4 μm in diameter. Exposure of freeze dried microbubbles to 1.5 bar of NO gas for 2 h resulted in NO loading within the microbubbles as verified via electron paramagnetic resonance spectroscopy. Approximately 3.6 μmol of NO was loaded per mg of microbubble with release durations in phosphate buffered saline (PBS) approaching 2 h. Of note, NO was released in the ambient as well indicating spontaneous loss via diffusion through the 0.4 μm thick shell. If stability in NO retention could be improved, the ideal release trigger for these microbubbles would be ultrasonic cracking. Indeed, preliminary studies found that the microbubbles can be sheared open by ultrasonic cracking to release their NO payload instantaneously.
Figure 1.

Representation of macromolecular vesicles encapsulating A) gaseous NO or B) LMW NO donor compounds (blue spheres). Nitric oxide freely diffuses from the scaffold when encapsulated as a gas, but water diffusion through the scaffold shell is necessary to initiate NO release when LMW NO donors are employed.
Liposome encapsulation of gaseous NO has also been investigated as a strategy for storing NO in the presence of scavenging molecules (e.g., hemoglobin).22 Huang et al. synthesized echogenic liposomes using 1,2-dipalmitoyl-sn-glycero-3-ethylphosphocholine, 1,2-dioleoyl-sn-glycero-3-phosphocholine, and cholesterol. The resulting lipid films were dried, rehydrated, and exposed to high pressures of NO. Freezing of the pressurized suspension and subsequent thawing resulted in NO-releasing liposomes. The amount of NO stored within the liposomes was tunable by co-encapsulating NO with argon in various proportions. For example, liposomes loaded with 10% NO and 90% argon released 0.045 μmol NO mg−1. Unfortunately, NO gas encapsulation is hindered by the lack of control over NO release rates. Until modifications to the vessels are possible for regulating NO diffusion, encapsulation of gaseous NO is of little clinical utility.
The encapsulation of LMW NO donor compounds was pursued as a solution to the rapid NO release kinetics characteristic of encapsulated gaseous NO (Figure 1B). Jeh et al. reported a double emulsion and solvent evaporation technique to encapsulate LMW PROLI/NO within poly-lactic-co-glycolic acid (PLGA) and polyethylene oxide-co-lactic acid (PELA) microparticles.23 The PROLI/NO encapsulation was dependent on the microparticle composition with only the PELA microparticles (~2.3 μm diameter) proving capable at trapping PROLI/NO and effectively storing 123 ± 7.6 nM NO per mg. The addition of gelatin as a hydrophilic binding agent only slightly increased the PROLI/NO entrapment efficiency for PELA, but facilitated PROLI/NO storage within the PLGA-based microparticles. Unfortunately, the gelatin and concomitant more hydrophilic structure resulted in accelerated NO release due to increased water uptake.
4. Polymeric organic scaffolds
The first tunable NO-releasing polymeric microparticles were reported by Meyerhoff and coworkers.24 Multiple secondary amine-functionalized methacrylate monomers were employed in free radical benzoyl peroxide-initiated polymerization to yield both homopolymers and methyl methacrylate-based copolymers. A suspension polymerization was utilized with methyl methacrylate, the amine-bearing monomer, and 1,6-hexanedioldimethacrylate as a cross-linking agent to yield polymeric microbeads. Scanning electron microscopy (SEM) indicated the size of the beads as 100–200 μm. Following a deprotection step, exposure to high pressures of NO in sodium methoxide resulted in NO-releasing microbeads with tunable NO storage based on the mol% of the amine-functionalized monomer (i.e., 20 and 40 mol%). A 40 mol% polymethacrylate microbead released 1.05 μmol NO mg−1 over 15 h in physiological conditions. Of note, the high density of amines within the particles was found to increase the local pH and slow diazeniumdiolate decomposition drastically, resulting in only partial (50%) release of the stored NO.
Due to precise control over size and the ability to multifunctionalize their structures to enable targeting and tracking, dendrimers have become ubiquitous as drug delivery vehicles in the biomedical arena.25 Defined generations of branching and corresponding exponential increase in end group surface functionalities are inherent to their chain growth synthesis. As such, their multifunctionality has been utilized to produce NO-releasing macromolecular scaffolds with large reservoirs of NO (Figure 2).26, 27 The potential of dendrimers as powerful NO storage/release vehicles was first reported by Stasko and Schoenfisch using commercially available generation 3 and 5 polypropylenimine dendrimers (DAB-Am-16 and DAB-Am-64 with 16 and 64 primary amine end groups, respectively).26 Since the primary amine-derived dendrimers stored only small amounts of NO (0.44–0.69 μmol mg−1) due to low NO donor conversion, secondary amines with alky tails were synthesized by reacting the dendrimers with heptanoyl chloride. Upon subsequent reduction and NO donor formation at the secondary amine sites, the NO storage levels were dramatically improved (3.2 and 3.5 μmol mg−1 for generation 3 and 5, respectively). Both the enhanced NO storage and NO release durations were attributed to the stability of secondary amine-based diazeniumdiolates and the hydrophobic nature of the alkyl tails, limiting proton-initiated diazeniumdiolate breakdown. The large concentration of amines regenerated upon initial diazeniumdiolate decomposition was postulated to increase local pH and slow further decomposition, resulting in sustained NO release. To further demonstrate the utility of the dendrimers, the DAB-Am-64 was reacted with propylene oxide to yield a scaffold with secondary amines and a more hydrophilic end group. The hydrophilic dendrimer adopted a fully extended conformation in solution that lead to increased diazeniumdiolate and NO storage (5.6 μmol mg−1), but at the expense of more rapid NO release kinetics.
Figure 2.
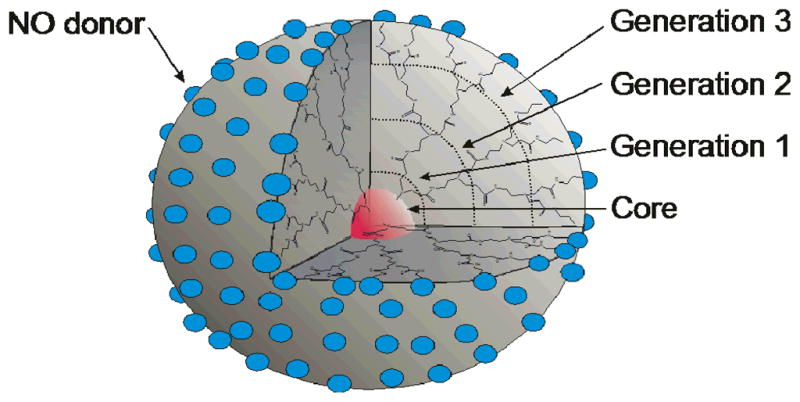
Representation of generation 3 polyamidoamine (PAMAM) dendrimer as a typical dendritic scaffold exhibiting highly branched and defined architecture with resulting surface functionalities modified as NO donors (blue spheres)..
In a subsequent study, Stasko et al. functionalized generation 4 polyamidoamine (PAMAM) dendrimers with either N-acetyl,-D,L-penicillamine or N-acetyl-L-cysteine to yield thiol-terminated dendrimers.27 Nitrosating the macromolecular scaffolds yielded S-nitrosothiol-modified dendrimers (G4-SNAP and G4-NACysNO). Both conjugates achieved appreciable total NO storage of ~2 μmol NO per mg of scaffold, yet their NO release kinetics varied based on the trigger (i.e., copper ion concentration, light). Although LMW tertiary RSNOs are generally regarded as more stable than their primary counterparts, the primary RSNO-derived G4-NACysNO exhibited greater stability/resilience than the tertiary G4-SNAP regardless of the NO release trigger. This effect was attributed to a more compact solution structure for G4-NACysNO that increased the tendency for radical recombination between the thiyl and NO. Overall, this work showcases significant evolution of NO release scaffolds with respect to NO payloads, release durations, and release kinetics.
5. Inorganic/Organic Hybrid Scaffolds
5.1. Metallic nanoparticles
Metallic clusters have become widely studied in biomedicine for their unique size-dependent properties that distinguish them from bulk materials.28 For example, many metallic particles offer the capability of surface plasmon resonance, hyperthermia, and magnetic targeting.29–31 Potential applications for these nanomaterials include molecular imaging diagnostics, drug delivery vehicles, and therapeutic agents.
Rothrock et al. first reported on the synthesis of NO-releasing monolayer-protected cluster (MPC) gold nanoparticles.32 The nanoparticles (~2 nm) were synthesized via the Brust-Schiffrin method and capped with hexanethiol ligands. These ligands were then exchanged with bromoalkane thiols. Subsequent reaction with ethylendiamine, butylamine, hexanediamine, or DETA resulted in secondary amine functionalization that upon exposure to NO gas resulted in NO-releasing gold nanoparticles. Both the NO storage and NO release kinetics were tunable by varying the amount and/or amine structure. While these particles represented the smallest nanometer-sized NO-releasing scaffold to date, the total NO storage was limited (~0.04 μmol NO per mg of scaffold). Due to both restricted NO storage and poor solubility in aqueous media, the potential of these materials as NO delivery agents was concluded to be lacking.
To design water soluble NO-releasing MPCs, Polizzi et al. synthesized tiopronin-protected gold clusters.33 N-Hydroxy-succinimide (NHS) and N-(3-dimethylaminopropyl)-N-ethylcarbodiimide (EDC) chemistry was then used to attach either DETA, tetraethylenepentamine, or pentaethylenehexamine to the carboxylic acid groups of the tiopronin-MPCs. Similar to the water-insoluble secondary amine MPCs, the diazeniumdiolate NO donor conversion efficiency was only ~1%. To increase the NO storage capacity of the gold nanoparticles, polyamine-stabilized MPCs (~5 nm) were synthesized directly (i.e., without tiopronin) with significantly enhanced NO release (up to 0.386 μmol NO per mg of scaffold) and durations (~16 h), depending on the polyamine. However, thermogravimetric analysis indicated that the polyamine ligands were attaching to the gold surface via both primary amine end groups, thus limiting NO donor conversion and overall NO storage.
The gold nanoparticles reported by Schoenfisch and co-workers32, 33 employed N-diazeniumdiolate-modified NO release materials characterized by spontaneous NO release in aqueous media (physiological pH). To enable control over NO release, Sortino and coworkers synthesized water-soluble platinum (Pt) MPCs with a photoactive NO release trigger.34 Thioglycolic acid ligands bound to the Pt nanoparticles (~1 nm) were used to render the particles water soluble. A partial phase exchange of these carboxy-terminated groups with a nitroaromatic-terminated alkane thiol ligand resulted in NO release capable, water soluble particles. While not a traditional NO donor class, the aromatic nitro group proved to be a novel NO-releasing substituent upon irradiation with visible light. Spectroscopic analysis revealed ~6 NO donors per Pt cluster. The NO storage of the material was thermally stable, but capable of liberating NO at a rate of 1.5 pmol s−1 upon visible light irradiation. In a subsequent report, a bifunctional NO release macromolecular scaffold was synthesized with mapping capacity via photoluminescence.35 A thiol-derivatized porphyrin and the NO photodonor ligand were immobilized on the Pt surface via place exchange with some of the carboxy ligands. Neither the NO-releasing nor porphyrin ligand influenced the photobehavior of the other when attached to the Pt surface. However, the porphyrin did influence particle size and more specifically induced particle aggregation (~10 nm aggregates). Water solubility was still retained due to the carboxy-terminated ligands. While the amount of porphyrin and NO photodonor ligand was tunable by varying the reaction time and/or molar ratio used in the exchange, the authors noted that reaction times longer than 30 min resulted in extensive replacement of the carboxy ligands, thus sacrificing the aqueous solubility of the particles. As expected, the NO storage of the Pt nanoparticle assemblies was stable in the dark at room temperature. Upon irradiation with visible light, nanomolar levels of NO were released at a rate of ~0.9 nM s−1. Unfortunately, the NO release kinetics were not normalized to mass of particles and thus direct comparison of these scaffolds to other NO-releasing macromolecular vehicles remains unclear.
5.2 Zeolites and molecular organic frameworks
Zeolites and metal organic frameworks (MOFs) are highly porous materials that have found wide utility in the fields of ion exchange, catalysis, and gas adsorption/storage.36–39 Morris and coworkers have pioneered the application of zeolites and MOFs to the field of NO release scaffolds.40 For zeolitic structures, NO is chemisorbed to cations associated with the inorganic framework. These nitrosyl complexes may then be displaced with an appropriate nucleophile (e.g., water) to initiate NO release. In an initial study, Wheatley et al. used Zeolite-A, an alternating alumina/silica network with cobalt cations to chemisorb NO.41 These materials were capable of storing ~1.2–1.3 μmol NO per mg of zeolite. Unfortunately, this material was not water soluble and the only way to reproducibly measure the NO release was by exposure to a steady stream of wet gas with controlled humidity. Using a stream of nitrogen at 11% relative humidity (RH), these materials were shown to release 1.02 μmol NO per mg of zeolite over 2500 s with an associated half-life of ~340 s. Altering the humidity of the gas influenced the NO kinetics. More humid gas (22% RH) decreased the half-life to 208 s, while a drier gas (1.5% RH) increased the NO release half-life to >3000 s. The identity and amount of the metal ion (e.g., Co, Ni, Cu, Mn, and Na) within the zeolite channels was also shown to influence NO release based on the affinity of each metal to bind NO, with Co storing the greatest amount.
Xiao et al. first reported the ability to store NO using a MOF consisting of copper benzene tricarboxylate referred to as HKUST-1.42 The material was synthesized by mixing Cu(NO3)2·3H2O and benzene 1,3,5-tricarboxylic acid in a 50:50 ethanol:water solution for 30 min at ambient temperature followed by heating at 383 K for 24 h. The dried material was then loaded with NO by exposure to pressurized NO gas (1 bar) at 196 K. The HKUST-1 could adsorb ~9 μmol of NO per mg of MOF. Desorption of the material revealed that most of the NO was physisorbed within the pores of the material, but roughly 2.21 μmol mg−1 was strongly chemisorbed to open metal sites (i.e., unsaturated with ligands) at approximately 1 NO equivalent per dicopper(II) tetracarboxylate group. Exposing the NO-loaded HKUST-1 to a stream of wet nitrogen gas resulted in water displacing only a fraction of the chemisorbed NO (~1 nmol NO per mg of MOF).41 The lack of dispersal in solution, the inaccessibility of the total NO stored payloads, and potential cytotoxicity to healthy cells circumvents therapeutic use or potential of this MOF scaffold at this stage. Of note, more recent NO-releasing MOFs have shown improved delivery behavior over HKUST-1 with nearly total release (~7 μmol NO per mg of MOF) achieved in one example.43
To overcome the finite reservoir of available NO, Böes et al. synthesized copper-containing zeolites that both released ~0.002 μmol of chemisorbed NO per mg of scaffold within ~60 min of solution immersion and then catalytically produced NO via reduction of nitrite at Cu(I) sites.44 However, the potential toxicity of the Cu(I) sites lessens the enthusiasm for these materials as therapeutics.
Other researchers have focused on overcoming the rapid release of chemisorbed NO coordinated to metal sites in conventional MOF structures.45–47 For example, Nguyen et al. attempted to incorporate NO donors within MOF scaffolds by employing 2-amino-1,4-benzenedicarboxylic acid as an organic linker between metal sites.46 By forming N-diazeniumdiolates on pendant amines of the linker, the resulting materials released up to ~0.51 μmol NO per mg of MOF. Unfortunately, the release was still rapid (~5 min) and the materials lost ~20% of their NO payload after 10 d of storage due to the well-known instability of primary amine-based diazeniumdiolates.9 Of note, the parent MOFs (i.e., IRMOF-3 and UMCM-1-NH2) used for these studies lack open metal sites characteristic of the aforementioned NO-releasing MOFs (e.g., HKUST-1). Thus, the NO was not chemisorbed at metal centers concomitantly with diazeniumdiolate formation on the organic linkers of these scaffolds (Figure 3). A MOF design that incorporates both features of NO functionalization may hold greater therapeutic potential.
Figure 3.

Representations of A) a MOF with chemisorbed NO at its metal sites (red spheres) and B) of IRMOF-3 with diazeniumdiolate formation on its organic bridging ligands.
5.3 Silica particles
Silica-based materials have become ubiquitous in the biomedical arena due to their straight forward synthesis that enables customization of size, morphology, and composition.48–51 Furthermore, silica is well regarded as a stable and nontoxic drug delivery vehicle. Among the first reports of silica as a macromolecular scaffold for NO release, Zhang et al. grafted aminosilanes to the surface of fumed silica particles (0.2–0.3 μm).52 Roughly, 50–70% of the surface silanols were functionalized. As a result, NO storage was rather limited (~0.6 μmol NO per mg of silica). While the structure of the precursor amine influenced the rate of NO release, the dissociation kinetics were complex with little correlation to structure. However, the NO release half-lives of the particles were significantly longer than their solution phase LMW analogs. This phenomenon was attributed to local surface increases in pH due to the large concentration of amines at the silica surface following NO donor decomposition, illustrating a benefit afforded solely by the macromolecular scaffold.
In subsequent work, Frost et al. grafted fumed silica particles (7–10 nm) with APTMS and then linked cysteine, N-acetyl-L-cysteine, or N-acetyl-DL-penicillamine to the particles using amide chemistry.53 The derivatization yielded free thiols up to ~0.142 μmol per mg of particles. Upon treatment with an organic nitrosating agent (i.e., t-butylnitrite), RSNOs were formed on the silica scaffolds, with the S-nitroso-N-acetylpenicillamine (SNAP) storing the greatest payload (~0.138 μmol NO per mg). The authors postulated that the smaller size of the protected thiolactone for the SNAP coupling led to more thiol and resulting NO loading. Similar to LMW RSNO analogs, the NO release levels of the particles were found to be dependent on the concentration of copper and light exposure.
Although capable of imparting NO release, a disadvantage of surface grafting as a strategy for functionalizing silica is the limited and low NO loading per particle.52, 53 In contrast, sol-gel chemistry is the hydrolysis and condensation of silanes that allows for the formation of silica scaffolds with organo functionalities throughout, enabling unparalleled tunability in terms of particle, size, morphology, and composition.48 Shin et al. first reported the co-condensation of a number of aminoalkoxysilane precursors including N-(6-aminohexyl)-3-aminopropyltrimethoxysilane (AHAP3), (aminoethylamino-methyl)phenethyltrimethoxysilane (AEMP3), and N-(2-aminoethyl)-3-aminopropyltrimethoxysilane (AEAP3) with either tetramethoxysilane (TMOS) or tetraethoxysilane (TEOS) in solutions of alcohol, water, and ammonia.54 As anticipated, both the aminosilane concentration and size of the particles were tunable from ~10–77 mol% and ~20–500 nm, respectively. Exposing the particles to NO gas (5 atm) in a mixture of N,N-dimethylformamide, methanol, and sodium methoxide for 3 d resulted in NO storage ranging from ~0.05–1.78 μmol per mg of silica, with the greatest NO storage attainable for 77 mol% AHAP3/TMOS. Of note, this value is roughly three times larger than fumed silica grafted with equivalent NO donors.52 In addition, the sol-gel-derived silica particles released NO for significantly longer periods (12 vs. 2.4 h half-lives for sol-gel vs. surface grafted fumed silica, respectively).52
Despite achieving NO release levels that were significantly greater than prior NO-releasing silica particle systems, the authors noted limited (~5–50 %) NO donor conversion efficiencies.54 Although sol-gel chemistry allows for incorporation of amine functionalities throughout the silica scaffold, the limited particle porosity and amine mobility result in lower than expected diazeniumdiolation and NO donor stabilization, respectively. In a subsequent paper, Shin and Schoenfisch improved the NO storage/release from these vehicles by forming the diazeniumdiolate groups on the aminosilane precursors before particle synthesis.55 As shown in Figure 4, this synthetic strategy enhances the degree of NO donor functionality incorporation within the particle when compared to surface grafting and post-synthesis diazeniumdiolate modification routes. In addition to NO donor conversion efficiencies approaching 99%, the total NO storage for these particles ranged from 0.5–11.3 μmol mg−1. The greatest NO storage (11.3 μmol mg−1) was achieved using N-diazeniumdiolated N-(6-aminohexyl)aminomethyltrimethoxysilane (AHAM3). While the NO release kinetics of AHAM3 particles were rapid (NO release half-lives of 4 min), other particle systems (e.g., AEMP3 and (3-trimethoxysilylpropyl)diethylenetriamine (DET3)) were shown to release NO for significantly greater periods (253 min and 101 h, respectively.) The assorted NO release characteristics for NO donor-modified silica exemplify the benefit of the sol-gel approach for creating diverse NO release scaffolds.
Figure 4.

Representations of NO-releasing silica particles modified with NO donors (blue spheres) A) via surface grafting, B) after particle synthesis and C) on silane precursors before particle synthesis via sol-gel chemistry. Cross section of particle interior depicts a lack of functionality within surface-grafted particles and the presence of non-modified precursor functionalities (white circles) within traditional sol-gel synthesized particles.
Riccio et al. also employed sol-gel chemistry to synthesize S-nitrosothiol-modified silica particles using 3-mercaptopropyltrimethoxysilane (MPTMS).56 The ratio of MPTMS to either TMOS or TEOS was tuned to achieve thiol concentrations up to 85 mol%. After exposure to acidified nitrite, the incorporated thiols were converted to S-nitrosothiol form with corresponding NO storage approaching 4.40 μmol per mgof particle. As anticipated, the particles exhibited NO release levels dependent on heat, light, and/or copper concentration. At 37 °C in PBS the particles released only ~37% of their total NO storage, illustrating the extended release duration achievable with S-nitrosothiol-modified silica. The MPTMS-based silica also exhibited much greater control over particle monodispersity, size, and morphology than the previous N-diazeniumdiolate systems54, 55 due in part to the controlled addition of silane precursors to the ethanol, water, and ammonia solution via a syringe pump. Additionally, only specific reaction conditions (e.g., high ammonia concentration, low total silane concentration) yielded spherical, non-aggregated particles ranging in size from ~250–700 nm depending on MPTMS, backbone silane (i.e., TEOS vs. TMOS), and water concentrations. Generally, greater amounts of water in the reaction mixture yielded smaller particles without significantly altering the sulfur weight percent and corresponding NO storage.
To control both particle size and monodispersity for aminosilane-derived silica particles, Carpenter et al. developed a reverse microemulsion sol-gel chemistry method in which reaction solvent, volume, and time were varied.57 Tuning particle size into the nanometer range while maintaining monodispersity necessitated a core/shell design. While this design restricted the aminosilane content to the shell, the concentration of aminosilane was consistent for three particle sizes (50, 100 and 200 nm in diameter) as verified by elemental analysis. Of note, the N-diazeniumdiolate conversion efficiency was greater for 50 nm particles, compared to the two larger particles. The authors attributed the enhanced NO donor formation to an increased surface area to volume ratio resulting in more amines accessible at the surface. Nevertheless, the NO release kinetics were comparable for all sizes with NO storage totals of ~1.0–1.5 μmol NO per mg of scaffold.
6. Nitric Oxide-Releasing Coatings
6.1 Coatings with noncovalently incorporated LMW NO donors
The vast NO release characteristics achievable with macromolecular scaffolds has enabled the development of NO release materials for specific biomedical applications (see Nitric Oxide Release Part II: Therapeutic Applications). For applications that necessitate NO release directed at an interface (e.g., an indwelling medical device), NO-releasing polymeric coatings have been synthesized to elicit localized NO release from a surface. In the first report of NO-releasing coatings, Smith et al. doped LMW NO donor compounds noncovalently within polymeric matrices (Figure 5A) to establish NO-releasing polymers.58 More specifically, N-diazeniumdiolated DETA and polycaprolactone were mixed to yield a biodegradable polymer that released NO upon breaking down (56 nmol of NO per mg over 1 week). In attempting to design hydrophobic NO-releasing polyurethanes and poly(vinyl chloride)s (PVC), Mowery et al. observed leaching of LMW NO donors, raising concerns about cytotoxicity and nonlocalized NO release.59 To avoid leaching, Batchelor et al. designed LMW NO donors to be more lipophilic (e.g., N-diazeniumdiolated dialkylhexamethylenediamines with alkyl groups ranging from methyl to didodecyl) and thus enhance their retention within hydrophobic polymers.60 In this work, a correlation between water uptake by dioctyl sebacate-plasticized PVC and NO release kinetics was reported. As expected, films with greater polymer-to-plasticizer ratio (2:1 vs. 1:2) were characterized by decreased water uptake and NO flux (~17 pmol cm−2 s−1 vs. ~100 pmol cm−2 s−1) with extended release duration (100 vs. 25 h). While leaching was not quantified, it was surmised to be negligible based on the NO donor structure and slowed NO release kinetics.
Figure 5.

Representations of strategies to fabricate NO-releasing surfaces including A) doping of LMW NO donors (small blue spheres), B) covalent tethering of NO donor functionalities to the polymer backbone, and C) doping of NO-releasing macromolecular scaffolds (large blue spheres) within the polymer. Leaching of LMW dopants and the limited NO reservoir of covalently-modified polymers are schematically depicted.
6.2 Coatings with covalently attached NO donors
As shown in Figure 5B, an alternative strategy for alleviating LMW dopant leaching is covalent binding of the NO donor or NO donor precursor to the polymer backbone. In the earliest report of NO-releasing polymers,58 Smith et al. crosslinked poly(ethylenimine) with 1,4-dibutanediol diglycidyl to form an insoluble amine-containing polymeric coating material. After NO donor formation on the secondary amines, the materials released NO for up to 5 weeks. While the NO release levels were low (i.e., ~0.33 pmol mg−1 s−1), larger levels approaching ~30 pmol mg−1 s−1 were achieved with thicker coatings. Additional examples of covalent coupling of diazeniumdiolates to polymeric scaffolds include PVC,61 silicone rubber,62 and polyurethane.63 Likewise, S-nitrosothiol NO donors have been covalently linked to polyester64, 65 and polyurethane.66 For a more detailed description of NO-releasing polymers the reader is directed to reviews by Frost et al.,11 Hetrick and Schoenfisch,12 and Varu et al.13
Akin to the synthesis of silica nanoparticles, sol-gel chemistry has been used to prepare NO-releasing xerogel films, with the earliest reports combining aminosilanes and isobutyltrimethoxysilane to yield amine-modified xerogels.67–69 Varying the aminosilane concentration resulted in xerogels with tunable NO release. As expected, the chemical environment around the aminosilane affected NO storage and release kinetics. For example, xerogels comprised of DET3 exhibited the greatest diazeniumdiolate conversion efficiency due to the enhanced deprotonation resulting from the additional amines. Nitric oxide fluxes approaching 400 pmol cm−2 s−1 were achieved using AHAP3-derived xerogels on stainless steel substrates, illustrating versatility of the sol-gel method (for coating almost any substrate).70 In some cases, the xerogel integrity was found to be compromised upon immersion in physiological solution (i.e., PBS). Nablo et al. applied thin (i.e., 10–30 μm) PVC films over the N-diazeniumdiolated xerogels to inhibit undesirable continued hydrolysis of the siloxane backbone, thus enhancing stability and reducing fragmentation.71 Additionally, the hydrophobic PVC was shown to hinder water diffusion, thus reducing the initial NO flux and prolonging NO release duration.
In a subsequent study, Riccio et al. designed thiol-modified xerogels derived from MPTMS and methyltrimethoxysilane.72 Subsequent nitrosation resulted in the formation of S-nitrosothiols. These xerogel films exhibited negligible network fragmentation and NO fluxes dependent on the MPTMS concentration, analogous to the diazeniumdiolate-modified xerogels. Furthermore, NO release could be triggered thermally and/or upon copper or light exposure. Under physiological conditions, the films released NO for up to 2 weeks with a maximum NO flux and total payload of ~600 pmol cm−2 s−1 and 1.31 μmol mg−1, respectively. While the enhanced stability and larger NO storage suggest these films may be highly promising for biomedical applications, the lack of commercially available mercaptosilane precursors limits the tunability of NO release kinetics from these materials. Furthermore, they must be stored at cold temperatures and in the dark to circumvent premature breakdown of the RSNO NO donor.
6.3 Macromolecular scaffold-doped coatings
Due to leaching of LMW NO donors and limited NO payloads for covalently bound NO donors, recent studies have evaluated the incorporation of macromolecular dopants within polymers whereby the larger size and greater reservoir of NO inherent to these scaffolds may circumvent their leaching and enhance NO storage/delivery from the coating (Figure 5C). Indeed, a number of the aforementioned macromolecular scaffolds (e.g., zeolites and silica particles) have been subsequently used as NO-releasing dopants within polymeric coatings.41, 52–54, 73, 74 For example, Koehler et al. encapsulated SNAP within ~150 nm liposome vesicles and then immobilized the liposomes within a TMOS xerogel to create a composite capable of releasing ~14 pmol NO cm−2 s−1 after photoirradiation.75 While the authors postulated that the liposome barrier would hinder SNAP leaching, the NO release studies indicated SNAP associated with the exterior of the vesicles during liposome preparation. Thus, additional washing of the liposomes was necessary to ensure removal of any non-encapsulated SNAP before incorporation within the composite film. In other work, Wheatley et al. reported on pressed poly(tetrafluoroethylene) and poly(dimethylsiloxane) discs using NO-releasing zeolites.41 The addition of the polymer matrix enabled zeolite immersion in water (not possible for zeolite powders) and extended NO release durations (half-lives approaching ~1 h). Of note, the zeolites were loaded with NO after polymer embedding thus sacrificing the total achievable NO payload (only up to ~0.02 μmol per mg).
Zhang et al. doped N-diazeniumdiolated surface-grafted silica within polyurethane at 20 wt% to enable NO release at a flux of ~7 pmol cm−2 s−1.52 Likewise, Frost and Meyerhoff doped RSNO-modified surface-grafted silica particles within trilayer polymeric films.53, 73 However, the hydrophobicity of the particle-doped silicone rubber blocked diffusion of copper ions into the film. As such, light irradiation was the only means of initiating NO release with fluxes approaching only ~1 pmol cm−2 s−1 depending on the light intensity, polymer film thickness, and identity and concentration of the grafted RSNO. Most recently, Koh et al. doped N-diazeniumdiolate- and S-nitrosothiol-based silica nanoparticles into a range of polyurethane blends to create an array of NO-releasing membranes.76 The reported NO release durations (16 h to 14 d) were dependent on both the type and concentration of the macromolecular dopant with NO fluxes approaching ~2500 pmol cm−2 s−1. As might be expected, the NO release kinetics from polymers doped with N-diazeniumdiolate-modified particles could be further tuned by altering the chemical identity and thickness of the polyurethane matrix, as water uptake by the polymer will dictate diazeniumdiolate decomposition.
7. Conclusions
As summarized in Table 1, the development of macromolecular NO release scaffolds is a burgeoning field with great therapeutic potential due to widely tunable NO release characteristics (e.g., payload, duration, and level of release) and scaffold properties (e.g., size, surface functionalities, chemical composition). While strategies to enhance the amount and sustain the duration of NO release have marked the early development of such materials, precise control over the delivery location is still lacking. Future research is focused on multifunctionalization of such scaffolds to enable more targeted NO delivery (Figure 6). Targeting strategies that have been developed for other therapeutics including the use of magnetic cores and antibody ligands should be feasible with NO-releasing macromolecular vehicles.77 Ultimately, the fate and interaction of the vehicles in vivo must be understood to fully realize the potential of NO-releasing macromolecular scaffolds. Fortunately, the chemical flexibility of these scaffolds also enables the ability to incorporate tracking moieties (e.g., fluorescent labels) to aid in such investigations.
Table 1.
Nitric oxide storage characteristics for notable examples within each macromolecular scaffold category.
| NO-releasing scaffold | NO donor modification | Category | Total NO storage | Ref. |
|---|---|---|---|---|
| serum albumin | N-diazeniumdiolate | protein | 40 mol per mol | [16] |
| serum albumin | S-nitrosothiol | protein | 20 mol per mol | [17] |
| polyethylene glycol-modified, polythiolated-serum albumin | S-nitrosothiol | protein | 10 mol per mol | [20] |
| poly(vinyl alcohol) microbubble | n/a (gaseous NO) | encapsulation | 3.6 μmol per mg | [21] |
| polyethylene oxide-co-lactic acid microparticle | N-diazeniumdiolate | encapsulation | 125 nM per mg | [23] |
| polymethacrylate microbead | N-diazeniumdiolate | polymeric organic scaffolds | 1.05 μmol per mg | [24] |
| generation 5 polypropylenimine dendrimer | N-diazeniumdiolate | polymeric organic scaffolds | 5.6 μmol per mg | [26] |
| generation 4 polyamidoamine dendrimer | S-nitrosothiol | polymeric organic scaffolds | 2.0 μmol per mg | [27] |
| monolayer-protected cluster (MPC) gold nanoparticle | N-diazeniumdiolate | metallic nanoparticles | 0.04 μmol per mg | [32] |
| polyamine-stabilized MPC gold nanoparticle | N-diazeniumdiolate | metallic nanoparticles | 0.39 μmol per mg | [33] |
| platinum (Pt) MPC | aromatic nitro group | metallic nanoparticles | 6 NO donors per Pt cluster | [34] |
| Zeolite-A | n/a (coordinated NO) | zeolites | 1.3 μmol per mg | [41] |
| HKUST-1 | n/a (coordinated NO) | molecular organic frameworks | 2.21 μmol per mg | [42] |
| IRMOF-3 | N-diazeniumdiolate | molecular organic frameworks | 0.51 μmol per mg | [46] |
| surface-grafted silica particle | N-diazeniumdiolate | silica particles | 0.6 μmol per mg | [52] |
| surface-grafted silica particle | S-nitrosothiol | silica particles | 0.14 μmol per mg | [53] |
| sol-gel synthesized silica particle (post-synthesis modification) | N-diazeniumdiolate | silica particles | 1.78 μmol per mg | [54] |
| sol-gel synthesized silica particle (pre-synthesis modification) | N-diazeniumdiolate | silica particles | 11.3 μmol per mg | [55] |
| sol-gel synthesized silica particle | S-nitrosothiol | silica particles | 4.40 μmol per mg | [56] |
Figure 6.

Representation of a multifunctionalized NO release scaffold outfitted with multiple NO donors (blue spheres), fluorescent labels (pink stars) for vehicle tracking, and targeting ligands (orange cubes) and a magnetic core (green sphere) for targeted NO delivery.
With respect to devices whereby the physiological interface is critical to biocompatibility, the design of polymeric coatings capable of releasing NO from a surface represents a fresh strategy for addressing biofouling-related problems (e.g., platelet and bacterial adhesion). The use of macromolecular NO release scaffolds as polymeric dopants represents a promising strategy for imparting NO release to the medical device with minimal impact on its function. Indeed, the size and NO storage amounts inherent to such NO release scaffolds enhances both material stability (i.e., no dopant leaching) and NO delivery. As the development of NO-releasing surfaces continues, future research may include materials capable of generating NO from endogenous sources (i.e., RSNOs). Meyerhoff and coworkers have recently derivatized polymers with Cu(II)-cyclen moieties that produce NO fluxes approaching ~75 pmol cm−2 s−1 in physiological solutions containing GSNO and glutathione.78
Arguably, NO-releasing materials with complete spatial and temporal control represent the ultimate goal in this field. Precisely controlled NO release, for instance when triggered by photoirradiation,15 would enable a clearer understanding of the NO release levels and durations required to elicit specific physiological responses. With the widespread and concentration dependent roles of NO in physiology and pathophysiology, such knowledge is paramount to harnessing the therapeutic potential of NO pharmacologically.
Acknowledgments
The authors acknowledge financial support from National Institute of Health (EB000708).
Biographies
 Daniel A. Riccio received his BA in Chemistry in 2006 from Drew University and his PhD in Chemistry in 2011 from the University of North Carolina at Chapel Hill. His dissertation research was focused on the design of S-nitrosothiol-derived nitric oxide release materials. He is currently a Postdoctoral Research Associate for the Department of Veterans Affairs in Durham, NC. His current research interests include endogenous S-nitrosothiols and their role in neonatal circulation as well as during red blood cell storage.
Daniel A. Riccio received his BA in Chemistry in 2006 from Drew University and his PhD in Chemistry in 2011 from the University of North Carolina at Chapel Hill. His dissertation research was focused on the design of S-nitrosothiol-derived nitric oxide release materials. He is currently a Postdoctoral Research Associate for the Department of Veterans Affairs in Durham, NC. His current research interests include endogenous S-nitrosothiols and their role in neonatal circulation as well as during red blood cell storage.
 Mark Schoenfisch is a Professor of Chemistry in the Department of Chemistry at the University of North Carolina at Chapel Hill (UNC-Chapel Hill). Dr. Schoenfisch received undergraduate degrees in Chemistry (BA) and Germanic Languages and Literature (BA) at the University of Kansas prior to attending the University of Arizona for graduate studies in Chemistry (PhD). Before starting at UNC-Chapel Hill, he spent two years as a National Institutes of Health Postdoctoral Fellow at the University of Michigan. His research interests include analytical sensors, biomaterials, and the development of nitric oxide release scaffolds as new therapeutics.
Mark Schoenfisch is a Professor of Chemistry in the Department of Chemistry at the University of North Carolina at Chapel Hill (UNC-Chapel Hill). Dr. Schoenfisch received undergraduate degrees in Chemistry (BA) and Germanic Languages and Literature (BA) at the University of Kansas prior to attending the University of Arizona for graduate studies in Chemistry (PhD). Before starting at UNC-Chapel Hill, he spent two years as a National Institutes of Health Postdoctoral Fellow at the University of Michigan. His research interests include analytical sensors, biomaterials, and the development of nitric oxide release scaffolds as new therapeutics.
References
- 1.Ignarro LJ. Angew Chem-Int Edit. 1999;38:1882–1892. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1882::AID-ANIE1882>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 2.Walford G, Loscalzo J. J Thromb Haemost. 2003;1:2112–2118. doi: 10.1046/j.1538-7836.2003.00345.x. [DOI] [PubMed] [Google Scholar]
- 3.Fang FC. J Clin Invest. 1997;99:2818–2825. doi: 10.1172/JCI119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo JD, Chen AF. Acta Pharmacol Sin. 2005;26:259–264. doi: 10.1111/j.1745-7254.2005.00058.x. [DOI] [PubMed] [Google Scholar]
- 5.Mocellin S, Bronte V, Nitti D. Med Res Rev. 2007;27:317–352. doi: 10.1002/med.20092. [DOI] [PubMed] [Google Scholar]
- 6.Ignarro LJ. Nitric Oxide: Biology and Pathobiology. Academic Press; 2000. [Google Scholar]
- 7.Hetrick EM, Schoenfisch MH. Annu Rev Anal Chem. 2009;2:409–433. doi: 10.1146/annurev-anchem-060908-155146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang PG, Xian M, Tang XP, Wu XJ, Wen Z, Cai TW, Janczuk AJ. Chem Rev. 2002;102:1091–1134. doi: 10.1021/cr000040l. [DOI] [PubMed] [Google Scholar]
- 9.Hrabie JA, Keefer LK. Chem Rev. 2002;102:1135–1154. doi: 10.1021/cr000028t. [DOI] [PubMed] [Google Scholar]
- 10.Williams DLH. Accounts Chem Res. 1999;32:869–876. [Google Scholar]
- 11.Frost MC, Reynolds MM, Meyerhoff ME. Biomaterials. 2005;26:1685–1693. doi: 10.1016/j.biomaterials.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 12.Hetrick EM, Schoenfisch MH. Chem Soc Rev. 2006;35:780–789. doi: 10.1039/b515219b. [DOI] [PubMed] [Google Scholar]
- 13.Varu VN, Tsihlis ND, Kibbe MR. Vasc Endovasc Surg. 2009;43:121–131. doi: 10.1177/1538574408322752. [DOI] [PubMed] [Google Scholar]
- 14.Seabra AB, Duran N. J Mater Chem. 2010;20:1624–1637. [Google Scholar]
- 15.Sortino S. Chem Soc Rev. 2010;39:2903–2913. doi: 10.1039/b908663n. [DOI] [PubMed] [Google Scholar]
- 16.Hrabie JA, Saavedra JE, Roller PP, Southan GJ, Keefer LK. Bioconjugate Chem. 1999;10:838–842. doi: 10.1021/bc990035s. [DOI] [PubMed] [Google Scholar]
- 17.Simon DI, Mullins ME, Jia L, Gaston B, Singel DJ, Stamler JS. Proc Natl Acad Sci U S A. 1996;93:4736–4741. doi: 10.1073/pnas.93.10.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ewing JF, Young DV, Janero DR, Garvey DS, Grinnell TA. J Pharmacol Exp Ther. 1997;283:947–954. [PubMed] [Google Scholar]
- 19.Ishima Y, Sawa T, Kragh-Hansen U, Miyamoto Y, Matsushita S, Akaike T, Otagiri M. J Pharmacol Exp Ther. 2007;320:969–977. doi: 10.1124/jpet.106.114959. [DOI] [PubMed] [Google Scholar]
- 20.Katsumi H, Nishikawa M, Yamashita F, Hashida M. J Pharmacol Exp Ther. 2005;314:1117–1124. doi: 10.1124/jpet.105.087429. [DOI] [PubMed] [Google Scholar]
- 21.Cavalieri F, Finelli I, Tortora M, Mozetic P, Chiessi E, Polizio F, Brismar TB, Paradossi G. Chem Mater. 2008;20:3254–3258. [Google Scholar]
- 22.Huang SL, Kee PH, Kim H, Moody MR, Chrzanowski SM, MacDonald RC, McPherson DD. J Am Coll Cardiol. 2009;54:652–659. doi: 10.1016/j.jacc.2009.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeh HS, Lu S, George SC. J Microencapsul. 2004;21:3–13. doi: 10.1080/02652040310001619767. [DOI] [PubMed] [Google Scholar]
- 24.Parzuchowski PG, Frost MC, Meyerhoff ME. J Am Chem Soc. 2002;124:12182–12191. doi: 10.1021/ja020268l. [DOI] [PubMed] [Google Scholar]
- 25.Mintzer MA, Grinstaff MW. Chem Soc Rev. 2011;40:173–190. doi: 10.1039/b901839p. [DOI] [PubMed] [Google Scholar]
- 26.Stasko NA, Schoenfisch MH. J Am Chem Soc. 2006;128:8265–8271. doi: 10.1021/ja060875z. [DOI] [PubMed] [Google Scholar]
- 27.Stasko NA, Fischer TH, Schoenfisch MH. Biomacromolecules. 2008;9:834–841. doi: 10.1021/bm7011746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhattacharya R, Mukherjee P. Adv Drug Deliv Rev. 2008;60:1289–1306. doi: 10.1016/j.addr.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 29.Cherukuri P, Glazer ES, Curleya SA. Adv Drug Deliv Rev. 2010;62:339–345. doi: 10.1016/j.addr.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao R, Xing RJ, Xu ZC, Hou YL, Gao S, Sun SH. Adv Mater. 2010;22:2729–2742. doi: 10.1002/adma.201000260. [DOI] [PubMed] [Google Scholar]
- 31.Liao HW, Nehl CL, Hafner JH. Nanomedicine. 2006;1:201–208. doi: 10.2217/17435889.1.2.201. [DOI] [PubMed] [Google Scholar]
- 32.Rothrock AR, Donkers RL, Schoenfisch MH. J Am Chem Soc. 2005;127:9362–9363. doi: 10.1021/ja052027u. [DOI] [PubMed] [Google Scholar]
- 33.Polizzi MA, Stasko NA, Schoenfisch MH. Langmuir. 2007;23:4938–4943. doi: 10.1021/la0633841. [DOI] [PubMed] [Google Scholar]
- 34.Caruso EB, Petralia S, Conoci S, Giuffrida S, Sortino S. J Am Chem Soc. 2007;129:480–481. doi: 10.1021/ja067568d. [DOI] [PubMed] [Google Scholar]
- 35.Barone M, Mascali A, Sortino S. New J Chem. 2008;32:2195–2200. [Google Scholar]
- 36.Ferey G. Chem Soc Rev. 2008;37:191–214. doi: 10.1039/b618320b. [DOI] [PubMed] [Google Scholar]
- 37.Natarajan S, Mahata P. Chem Soc Rev. 2009;38:2304–2318. doi: 10.1039/b815106g. [DOI] [PubMed] [Google Scholar]
- 38.Rosseinsky MJ. Microporous Mesoporous Mat. 2004;73:15–30. [Google Scholar]
- 39.McKinlay AC, Morris RE, Horcajada P, Ferey G, Gref R, Couvreur P, Serre C. Angew Chem-Int Edit. 2010;49:6260–6266. doi: 10.1002/anie.201000048. [DOI] [PubMed] [Google Scholar]
- 40.Hinks NJ, McKinlay AC, Xiao B, Wheatley PS, Morris RE. Microporous Mesoporous Mat. 2010;129:330–334. [Google Scholar]
- 41.Wheatley PS, Butler AR, Crane MS, Fox S, Xiao B, Rossi AG, Megson IL, Morris RE. J Am Chem Soc. 2006;128:502–509. doi: 10.1021/ja0503579. [DOI] [PubMed] [Google Scholar]
- 42.Xiao B, Wheatley PS, Zhao XB, Fletcher AJ, Fox S, Rossi AG, Megson IL, Bordiga S, Regli L, Thomas KM, Morris RE. J Am Chem Soc. 2007;129:1203–1209. doi: 10.1021/ja066098k. [DOI] [PubMed] [Google Scholar]
- 43.McKinlay AC, Xiao B, Wragg DS, Wheatley PS, Megson IL, Morris RE. J Am Chem Soc. 2008;130:10440–10444. doi: 10.1021/ja801997r. [DOI] [PubMed] [Google Scholar]
- 44.Boës AK, Xiao B, Megson IL, Morris RE. Top Catal. 2009;52:35–41. [Google Scholar]
- 45.Ingleson MJ, Heck R, Gould JA, Rosseinsky MJ. Inorg Chem. 2009;48:9986–9988. doi: 10.1021/ic9015977. [DOI] [PubMed] [Google Scholar]
- 46.Nguyen JG, Tanabe KK, Cohen SM. Crystengcomm. 2010;12:2335–2338. [Google Scholar]
- 47.Wei F, Yang JY, Hou QA, Zhu JH. New J Chem. 2010;34:2897–2905. [Google Scholar]
- 48.Gupta R, Kumar A. Biomed Mater. 2008;3 doi: 10.1088/1748-6041/3/3/034005. [DOI] [PubMed] [Google Scholar]
- 49.Rosenholm JM, Sahlgren C, Linden M. Nanoscale. 2010;2:1870–1883. doi: 10.1039/c0nr00156b. [DOI] [PubMed] [Google Scholar]
- 50.Slowing, Vivero-Escoto JL, Wu CW, Lin VSY. Adv Drug Deliv Rev. 2008;60:1278–1288. doi: 10.1016/j.addr.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 51.Vivero-Escoto JL, Slowing, Trewyn BG, Lin VSY. Small. 2010;6:1952–1967. doi: 10.1002/smll.200901789. [DOI] [PubMed] [Google Scholar]
- 52.Zhang HP, Annich GM, Miskulin J, Stankiewicz K, Osterholzer K, Merz SI, Bartlett RH, Meyerhoff ME. J Am Chem Soc. 2003;125:5015–5024. doi: 10.1021/ja0291538. [DOI] [PubMed] [Google Scholar]
- 53.Frost MC, Meyerhoff ME. J Biomed Mater Res Part A. 2005;72A:409–419. doi: 10.1002/jbm.a.30275. [DOI] [PubMed] [Google Scholar]
- 54.Shin JH, Metzger SK, Schoenfisch MH. J Am Chem Soc. 2007;129:4612–4619. doi: 10.1021/ja0674338. [DOI] [PubMed] [Google Scholar]
- 55.Shin JH, Schoenfisch MH. Chem Mater. 2008;20:239–249. doi: 10.1021/cm702526q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Riccio DA, Nugent JL, Schoenfisch MH. Chem Mater. 2011;23:1727–1735. doi: 10.1021/cm102510q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carpenter AW, Slomberg DL, Rao KS, Schoenfisch MH. ACS Nano. 2011 doi: 10.1021/nn202054f. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smith DJ, Chakravarthy D, Pulfer S, Simmons ML, Hrabie JA, Citro ML, Saavedra JE, Davies KM, Hutsell TC, Mooradian DL, Hanson SR, Keefer LK. J Med Chem. 1996;39:1148–1156. doi: 10.1021/jm950652b. [DOI] [PubMed] [Google Scholar]
- 59.Mowery KA, Schoenfisch MH, Saavedra JE, Keefer LK, Meyerhoff ME. Biomaterials. 2000;21:9–21. doi: 10.1016/s0142-9612(99)00127-1. [DOI] [PubMed] [Google Scholar]
- 60.Batchelor MM, Reoma SL, Fleser PS, Nuthakki VK, Callahan RE, Shanley CJ, Politis JK, Elmore J, Merz SI, Meyerhoff ME. J Med Chem. 2003;46:5153–5161. doi: 10.1021/jm030286t. [DOI] [PubMed] [Google Scholar]
- 61.Saavedra JE, Booth MN, Hrabie JA, Davies KM, Keefer LK. J Org Chem. 1999;64:5124–5131. doi: 10.1021/jo9901539. [DOI] [PubMed] [Google Scholar]
- 62.Zhang HP, Annich GM, Miskulin J, Osterholzer K, Merz SI, Bartlett RH, Meyerhoff ME. Biomaterials. 2002;23:1485–1494. doi: 10.1016/s0142-9612(01)00274-5. [DOI] [PubMed] [Google Scholar]
- 63.Reynolds MM, Hrabie JA, Oh BK, Politis JK, Citro ML, Keefer LK, Meyerhoff ME. Biomacromolecules. 2006;7:987–994. doi: 10.1021/bm060028o. [DOI] [PubMed] [Google Scholar]
- 64.Coneski PN, Rao KS, Schoenfisch MH. Biomacromolecules. 2010;11:3208–3215. doi: 10.1021/bm1006823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seabra AB, da Silva R, de Oliveira MG. Biomacromolecules. 2005;6:2512–2520. doi: 10.1021/bm050216z. [DOI] [PubMed] [Google Scholar]
- 66.Coneski PN, Schoenfisch MH. Polym Chem. 2011;2:906–913. doi: 10.1039/C0PY00269K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marxer SM, Rothrock AR, Nablo BJ, Robbins ME, Schoenfisch MH. Chem Mater. 2003;15:4193–4199. [Google Scholar]
- 68.Nablo BJ, Chen TY, Schoenfisch MH. J Am Chem Soc. 2001;123:9712–9713. doi: 10.1021/ja0165077. [DOI] [PubMed] [Google Scholar]
- 69.Nablo BJ, Schoenfisch MH. J Biomed Mater Res, Part A. 2003;67A:1276–1283. doi: 10.1002/jbm.a.20030. [DOI] [PubMed] [Google Scholar]
- 70.Nablo BJ, Rothrock AR, Schoenfisch MH. Biomaterials. 2005;26:917–924. doi: 10.1016/j.biomaterials.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 71.Nablo BJ, Schoenfisch MH. Biomacromolecules. 2004;5:2034–2041. doi: 10.1021/bm049727w. [DOI] [PubMed] [Google Scholar]
- 72.Riccio DA, Dobmeier KP, Hetrick EM, Privett BJ, Paul HS, Schoenfisch MH. Biomaterials. 2009;30:4494–4502. doi: 10.1016/j.biomaterials.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frost MC, Meyerhoff ME. J Am Chem Soc. 2004;126:1348–1349. doi: 10.1021/ja039466i. [DOI] [PubMed] [Google Scholar]
- 74.Fox S, Wilkinson TS, Wheatley PS, Xiao B, Morris RE, Sutherland A, Simpson AJ, Barlow PG, Butler AR, Megson IL, Rossi AG. Acta Biomater. 2010;6:1515–1521. doi: 10.1016/j.actbio.2009.10.038. [DOI] [PubMed] [Google Scholar]
- 75.Koehler JJ, Zhao J, Jedlicka SS, Porterfield DM, Rickus JL. J Phys Chem B. 2008;112:15086–15093. doi: 10.1021/jp803276u. [DOI] [PubMed] [Google Scholar]
- 76.Koh A, Riccio DA, Sun B, Carpenter AW, Nichols SP, Schoenfisch MH. Biosens Bioelectron. 2011;28:17–24. doi: 10.1016/j.bios.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang M, Thanou M. Pharmacol Res. 2010;62:90–99. doi: 10.1016/j.phrs.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 78.Puiu SC, Zhou ZR, White CC, Neubauer LJ, Zhang ZF, Lange LE, Mansfield JA, Meyerhoff ME, Reynolds MM. J Biomed Mater Res, Part B. 2009;91B:203–212. doi: 10.1002/jbm.b.31391. [DOI] [PubMed] [Google Scholar]