

Abstract
The heterodimeric IL-12 cytokine family is characterized by the sharing of three α (p19, p28, p35) and two β (p40 and Ebi3) subunits, and includes IL-12 (p35/p40), IL-23 (p19/p40), IL-27 (p28/Ebi3) and IL-35 (p35/Ebi3). In this study, the dimerization interfaces of IL-12 family members were characterized, with emphasis on IL-35. Ebi3 and p35 subunits from human and mouse paired effectively with each other, indicating there is no species barrier to IL-35 dimerization and suggesting a conserved dimerization interface. Specific p35 residues that contribute to formation of the IL-12 interface were assessed for their contribution to the IL-35 interface, and candidate Ebi3 residues were screened for their contribution to both IL-27 and IL-35 interfaces. Several residues were identified as critical to the IL-12 or IL-27 interfaces. Conversely, no single mutation was identified that completely disrupts p35/Ebi3 pairing. Linear alanine scanning mutagenesis on both p35 and Ebi3 subunits was performed, focusing on residues that are conserved between the mouse and human proteins. Additionally, a structure-based alanine-scanning approach in which mutations were clustered based on proximitiy was performed on the p35 subunit. Both approaches suggest that IL-35 has distinct criteria for subunit pairing and is remarkabley less sensitive to structural perturbation than IL-12 and IL-27. Additionally, studies using a panel of anti-p35 and anti-Ebi3 antibodies indicate differential availability of epitopes within IL-12 family members that share these subunits, suggesting that IL-35 has distinct structural features, relative to IL-12 and IL-27. These results may be useful in future directed therapeutic targeting of IL-12 family members.
Keywords: Cytokines, Structure, IL-35, IL-12, IL-27
1. INTRODUCTION
The IL-12 cytokine family consists of IL-12, IL-23, IL-27 and IL-35, which have diverse functions and play a role in both pro- and anti-inflammatory responses. IL-12, IL-23, and IL-27 are all expressed by activated antigen presenting cells (APCs). IL-12 promotes Th1 differentiation and IFN-γ production by T cells, and plays a role in induction of anti-tumor responses (Egilmez and Kilinc, 2010; Trinchieri, 2003; Watford et al., 2003). IL-23 is a key mediator of Th17 responses, promoting IL-17/IL-22 production, and expansion and maintenance of Th17 cells in autoimmune disorders (Ahern et al., 2010; Awasthi and Kuchroo, 2009; Cua et al., 2003; McGeachy et al., 2009). IL-23 is also known to play a role in induction and maintenance of T cell memory (Oppmann et al., 2000). IL-27 can synergize with IL-12 to promote IFN-γ production early in the Th1 response, and can also suppress the formation of TGF-β induced Tregs (Neufert et al., 2007; Pflanz et al., 2002). IL-27 also acts in an anti-inflammatory manor, promoting induction of Foxp3− IL-10 producing Tr1 cells, limiting IL-2 production, and blocking lineage commitment and induction of Th17 responses (Awasthi et al., 2007; Diveu et al., 2009; Villarino et al., 2006). The newest IL-12 family member, IL-35, is expressed by regulatory T cells (Tregs) and has been shown to contribute to their suppressive capacity and has been suggested to suppress Th17 cells (Chaturvedi et al., 2011; Collison et al., 2007; Niedbala et al., 2007). IL-35 can also induce its own expression, generating an inhibitory population of Foxp3− IL-35+ cells (iTr35s) that suppress inflammation in IBD and EAE models (Collison et al., 2010). More recently, IL-35 has also been shown to suppress airway inflammation in asthma model and to block IL-2-mediated CNS demyelination, highlighting its potential as a therapeutic reagent in multiple autoimmune disorders (Huang et al., 2011; Zandian et al., 2011). Conversely, IL-35 expression by Tregs and iTr35s in a tumor microenvironment leads to reduced tumor immunity and blockade of IL-35 function may promote tumor clearance (Collison et al., 2010).
The IL-12 family cytokines are α/β heterodimers consisting of one of three α subunits (p35, p28, p19) and one of two β subunits (p40, Ebi3), and this interaction is structurally homologous to type-I IL-6 cytokine/receptor interactions. IL-12 family α subunits are four-helix bundle long-chain cytokines, similar to IL-6 family cytokines (IL-6, IL-11, LIF, OSM, CNTF). The β subunits, p40 and Ebi3, are homologous to non-signaling receptors of the IL-6 family (IL-6R, IL-11R, CNTFR). These β-subunits/receptors consist of two tandem fibronectin type III domains (FNIII) that form a cytokine-binding homology region (CHR), as well as an N-terminal Ig domain. Ebi3 is unique among IL-6/IL-12 family members in that it is lacking the N-terminal Ig domain (Jones and Vignali, 2011; Wang et al., 2009). Unlike IL-6-like cytokines, which are secreted and bind to their receptors extracellularly, IL-12 family members are secreted as functional heterodimers.
Insight into subunit pairing and sharing has been gained by several structural studies of IL-12 family members. A crystal structure of human IL-12 showed that charge interactions are important within the p40/p35 interface (Yoon et al., 2000). A key arginine residue on the p35 subunit (R189) interacts with an aspartic acid on the p40 subunit (D290) and mutation of either residue to alanine completely abrogates dimer formation. Furthermore, substitution of either residue with another of a conserved charge partially restores the interaction, indicating the importance of this interaction (Yoon et al., 2000). The more recent structure of IL-23, which shares the p40 β subunit with IL-12, has improved understanding of the molecular basis of subunit sharing within the IL-12 family (Lupardus and Garcia, 2008). This structure showed that some key interactions are conserved between IL-12 and IL-23. The p19 subunit contains an arginine residue homologous to the critical arginine in p35 and the arginine interaction with the p40 aspartic acid residue is conserved in both cytokines. The IL-23 and IL-12 structures contained areas of p40 involved in distinct interactions as well, suggesting that it might be possible to target these two cytokines independently with small molecule inhibitors. It is not known if this is a feature common to all IL-12 family members, or unique to the p40 subunit. There is no crystal structure of IL-27, however, a recent mutagenesis study on human IL-27 identified a conserved tryptophan residue in the p28 subunit, as well as two acidic Ebi3 residues that are involved in the IL-27 dimer interface (Rousseau et al., 2010).
IL-35 has shown early promise as a potential therapeutic target, but nothing is known about the nature of the dimer interface and whether the p35 and Ebi3 subunits use the same interface for IL-35 formation as they do for IL-12 and IL-27, respectively. Given the nature of subunit sharing within the IL-12 cytokine family, a thorough understanding of these interactions at the molecular level is essential for therapeutic development of IL-35 reagents that do not interfere with IL-12 and IL-27. In this study we characterized the dimerization interfaces of IL-12 family members, with an emphasis on IL-35, using both targeted mutagenesis and alanine-scanning mutagenesis approaches. These results elucidate the molecular basis of subunit sharing within the IL-12 family and will be useful for developing targeted inhibitors of IL-12 family members.
2. MATERIALS AND METHODS
2.1. Mice and cord blood
C57BL/6 mice were purchased from the Jackson Laboratory. All animal experiments were performed in American Association for the Accreditation of Laboratory Animal Care-accredited, specific-pathogen-free facilities in the St. Jude Animal Resource Center following national, state and institutional guidelines. Animal protocols were approved by the St Jude Animal Care and Use Committee. Human umbilical cord samples were provided by Brandon Triplett, Michelle Howard and Melissa McKenna at the St. Louis Cord Blood Bank, and were obtained from the umbilical vein immediately after vaginal delivery with the informed consent of the mother and approved by St. Louis Cord Blood Bank Institutional Review Board. Use at St. Jude was approved by the St. Jude IRB.
2.2. Cell lines and antibodies
HEK293T (ATCC catalog # CRL-11268) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FCS, 2mM glutamine, 1mM pyruvate, 100μM MEM non-essential amino acids (Gibco), 5mM HEPES, 5.5×10−5M β-mercaptoethanol, 100U/ml penicillin and 100μg/ml streptomycin. The following antibodies were purchased from Sigma-Aldrich Biotechnology: purified mouse monoclonal anti-HA biotin clone HA-7, agarose-conjugated purified mouse monoclonal anti-HA clone HA-7, purified mouse monoclonal anti-FLAG biotin clone M2, and agarose-conjugated purified mouse monoclonal anti-FLAG clone M2. Purified rat anti-mouse p35 clone C18.2 was purchased from eBioscience. Monoclonal mouse anti-mouse p35 clones MMp35.8G7 (8G7) and MMp35.A1.6 (A1.6) were generated in C57BL/6 mice vaccinated respectively with mouse p35 N-ter peptide (RVIPVSGPARCLSQSRNLLKT)-ovalbumin or mouse IL-12 protein-ovalbumin conjugates produced according to previously described protocol (Uyttenhove et al., 2011). Monoclonal hamster anti-mouse p35 clone RedT was a generous gift from E. R. Unanue (Washington University, St. Louis, MO) (Tripp et al., 1995). Monoclonal rat anti-mouse p35 clone 45806 was provided by R&D Systems. Biotinylated rat-anti-mouse p40 clone C17.8 was purchased from Biolegend. All purified mouse monoclonal anti-Ebi3 antibodies were generated as previously described (Collison et al., 2010). Mouse anti-GFP clone B2 was obtained from Santa Cruz Biotechnology. Peroxidase-conjugated goat anti-mouse polyclonal IgG was purchased from Thermo Fisher Scientific. Streptavidin-biotinylated horseradish peroxidase complex was obtained from GE Healthcare UK Limited.
2.3. IL-35-mediated suppression assays
Culture supernatants containing mouse and human IL-35 were generated as previously described (Collison et al., 2007). Purified CD4+ CD45RBhi CD25− T cells from C56BL/6 mice were purified by FACS and activated with anti-CD3/CD28 beads in the presence and absence of mIL-35, hIL-35 or control supernatants as previously described (Collison et al., 2007). For suppression of human T cell proliferation, CD4+ CD25− T cells from human cord blood were purified by FACS and activated with anti-CD3/CD28 beads and 10 IU/ml IL-2 in the presence and absence of mIL-35, hIL-35 or control supernatants as previously described (Chaturvedi et al., 2011; Collison et al., 2010). Cells were cultured in the presence of IL-35 for 72 hours for mouse T cells and 5d for human T cells and proliferation was measured by incorporation of 3H-thymidine during the last 8 hours.
2.4. Amino acid sequence alignments and structure predictions
Amino acid sequences were aligned using ClustalW as previously described (Goujon et al., 2010; Larkin et al., 2007). Alignments were generated in Microsoft Excel and identical residues are colored black. Residues with a similar character (i.e. hydrophobic, polar, charged) are colored grey. Secondary structure of aligned sequences were predicted with PSIpred as previously described (Bryson et al., 2005). In addition, the structure of human IL-12 and a model of human IL-27 were used to further predict structural features of murine p35 and Ebi3 (Rousseau et al., 2010; Yoon et al., 2000).
2.5. Generation of IL-12 family subunit constructs
Construct in which the native signal peptides of human Ebi3 and p35, and murine p40, Ebi3, p35 and p28 were replaced with the CD33 signal peptide for targeted secretion (MPLLLLLPLLWAGALA) were used as templates for PCR addition of epitope tags to the IL-12 family subunits. Briefly, a triple-FLAG epitope tag (DYKDDDDKDYKDHDIDYKDDDDK) followed by a GSS linker was added to the N-terminus of IL-12 family β subunits (human Ebi3, murine Ebi3, murine p40) and cloned into pIYneo, a variant of pCI-neo (Promega) containing an IRES-YFP, as NheI-EcoRI fragments. A triple-HA epitope tag (YPYDVPDYAGYPYDVPDYAGYPYDVPDYA) was added to the C-terminus of IL-12 family α subunits (human p35, murine p35, murine p28), separated by a GSG linker and cloned into pIGneo, a pCI-neo variant containing an IRES-GFP, as NheI-EcoRI fragments. Similar constructs lacking epitope tags and containing native signal peptides were used for all ELISAs.
2.6. Mutagenesis
All point mutations to p35 and Ebi3 were generated using the Quikchange Site-Directed Mutagenesis kit (Stratagene). All residues referred to in the text are numbered such that residue one is the first amino acid of the full length secreted protein, following cleavage of the signal peptide. For p35 and Ebi3 linear Alanine/Serine-scan mutants, constructs were designed to include all residues that are conserved or semi-conserved between murine and human p35 or Ebi3. Residues that were investigated as individual point mutations and those that did not have surface-exposed side-chains in the IL-12 structure (Yoon et al., 2000) were excluded from analysis of p35. Residues predicted to be involved in intra-chain disulfide bonds were excluded from the Ebi3 linear Alanine/Serine-scan mutants. For p35 structure-based Alanine/Serine-scan constructs, all conserved, surface exposed residues were included. Mutations to p35 and Ebi3 were generated by overlap-extension PCR and cloned into either the pIGneo (p35) or pIYneo (Ebi3) vectors as described above. Detailed information regarding primer sequence and construct generation will be made available upon request.
2.7. Transient transfection of HEK293T cells
Cells were cultured in 6-well plates at a concentration of 1.7×105 cells/well. After 24 hours, cells were transfected with 1μg total plasmid DNA (0.5ug of each in cases requiring two vectors) using TransIT-LT1 transfection reagent (Mirius Bio LLC) according to manufacturer instructions. Culture medium was changed 24 hours post-transfection and cells and supernatants were harvested 48 hours later.
2.8. Immunoprecipitation and Immunoblotting
Transfected HEK293T cells were harvested and lysed in buffer containing 50mM HEPES, 150mM NaCl, 1mM EDTA, 0.1% Tween-20, and Roche complete protease inhibitor cocktail (Roche). Lysates were quantified using BCA protein assay (Pierce) and normalized to 100μg (IL-12 and IL-27) or 50μg (IL-35) of total protein in 500μl of lysis buffer. Samples were immunoprecipitated overnight at 4°C with the indicated antibody plus protein G agarose (GE Healthcare). Agarose was washed with lysis buffer, boiled in SDS-PAGE sample loading buffer (with 10% b-mercaptoethanol), and run on SDS-PAGE. Gels were transferred to nitrocellulose membranes, blocked with TBS plus 0.002% Tween-20 (TBS-T) and 3% BSA and probed with indicated primary antibodies. After incubation with HRP-conjugated secondary antibody or HRP-streptavidin, proteins were detected with Western Lightening ECL system (PerkinElmer). Lysate samples prior to IP were also run and probed for GFP as a control for variability in transfection efficiency. Blots were quantified using ImageJ software (NIH).
2.9. ELISAs
For IL-12 detection, plates were coated with 2μg/ml of anti-p35 clone C18.2 (eBioscience) overnight. Plates were blocked with PBS-1% BSA for 2 hours and supernatants or standards were added and incubated overnight at 4°C. Biotinylated anti-p40 (Biolegend) was added for 1 hour, followed by 1:2000 streptavidin-HRP for 20 minutes. TMB substrate (Thermo) was added and color was allowed to develop for 20 minutes at room temperature. Reactions were stopped with 1N H2SO4 and absorbance at 450nm was read. Plates were washed 4 times with PBS-0.05% Tween-20 between each step. For detection of IL-27 and IL-35, a similar protocol was used, except anti-p35 clone 8G7 (capture) and biotinylated anti-Ebi3 V1.4F5.29 (detection) were used for IL-35, and anti-Ebi3 4E2 (capture) and biotinylated anti-Ebi3 V1.4C4.22 (detection) were used for IL-27.
2.10. Statistical Analysis
All data from cytokine mutants is shown as percent of wild type cytokine levels. For statistical analysis, one-sample t-tests (two tailed, CI of 95%) were performed using a theoretical mean of 100 to determine if mutants were significantly different from wild type. All analysis was performed with GraphPad Prism Version 4.
3. RESULTS
3.1. The Ebi3/p35 dimer interface and receptor interaction sites are conserved between mouse and human IL-35
We assessed the degree of sequence identity between mouse and human p35 and Ebi3 and found 58% identity for p35 (Supplemental Fig. 1A) and 63% identity for Ebi3 (Supplemental Fig. 1B). Conservation of mouse and human IL-35 subunits led us to determine if there is a species barrier for IL-35 function. Proliferation of mouse CD4+ T cells was suppressed comparably by mouse and human IL-35 (Fig. 1A). Similarly, proliferation of human CD4+ T cells was also suppressed by mouse and human IL-35 (Fig. 1B), indicating that there is no species barrier for IL-35 function, and suggesting that the receptor interaction sites of both mouse and human IL-35 are functionally conserved.
Fig. 1. IL-35 receptor interaction sites and heterodimer interface is conserved from mouse to human.

(A) CD4+ CD45RBhi CD25− T cells from C56Bl/6 mice were FACS purified and activated with anti-CD3/CD28 beads in the presence and absence of mIL-35, hIL-35 or control supernatants. Counts per minute of activated T cells alone were 30,000–50,000. (B) CD4+CD25− T cells from human cord blood were FACs purified and activated with anti-CD3/CD28 beads and 10 IU/ml IL-2 in the presence and absence of mIL-35, hIL-35 or control supernatants. Counts per minute of activated T cells alone were 50,000–100,000. Data represents mean ± SEM of 3 experiments done in duplicate. (C) 293Ts were transfected with indicated combination of mouse and human FLAG-Ebi3 and p35-HA subunits. Lysate samples were immunoprecipitated using an anti-HA antibody and immunoblotted with anti-FLAG to assess the degree of subunit pairing. Multiple bands for IL-35 are a result of multiple intracellular glycosylation species. Lysates were immunoblotted for GFP as a loading control. (D) Western blots were quantified and plotted normalized to GFP. Data represent the mean ± SEM of 4 independent experiments.
We then asked if the Ebi3/p35 subunit interface was also conserved between mouse and human IL-35. Co-transfection of cells with mouse Ebi3 (mEbi3) and human p35 (hp35), followed by co-immunoprecipitation to identified paired complexes showed that these proteins are still able to form an IL-35 dimer. Likewise, human Ebi3 (hEbi3) and murine p35 (mp35) also formed heterodimers, although formation of this pair was less efficient (Fig. 1C, 1D). Thus the dimer interface between Ebi3 and p35 is conserved between mouse and human, and serves as a starting point for identification of residues involved in the heterodimer interface.
3.2. Site-directed mutagenesis of p35
In order to identify specific residues that are involved in IL-35 dimer formation, we focused on p35 residues that are conserved between the murine and human protein. We chose to detect cytokine dimerization in the lysates of transfected cells, rather than in the supernatants for two reasons. First, mutations that affect cytokine secretion but not necessarily heterodimer formation will not interfere with our study. Second, secretion of IL-35 is relatively poor, compared to that of IL-12 and IL-27, whereas examining pairing in lysates gave a more robust signal.
The crystal structure of human IL-12 previously identified several p35 residues that are at the IL-12 interface and critical to formation of the IL-12 dimer (Yoon et al., 2000). We assessed four groups of p35 mutations, (23 total point mutations) that were examined collectively: (1) residues that are critical for IL-12 formation; (2) residues at the IL-12 dimer interface, but not critical for dimer formation; (3) residues near, but not at the IL-12 dimer interface; and (4) residues on the opposite side of p35. Human p35 residues C74, R189 and Y193 have been shown to be critical to p35 association with p40, and are conserved in mp35 (C70, R185 and Y189).
As expected, mutation of R185 and Y189 to alanine significantly reduced the formation of IL-12 dimer in transfected cells. We also observed consistent, but not significant decrease in the formation of IL-12 in the absence of the inter-chain disulfide bond (C70S) (Fig. 2A, 2C). Conversely, these mutations had no effect on IL-35 dimer formation (Fig. 2B, 2C). Next we focused on p35 residues that are at the interface of the IL-12 dimer, but have been shown to have little to no effect on IL-12 dimer formation (hp35 residues R34, L68 and D188; mp35 residues R30, L64 and N184) (Yoon et al., 2000). As expected, mutation of these residues to alanine did not inhibit IL-12 dimer formation. However, mutation of N184 to tryptophan was able to disrupt formation of the IL-12 dimer (Fig. 2A, 2C). We also examined total lysates levels of all p35 point mutants to ensure that any differences seen were a result of pairing deficiency and not due to reduced levels of total p35 protein (Supplemental Fig. 1C). Once again, no mutations had a significant effect on IL-35 formation (Fig. 2B, 2C). Thus the IL-35 subunit Ebi3 interacts with p35 in a manner that is distinct from the IL-12 subunit p40.
Fig. 2. Contribution of selected p35 residues to IL-12 and IL-35 formation.

HEK293T cells were transfected with point mutants of p35 and either (A) p40 or (B) Ebi3 and lysates were analyzed for paired cytokines by immunoprecipitation and western blot. Lysates were immunoblotted for GFP as a loading control. Residues at the IL-12 interface (R30, L64, N184, R185, Y189), near IL-12 interface (Y163, R164, H174), opposite face of p35 (Y102, E103, K106, D138, E139, Q142) were selected based on the crystal structure of IL-12. Residues in an unresolved loop of p35 (D44, E46, D47, R50) were also selected. (C) Western blots from at least 3 independent experiments were quantified and are shown as percent of wt IL-12 or IL-35 (mean ± SEM). *p< 0.05, ** p< 0.01, *** p< 0.001
Structural analysis of IL-12 and IL-23, which share the p40 subunit, has shown that rotation of p19 or p35 relative to the common p40 subunit results in p40 residues that are exclusively involved in IL-12 or IL-23 dimerization (Lupardus and Garcia, 2008). Thus we reasoned that p35 residues that are near, but not at, the IL-12 interface may play a similar role in IL-35 (eg. murine p35 residues Y163, R164 and H174; human p35 residues Y167, K168 and H178). It is also possible that regions of p35 involved in IL-35 formation are not utilized in IL-12 and may be conformationally flexible until stabilized by interaction with Ebi3. We examined several residues within a region between the A and B helices of p35 that is unresolved in the IL-12 structure, but highly conserved between mouse and human p35 (murine p35 residues D44, E46, D47 and R50; human p35 residues D48, E50, D51 and K54). Surprisingly, several of these residues had subtle effects on IL-12 dimer formation when mutated to alanine, although they are not implicated in the IL-12 dimer interface (E46, R50) (Yoon et al., 2000) (Fig. 2A, 2C). Moreover, none of these residues had any significant impact on IL-35 dimer formation (Fig. 2B, 2C). Although less likely, it is possible that the Ebi3/p35 interaction utilizes a completely different surface of p35 than the p40/p35 interaction. Therefore we also examined several conserved residues that are located on the opposite side of p35 as the IL-12 interface (murine p35 residues Y102, E103, K106, D138, E139 and Q142; human p35 residues Y106, E107, K110, D142, E143 and Q146). As expected, none of these mutations had significant effects on IL-12 dimer formation (Fig. 2A, 2C). Although some residues (Y102A, E103A) had slightly reduced IL-35 levels, these reductions were not significant or consistent over several experiments (Fig. 2B, 2C). Thus, no individual p35 point mutation was able to abrogate or improve IL-35 heterodimer formation.
3.3. Site-directed mutagenesis of Ebi3
Although there is no crystal structure of Ebi3, we utilized sequence homology and the IL-12 p40 structure to identify 7 Ebi3 residues that could potentially be involved in the IL-27 or IL-35 interface. Residues D290 and Y293 of the human IL-12 p40 subunit were shown to be critical for formation of IL-12. D290 is conserved in mEbi3 (D187). Although Y293 is not strictly conserved in Ebi3, there is a tyrosine positioned one residue over that could potentially play a role in Ebi3 interface formation (Y191). In addition, a recent modeling and mutagenesis study of IL-27 identified several human Ebi3 residues that disrupt IL-27 dimer formation (F77, E139 and D190), and these residues are conserved in mEbi3 (F78, D139, D190) (Rousseau et al., 2010). Finally, upon examination of the IL-12 p40 structure and p40:Ebi3 sequence alignments, we noticed that the B′–C′ loop of p40 may be slightly extended in Ebi3, allowing for closer contact with p35 in this region. We selected two residues that are conserved in mEbi3 and hEbi3 from this region (F141 and K144 in both mouse and human Ebi3). These seven residues were mutated to alanine and assessed for their contribution to IL-27 and IL-35 dimer formation.
Six out of the seven point mutations significantly disrupted IL-27 dimer formation (Fig. 3A, 3C). We did not observe a defect in IL-27 pairing with the D139A mutant as has been previously reported, although the previous study was performed using human IL-27 this residue is a glutamic acid in human Ebi3 (Rousseau et al., 2010). Total levels of mutant Ebi3 were similar to wild-type Ebi3 levels, indicating that decreased IL-27 levels were due to deficiencies in pairing and not reduced Ebi3 levels (Supplemental Fig. 1D). Once again, no single point mutation to mEbi3 was able to significantly abrogate or improve IL-35 dimerization, suggesting that IL-35 has a distinct Ebi3 interface, relative to IL-27 (Fig. 3B, 3C).
Fig. 3. Contribution of selected Ebi3 residues to IL-27 and IL-35 formation.

HEK293T cells were transfected with point mutants of Ebi3 and either (A) p28 or (B) p35 and lysates were analyzed for paired cytokines by immunoprecipitation and western blot. Lysates were immunoblotted for GFP as a loading control. Ebi3 residues were selected based on sequence homology to p40 and the IL-12 crystal structure. (C) Western blots from at least 3 independent experiments were quantified and are shown as percent of wt IL-27 or IL-35 (mean ± SEM). ** p< 0.01, *** p< 0.001
3.4. Alanine-scanning mutagenesis of p35
It is possible that we were unable to identify specific mutations to Ebi3 or p35 that affect IL-35 pairing because multiple weak interactions are collectively required to mediate Ebi3:p35 pairing and thus single point mutations are insufficient. Thus multiple mutations within a region may be required to exert an effect. In an effort to more thoroughly examine potential Ebi3:p35 interaction sites, we performed alanine-scanning mutagenesis of p35. Residues that were conserved between mp35 and hp35, and surface exposed in the IL-12 p35 structure were included in the alanine scan. Fifteen different mutant constructs (A1-A15) with between four and seven mutations in each construct where generated, for a total of 78 mutations (Supplemental Fig. 2A). In an effort to prevent misfolding of the mutant proteins, hydrophobic residues were replaced with alanine, while polar or charged residues were replaced with serine.
Co-expression of the IL-12 alanine/serine-scan p35 mutants with p40 identified 10 out of 15 mutants with significant effects on IL-12 dimer formation (Fig. 4A, 4C). Mutant A15 contains alanine or serine mutations to residues V181, T182, R185 and Y189, which have been previously shown to be involved in human IL-12 interface formation (human p35 V185, T186, R189 and Y193), consistent with our observed decrease in IL-12 levels. Mutant A7 contains the cysteine involved in the inter-chain disulfide bond, which although not critical to IL-12 formation, leads to decreased IL-12 stability and secretion, consistent with our results here. Mutant A6 contains an inter-chain disulfide bond, which presumably disrupts p35 folding or stability. Unexpectedly, mutants A11 and A13 exert negative effects on IL-12 formation, although they are located away from the interface on the opposite side of p35. It is possible that these mutations disrupt protein folding such that p40 can no longer associate, or cause allosteric changes that disrupt dimer formation. Finally, several mutants contain residues that are unresolved in the IL-12 crystal structure (mutants A1, A2, A5 and A9), and thus defining the mechanism by which they affect IL-12 dimerization remains to be determined.
Fig. 4. Alanine/Serine scan of p35 subunit and contribution to IL-12 and IL-35 formation.

Lysates from HEK293T cells transfected with wild type or mutant p35 and either (A) p40 or (B) Ebi3 were assessed for cytokine formation by immunoprecipitation and western blot. Lysates were immunoblotted for GFP as a loading control. (C) Western blots from at least 3 independent experiments were quantified and plotted as percent of wt IL-12 or IL-35 (mean ± SEM). *p< 0.05, ** p< 0.01, *** p< 0.001
Strikingly, IL-35 was largely unaffected by this panel of p35 mutations. Significant decreases in IL-35 formation were observed for mutants A7 and A14, although this was most likely due to decreased total p35 protein levels in these mutants (Fig. 4B, 4C; Supplemental Fig. 2B). A slight decrease in IL-35 dimer formation (~30%) was observed for mutant A13. This mutant contains residues that are located on the opposite face of p35, as well as several residues that are unresolved in the IL-12 crystal structure.
3.5. Alanine-scanning mutagenesis of Ebi3
We performed similar alanine/serine-scanning mutagenesis studies on the Ebi3 subunit of IL-35, utilizing a strategy similar to that describe above for p35. As there is no structural information for Ebi3, all residues conserved between mouse and human Ebi3 were included in the mutants. A total of 21 mutants were generated spanning the entire Ebi3 protein (B1-B21), each containing between four and seven mutations (Supplemental Fig. 3A). This panel contained mutations of 107 out of the 209 Ebi3 residues. Ebi3 mutants were co-transfected into cells with p28 or p35 and assessed for their ability to form IL-27 and IL-35 heterodimers by immunoprecipitation and western blot. In two cases, a predicted N-linked glycosylation site was mutated, causing Ebi3 to migrate further on the gel, relative to wild type Ebi3 (mutants B4 and B10). In the case of Ebi3 mutant B21, an additional O-linked glycosylation site was introduced near the C-terminus of Ebi3, resulting in retarded migration relative to wild type Ebi3 (Fig. 5A, 5B).
Fig. 5. Alanine/Serine scan of Ebi3 subunit and contribution to IL-27 and IL-35 formation.

Lysates from HEK293T cells transfected with wild type or mutant Ebi3 and either (A) p28 or (B) p35 were assessed for cytokine formation by immunoprecipitation and western blot. Lysates were immunoblotted for GFP as a loading control. (C) Western blots from at least 3 independent experiments were quantified and plotted as percent of wt IL-27 or IL-35 (mean ± SEM) * p<0.05, ** p<0.01
Similar to IL-12 p35 mutants, we found 11 out of 21 Ebi3 mutants that had significant detrimental effects on IL-27 dimer formation (Fig. 5A, 5C). The effects of some mutants are likely due to structural perturbations, as the absence of IL-27/Ebi3 structural information prevented us from restricting mutagenesis to surface exposed residues. Ebi3 mutant B3 contains cysteine and tryptophan residues that are conserved in many FN3 domains (Halaby et al., 1999). Mutants B13 and B15 contain proline rich regions that may be important for protein folding or loop stability. Mutant 21 contains the “WSXWS” motif (LSDWS in mEbi3), which is important for cytokine binding and may contribute to protein folding (Hilton et al., 1996). Mutants B16 and B20 contain several point mutations already shown to inhibit IL-27 dimer formation (F141A, K144A, D187A, D190A, Y191A), although they do not completely abrogate p28 binding in the context of the alanine scan mutants, suggesting other mutations may compensate somewhat for their negative effects. Reduction in IL-27 heterodimer detection was not due to reduced Ebi3 levels, as all alanine scan mutants were expressed at normal to elevated levels in transfected cells (Supplemental Fig. 3B). Once again, we failed to identify any mutations to Ebi3 that had a significant impact on IL-35 dimer formation, even in instances where overall protein folding could be affected (Fig. 5B, 5C).
3.6. Structure-based mutagenesis of p35
While alanine-scans are generally useful in identifying residues involved in protein-protein interactions, they fail to take into account protein structure. If mutations to multiple residues are required to disrupt the IL-35 interface, proximal residues may not necessarily be adjacent in the linear sequence and thus would not be detected by a sequence-based approach. Therefore, we generated a series of mutants based on proximity within the p35 structure. We divided p35 into six regions or “patches”, and introduced mutations to residues that are conserved between mouse and human and have surface-exposed side-chains in the crystal structure of IL-12 (Supplemental Fig. 1A, 4). These patch mutants (C1-C6) were co-transfected with either p40 or Ebi3 and assessed for IL-12 and IL-35 heterodimer formation by immunoprecipitation and western blot.
Similar to results with previous alanine scans, we identified several mutants that had detrimental effects on IL-12 pairing (Fig. 6A, 6C). Mutant C1, which contains the IL-12 interface, completely abrogated IL-12 dimer formation, as expected. In addition, mutants C2, C3 and C4 also disrupt IL-12 formation, although they contain mutations to residues not known to be involved in IL-12 pairing. Total p35 levels are similar to wild type for all mutants, indicating mutant p35 protein is not degraded (Supplemental Fig. 4C). However, the number of mutations in these proteins (21 in mutant C1, 7 in mutant C2, 21 in mutant C3 and 13 in mutant C4) likely generates some degree of misfolding of p35, which could inhibit IL-12 formation via allosteric mechanisms. Strikingly, IL-35 dimer formation is completely unaffected by extensive mutagenesis of p35 (Fig. 6B, 6C). We were unable to detect IL-35 dimer in cells transfected with Ebi3 or p35 alone, indicating that these results are not due to non-specific immunoprecipitation of over-expressed Ebi3 by the anti-HA antibody, or non-specific detection of p35 by the anti-FLAG antibody used for blotting (Fig. 6B).
Fig. 6. Structure-based Alanine/Serine scan of p35 and contribution to IL-12 and IL-35 formation.

Lysates from HEK293T cells transfected with wild type or mutant p35 and either (A) p40 or (B) Ebi3 were assessed for cytokine formation by immunoprecipitation and western blot. Lysates were immunoblotted for GFP as a loading control. In addition, p35, p40 or Ebi3 only transfected cells were included as negative controls. (C) Western blots from at least 3 independent experiments were quantified and plotted as percent of wt IL-12 or IL-35 (mean ± SEM). * p<0.05, ** p<0.01, *** p< 0.001
3.7. Confirmation of selected mutants by ELISA
We wanted to confirm that some of our more intriguing mutants could also be secreted by cells. Also, we wanted to confirm that the epitope tags we used for immunoblot experiments do not affect IL-12 family pairing. Thus we measured the amount of native wild type and mutant cytokine secreted by transfected 293Ts by ELISA. We developed a p35/Ebi3 sandwich ELISA that is specific to IL-35 and does not cross-react with unpaired Ebi3, IL-12 or IL-27 (data not shown). We also utilized an ELISA for IL-12 p70 heterodimer that does not react to p40 heterodimers to determine IL-12 concentrations. Supernatants from cells transfected with p35 point mutants that showed significant differences in pairing for IL-12, but not IL-35 were generated (N184W, R185A and Y189A). We also included a point mutant that had no effect on IL-12 (R30A) as a control. ELISA results confirmed our previous observations that p35 mutants N184W, R185A and Y189A disrupt IL-12 pairing, but have no effect on IL-35 (Fig. 7A).
Fig. 7. ELISA analysis of selected p35 and Ebi3 mutants.
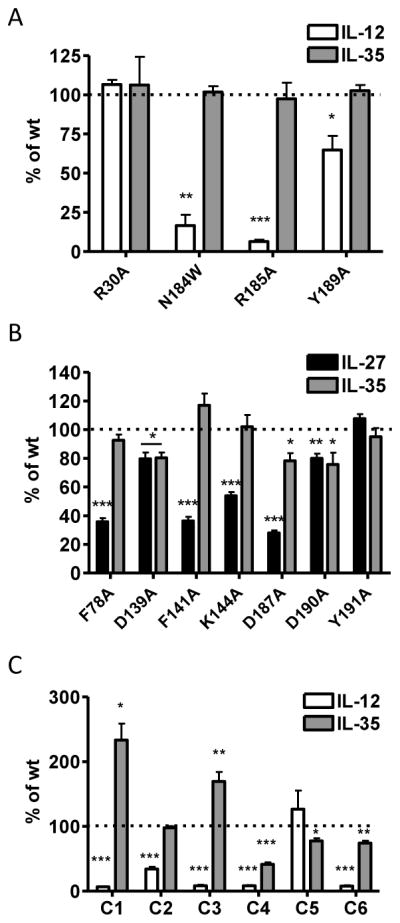
(A) Supernatants from HEK293T cells transfected with wild type or mutant p35 and either p40 or Ebi3 were assessed for cytokine levels by ELISA for IL-12 (anti-p35 C18.2 and anti-p40 biotin C17.8) and IL-35 (anti-p35 MMp35.8G7 and biotin anti-Ebi3 DV29). (B) Supernatants from HEK293T cells transfected with wild type or mutant Ebi3 and either p35 or p28 were assessed for soluble IL-27 or IL-35 levels by ELISA. (C) Supernatants from HEK293T cells transfected with wild type or patch mutant p35 and either p40 or Ebi3 were assessed for cytokine levels by ELISA for IL-12 or IL-35. Mean ± SEM from at least 3 independent experiments are shown as percent of wt IL-12 or IL-35. *p< 0.05, ** p< 0.01, *** p< 0.001
We also screened Ebi3 point mutations that resulted in significant reduction in IL-27 pairing, but not in IL-35 pairing (F78A, F141A, K144A, D187A, D190A and Y191A). Ebi3 mutant D139A, which had no effect on either IL-27 or IL-35, was included as a control. We developed an Ebi3/Ebi3 ELISA to detect secreted IL-27, as commercially available ELISAs for IL-27 only detect the p28 subunit, which can be secreted independently of Ebi3 (Pflanz et al., 2002; Shimozato et al., 2009; Stumhofer et al., 2010). Mutations F78A, F141A, K144A, D187A and D190A significantly reduced the amount of IL-27 detected by ELISA, as expected. Mutant Y191A reduced IL-27 pairing by western blot analysis, but did not significantly reduce the amount of IL-27 detected in supernatants (Fig. 7B). Assessment of IL-35 supernatant levels with these mutants largely confirmed our western blot results, with the majority of mutants having no effect on IL-35 levels. We did observe a slight, but significant decrease in IL-35 levels with D190A that was not seen in our pairing analysis, suggesting that this residue may play a role in IL-35 secretion. We also noted a significant decrease in both IL-27 and IL-35 with mutant E139A, which was not seen in our pairing studies that utilized epitope tags to detect Ebi3. The differences observed between immunoblot and ELISA results may be a due to changes in anti-Ebi3 antibody affinity for these mutants.
Finally, we screened the p35 patch mutants (C1-C6) for their effect on IL-12 and IL-35 secretion. As expected, significantly less IL-12 was detected for mutants C1-C4, and normal IL-12 levels were observed for mutant C5 (Fig. 7C). Mutant C6 paired effectively in our western blot analysis, but was not detectable at normal levels in supernatants by ELISA. Surprisingly, not only were all IL-35 patch mutants secreted at detectable levels, for C1 and C3 the p35 mutations appear to improve IL-35 secretion (Fig. 7C). Mutants C5 and C6 had slightly decreased amounts of IL-35 produced. For both cytokines there was one mutant (C6 for IL-12 and C4 for IL-35) that did not agree with results obtained for western blot pairing analysis using epitope tagged constructs. The most likely explanation for this is that C4 and C6 contain mutations that disrupt the epitopes of the anti-p35 antibodies used in IL-12 and IL-35 ELISAs. Taken together, these data indicate that IL-35 has a pairing interface that is distinct from IL-12 and IL-27, and that it is largely unaffected by substantial amino acid substitutions.
3.8. Differential antibody epitope availability in IL-12 family cytokines
These mutagenesis studies above suggest that IL-35 has distinct pairing criteria from IL-12 and IL-27. To provide additional support for this notion, we utilized a panel of p35 and Ebi3 antibodies to determine if there were differences in their ability to recognize IL-12 family cytokines. HEK293T cells were co-transfected with native, tag-less mutant p35 and either p40 or Ebi3 in separate vectors, and a panel of p35 antibodies was used to immunoprecipitate either IL-12 or IL-35. Interestingly, most of the antibodies tested preferentially immunoprecipitate either IL-12 or IL-35 (Fig. 8A, 8B). Only one clone, 8G7, was able to bind to both IL-12 and IL-35. Similar results were obtained using a panel of anti-Ebi3 antibodies and supernatants from cells transfected with either IL-27 or IL-35 (Fig. 8C, 8D). All Ebi3 antibody clones were obtained from Ebi3 knock-out mice immunized with recombinant Ebi3 protein (Shenandoah Biotechnology). In this instance, several clones had a clear preference for IL-27 and did not recognize IL-35 well, whereas other clones recognized both cytokines similarly. Together, these results clearly show the differential accessibility of antibody epitopes within IL-12 family cytokines that utilize the same subunits, and further suggest that IL-35 has distinct subunit pairing criteria relative to IL-12 and IL-27.
Fig. 8. Differential antibody epitope availability in IL-12 family cytokines.

(A) Supernatants from HEK293T cells transfected with wild type IL-12 or IL-35 were subjected to immunoprecipitation by indicated anti-p35 monoclonal antibodies. Western blot analysis was performed using anti-p40 biotin (IL-12) or anti-Ebi3 biotin (IL-35). (B) Western blots from 3 independent experiments were quantified and plotted as percent of maximal signal. (C) Supernatants from cells transfected with wild type IL-27 or IL-35 were subjected to immunoprecipitation by indicated anti-Ebi3 monoclonal antibodies. The lower band in all Ebi3 western blots represents the light chain of the antibody used for immunoprecipitation. (D) Western blots from 3 independent experiments were quantified and plotted as percent of maximal signal.
4. DISCUSSION
IL-6 family cytokines have clearly defined interaction sites between the helical cytokine and its primary receptor, termed Site 1 (i.e. IL-6 and IL-6Rα) (Wang et al., 2009). Similarly, these interactions are relatively conserved within the IL-12 family members, which utilize soluble β subunits instead of receptors. The crystal structures and mutagenesis studies of IL-12 and IL-23, as well IL-6 and CNTF confirmed that specific interactions between conserved residues are common to most if not all IL-6 and IL-12 family Site 1 interactions (Boulanger et al., 2003; Rousseau et al., 2008). These residues are present in p35 and Ebi3, and mutagenesis confirmed their involvement in mouse IL-12 and IL-27 dimer interfaces. The extensive mutagenesis performed here also indicates that both IL-12 and IL-27 pairing are sensitive to mutagenesis of residues that do not directly mediate subunit dimerization, suggesting structural perturbations introduced in areas distal to the interface can exert allosteric effects on dimer formation.
Given the degree of conservation of Site 1 interactions within the IL-6/IL-12 cytokine family, it is surprising that these residues have no impact on p35/Ebi3 heterodimer formation, suggesting that IL-35 does not conform to this paradigm. Furthermore, we were unable to identify any residues or regions that had significant impact on IL-35 heterodimer formation. This may not be surprising for point mutations, as multiple mutations can be required to disrupt certain interactions, particularly when the protein-protein interface is made up of a network of weak interactions. We utilized a structure-based mutagenesis approach in anticipation that we would disrupt the entire interface; however IL-35 was still unaffected. We cannot rule out the possibility that a large interface that may mediate Ebi3:p35 interaction straddles two or more of the mutants and thus may not be affected by only one.
This approach relied on mutagenesis of amino acid side chains, and will thus only identify regions of the interface in which side chains play a significant role. It is possible that the negligible effect of these mutations on IL-35 is because the Ebi3:p35 interface relies heavily on the use of backbone carbonyl interactions. While this would be an unconventional mode of binding within the IL-12 family, such interactions have been described. For instance, MHC class II peptide binding is mediated in part through a network of hydrogen bonds between the MHC helices and the main chain of the peptide (Stern et al., 1994; Stern and Wiley, 1994). Another possibility is that the interaction between mouse p35:human Ebi3 and vice versa are mediated not by conserved amino acid residues, but rather by conserved secondary structural elements, which may be generated with significant variation in amino acid sequence. For example, the DDB1 ubiquitin ligase recognizes short viral peptides that form α-helical structures, despite their divergent sequences (Li et al., 2010). If p35:Ebi3 interactions were mediated in a similar manor, the present study may fail to identify interaction sites that are not conserved between the mouse and human subunits. It is also possible that the functional secreted form of IL-35 is not in fact a heterodimer, but some higher ordered multimer. In this case, multiple redundant binding sites may make it difficult to identify residues involved in protein-protein interactions by this methodology. However, this explanation seems less likely given current knowledge of heterodimer formation within the IL-6/IL-12 cytokine family.
While it is possible that some of the residues mutated could contribute to protein folding (i.e. the Ebi3 WSXWS motif in mutant B21 or p35 mutant C3 with 21 mutations), it is unlikely that most of the mutants tested are completely misfolded, as most were not degraded and levels remain similar to that of their wild type counterparts. Additionally, at least for the p35 mutants C1-C6, p35 and Ebi3 not only pair effectively, but are also secreted to the same extent or better than wild type IL-35, suggesting that they are properly folded. It remains to be seen if these mutants retain IL-35 functional activity, but such analysis would also be compounded by mutations that affect receptor interaction (Collison, 2012). It is also possible that Ebi3 and p35 proteins are sticky, and our inability to identify mutations that abrogate binding is an artifact of the over-expression system used in this study. However, this seems unlikely given that IL-12 and IL-27 were over-expressed to similar, if not higher levels, and multiple mutations affecting dimerization were identified for these IL-12 family members that share subunits with IL-35.
IL-6/IL-12 family members also contain conserved sites for interaction with their receptors (Jones and Vignali, 2011; Wang et al., 2009). The large array of Ebi3 and p35 mutants generated in this study will also be useful in mapping IL-12, IL-27 and IL-35 receptor interactions. Any differences in receptor utilization by these cytokines will be useful in developing targeted therapies that inhibit specific IL-12 family members. In addition, many in vivo and in vitro studies of IL-12 family members have mainly relied on the use of mutant mice that are deficient in a single subunit. However these mice often lack more than one cytokine, making interpretation of data difficult (Collison and Vignali, 2008). We have identified mutations that clearly disrupt IL-12 and IL-27 dimer formation, but appear to leave the IL-35 heterodimer intact. Although functional analysis will be needed to confirm IL-35 activity, these mutants could potentially be used for the generation of targeted knock-in mice that specifically lack IL-12 or IL-27, but not IL-35, making in vivo analysis of the role of IL-12 and IL-27 in specific disease models more definitive.
Supplementary Material
HIGHLIGHTS.
Dimer interfaces of IL-12, IL-27 and IL-35 are characterized by extensive mutagenesis
Residues critical to IL-12 and IL-27 dimer formation do not affect IL-35 dimerization
IL-35 has distinct subunit pairing criteria relative to IL-12 and IL-27
Antibody epitope availability suggests that IL-35 has distinct structural features
Acknowledgments
This work was supported by the National Institutes of Health (R01 AI091977; D.A.A.V.), American Asthma Foundation (10-0128; D.A.A.V.), an Individual NRSA (F32 AI084330; L.L.J.), NCI Comprehensive Cancer Center Support CORE grant (CA21765; D.A.A.V.), the American Lebanese Syrian Associated Charities (ALSAC; D.A.A.V.) and by the Fonds National de la Recherche Scientifique Médicale (FRSM, Belgium; C.U, J.V.S.).
We would like to thank Emil Unanue and Jessie Ni for antibodies anti-p35 antibodies, Hugues Gascan for the structural model of human IL-27, and Brandon Triplett, Michelle Howard and Melissa McKenna at St. Louis Cord Blood Bank for cord blood samples. We are also grateful to Kate Vignali for technical assistance, Scott Brown, Creg Workman and Karen Forbes for generation, screening and purification of Ebi3 monoclonal antibodies and Dominique Donckers for help with anti-p35 vaccinations. We also thank Richard Cross, Greig Lennon and Stephanie Morgan for FACS, Karen Forbes, Ashley Castellaw, Amy Krause and Chris Dillon for maintenance, breeding and genotyping of mouse colonies, and the staff of the St. Jude Animal Resource Center for the animal husbandry, the staff of the Hartwell Center for Biotechnology and Bioinformatics at St Jude for PCR primers and sequencing.
Abbreviations used in the paper
- Treg
Regulatory T cell
- FNIII
Fibronectin type-III domain
- CHR
Cytokine-binding homology region
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ, Powrie F. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity. 2010;33:279–88. doi: 10.1016/j.immuni.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, Kuchroo VK, Oukka M, Weiner HL. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol. 2007;8:1380–9. doi: 10.1038/ni1541. [DOI] [PubMed] [Google Scholar]
- Awasthi A, Kuchroo VK. Th17 cells: from precursors to players in inflammation and infection. Int Immunol. 2009;21:489–98. doi: 10.1093/intimm/dxp021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger MJ, Chow DC, Brevnova EE, Garcia KC. Hexameric structure and assembly of the interleukin-6/IL-6 alpha-receptor/gp130 complex. Science. 2003;300:2101–4. doi: 10.1126/science.1083901. [DOI] [PubMed] [Google Scholar]
- Bryson K, McGuffin LJ, Marsden RL, Ward JJ, Sodhi JS, Jones DT. Protein structure prediction servers at University College London. Nucleic Acids Res. 2005;33:W36–8. doi: 10.1093/nar/gki410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi V, Collison LW, Guy CS, Workman CJ, Vignali DA. Cutting Edge: Human Regulatory T Cells Require IL-35 To Mediate Suppression and Infectious Tolerance. J Immunol. 2011;186:6661–6. doi: 10.4049/jimmunol.1100315. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, Finkelstein D, Forbes K, Workman CJ, Brown SA, Rehg JE, Jones ML, Ni HT, Artis D, Turk MJ, Vignali DA. IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol. 2010;11:1093–101. doi: 10.1038/ni.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather D, Satoskar AR, Garcia KC, Hunter CA, Drake CG, Murray PJ, Vignali DAA. Unconventional interleukin-35 receptor composition and signaling. Nat Immunol. 2012 doi: 10.1038/ni.2227. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison LW, Vignali DA. Interleukin-35: odd one out or part of the family? Immunol Rev. 2008;226:248–62. doi: 10.1111/j.1600-065X.2008.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–9. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–8. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- Diveu C, McGeachy MJ, Boniface K, Stumhofer JS, Sathe M, Joyce-Shaikh B, Chen Y, Tato CM, McClanahan TK, de Waal Malefyt R, Hunter CA, Cua DJ, Kastelein RA. IL-27 blocks RORc expression to inhibit lineage commitment of Th17 cells. J Immunol. 2009;182:5748–56. doi: 10.4049/jimmunol.0801162. [DOI] [PubMed] [Google Scholar]
- Egilmez NK, Kilinc MO. Tumor-resident CD8+ T-cell: the critical catalyst in IL-12-mediated reversal of tumor immune suppression. Arch Immunol Ther Exp (Warsz) 2010;58:399–405. doi: 10.1007/s00005-010-0097-7. [DOI] [PubMed] [Google Scholar]
- Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010;38:W695–9. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaby DM, Poupon A, Mornon J. The immunoglobulin fold family: sequence analysis and 3D structure comparisons. Protein Eng. 1999;12:563–71. doi: 10.1093/protein/12.7.563. [DOI] [PubMed] [Google Scholar]
- Hilton DJ, Watowich SS, Katz L, Lodish HF. Saturation mutagenesis of the WSXWS motif of the erythropoietin receptor. J Biol Chem. 1996;271:4699–708. doi: 10.1074/jbc.271.9.4699. [DOI] [PubMed] [Google Scholar]
- Huang CH, Loo EX, Kuo IC, Soh GH, Goh DL, Lee BW, Chua KY. Airway Inflammation and IgE Production Induced by Dust Mite Allergen-Specific Memory/Effector Th2 Cell Line Can Be Effectively Attenuated by IL-35. J Immunol. 2011;187:462–71. doi: 10.4049/jimmunol.1100259. [DOI] [PubMed] [Google Scholar]
- Jones LL, Vignali DA. Molecular interactions within the IL-6/IL-12 cytokine/receptor superfamily. Immunol Res. 2011 doi: 10.1007/s12026-011-8209-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Li T, Robert EI, van Breugel PC, Strubin M, Zheng N. A promiscuous alpha-helical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nat Struct Mol Biol. 2010;17:105–11. doi: 10.1038/nsmb.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupardus PJ, Garcia KC. The structure of interleukin-23 reveals the molecular basis of p40 subunit sharing with interleukin-12. J Mol Biol. 2008;382:931–41. doi: 10.1016/j.jmb.2008.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O’Shea JJ, Cua DJ. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314–24. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufert C, Becker C, Wirtz S, Fantini MC, Weigmann B, Galle PR, Neurath MF. IL-27 controls the development of inducible regulatory T cells and Th17 cells via differential effects on STAT1. Eur J Immunol. 2007;37:1809–16. doi: 10.1002/eji.200636896. [DOI] [PubMed] [Google Scholar]
- Niedbala W, Wei XQ, Cai B, Hueber AJ, Leung BP, McInnes IB, Liew FY. IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. Eur J Immunol. 2007;37:3021–9. doi: 10.1002/eji.200737810. [DOI] [PubMed] [Google Scholar]
- Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–25. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, Hibbert L, Churakova T, Travis M, Vaisberg E, Blumenschein WM, Mattson JD, Wagner JL, To W, Zurawski S, McClanahan TK, Gorman DM, Bazan JF, de Waal Malefyt R, Rennick D, Kastelein RA. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4(+) T cells. Immunity. 2002;16:779–90. doi: 10.1016/s1074-7613(02)00324-2. [DOI] [PubMed] [Google Scholar]
- Rousseau F, Basset L, Froger J, Dinguirard N, Chevalier S, Gascan H. IL-27 structural analysis demonstrates similarities with ciliary neurotrophic factor (CNTF) and leads to the identification of antagonistic variants. Proc Natl Acad Sci U S A. 2010;107:19420–5. doi: 10.1073/pnas.1005793107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F, Chevalier S, Guillet C, Ravon E, Diveu C, Froger J, Barbier F, Grimaud L, Gascan H. Ciliary neurotrophic factor, cardiotrophin-like cytokine, and neuropoietin share a conserved binding site on the ciliary neurotrophic factor receptor alpha chain. J Biol Chem. 2008;283:30341–50. doi: 10.1074/jbc.M803239200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimozato O, Sato A, Kawamura K, Chiyo M, Ma G, Li Q, Tagawa M. The secreted form of p28 subunit of interleukin (IL)-27 inhibits biological functions of IL-27 and suppresses anti-allogeneic immune responses. Immunology. 2009;128:e816–25. doi: 10.1111/j.1365-2567.2009.03088.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern LJ, Brown JH, Jardetzky TS, Gorga JC, Urban RG, Strominger JL, Wiley DC. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature. 1994;368:215–21. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- Stern LJ, Wiley DC. Antigenic peptide binding by class I and class II histocompatibility proteins. Structure. 1994;2:245–51. doi: 10.1016/s0969-2126(00)00026-5. [DOI] [PubMed] [Google Scholar]
- Stumhofer JS, Tait ED, Quinn WJ, 3rd, Hosken N, Spudy B, Goenka R, Fielding CA, O’Hara AC, Chen Y, Jones ML, Saris CJ, Rose-John S, Cua DJ, Jones SA, Elloso MM, Grotzinger J, Cancro MP, Levin SD, Hunter CA. A role for IL-27p28 as an antagonist of gp130-mediated signaling. Nat Immunol. 2010;11:1119–26. doi: 10.1038/ni.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- Tripp CS, Kanagawa O, Unanue ER. Secondary response to Listeria infection requires IFN-gamma but is partially independent of IL-12. J Immunol. 1995;155:3427–32. [PubMed] [Google Scholar]
- Uyttenhove C, Marillier RG, Tacchini-Cottier F, Charmoy M, Caspi RR, Damsker JM, Goriely S, Su D, Van Damme J, Struyf S, Opdenakker G, Van Snick J. Amine-reactive OVA multimers for auto-vaccination against cytokines and other mediators: perspectives illustrated for GCP-2 in L. major infection. J Leukoc Biol. 2011;89:1001–7. doi: 10.1189/jlb.1210699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarino AV, Stumhofer JS, Saris CJ, Kastelein RA, de Sauvage FJ, Hunter CA. IL-27 limits IL-2 production during Th1 differentiation. J Immunol. 2006;176:237–47. doi: 10.4049/jimmunol.176.1.237. [DOI] [PubMed] [Google Scholar]
- Wang X, Lupardus P, Laporte SL, Garcia KC. Structural biology of shared cytokine receptors. Annu Rev Immunol. 2009;27:29–60. doi: 10.1146/annurev.immunol.24.021605.090616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watford WT, Moriguchi M, Morinobu A, O’Shea JJ. The biology of IL-12: coordinating innate and adaptive immune responses. Cytokine Growth Factor Rev. 2003;14:361–8. doi: 10.1016/s1359-6101(03)00043-1. [DOI] [PubMed] [Google Scholar]
- Yoon C, Johnston SC, Tang J, Stahl M, Tobin JF, Somers WS. Charged residues dominate a unique interlocking topography in the heterodimeric cytokine interleukin-12. EMBO J. 2000;19:3530–41. doi: 10.1093/emboj/19.14.3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandian M, Mott KR, Allen SJ, Dumitrascu O, Kuo JZ, Ghiasi H. Use of cytokine immunotherapy to block CNS demyelination induced by a recombinant HSV-1 expressing IL-2. Gene Ther. 2011;18:734–42. doi: 10.1038/gt.2011.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.