

Abstract
The G-protein-coupled receptors (GPCRs) are a ubiquitous family of signaling proteins of exceptional pharmacological importance. The recent publication of structures of several GPCRs cocrystallized with ligands of differing activity offers a unique opportunity to gain insight into their function. To that end, we performed microsecond-timescale simulations of the A2A adenosine receptor bound to either of two agonists, adenosine or UK432097. Our data suggest that adenosine is highly dynamic when bound to A2A, in stark contrast to the case with UK432097. Remarkably, adenosine finds an alternate binding pose in which the ligand is inverted relative to the crystal structure, forming relatively stable interactions with helices I and II. Our observations suggest new experimental tests to validate our predictions and deepen our understanding of GPCR signaling. Overall, our data suggest an intriguing hypothesis: that the 100- to 1000-fold greater efficacy of UK432097 relative to adenosine arises because UK432097 stabilizes a much tighter neighborhood of active conformations, which manifests as a greater likelihood of G-protein activation per unit time.
Introduction
The G-protein-coupled receptor (GPCR) family comprises some 800 distinct members that transduce signals in an astonishing variety of physiological contexts. The rational design of therapeutic ligands targeting GPCRs is hotly pursued for the treatment of a wide array of conditions; indeed, it is estimated that half of all drugs target GPCRs (1). Our understanding of ligand binding and GPCR activation is advancing rapidly, thanks to recent successes in determining the structures of several members of the family (for a review highlighting recent progress in this field and its utility for designing ligands to modulate function, see Congreve and Marshall (2)).
In this study, we focus our attention on the A2A adenosine receptor. The adenosine receptor subfamily (comprising four subtypes) is actively targeted for the treatment of Parkinson's disease, schizophrenia, inflammation, and cardiac ischemia (3–5). In the attempt to elucidate the connection between ligand binding and adenosine receptor activation, investigators have taken a major step forward with the publication of several antagonist-bound structures (6–8) and, more recently, structures bound to three agonists: UK432097 (9), adenosine, and NECA (10). Although the crystal structures represent a crucial and timely advance, several lines of evidence suggest that we must reckon with ligand dynamics and conformational heterogeneity before we can gain a comprehensive understanding of receptor function. In this context, computational work has indicated a direct connection between cholesterol binding and the conformation of A2A (11), and dramatic consequences for ligand binding have been confirmed independently by experiment (12). A comparison of the dynamics of the A2A and A2B subtypes provided insight into subtype specificity (13). Published simulations of rhodopsin have revealed ligand conformational heterogeneity (14), and the role of binding-pocket hydration in receptor activation has been studied in simulations of bovine rhodopsin (15) and, more recently, in simulations of squid rhodopsin (16) and the β2 adrenergic receptor (17). Extensive simulations of the β2 adrenergic receptor have highlighted conformational variability and its role in ligand binding (18) and G-protein activation (17,19). Simulations of a cannabinoid receptor have revealed binding of the ligand followed by conformational changes consistent with current models of GPCR activation (20).
Here, we present evidence from microsecond simulations of A2A bound to two different agonists for conformational variability of the receptor, which manifests as multiple binding modes of the native ligand adenosine. This contrasts starkly with the binding of a highly potent synthetic agonist, UK432097, that is much less dynamic in the binding pocket and therefore stabilizes the protein in a much tighter neighborhood of conformations, perhaps explaining the 100- to 1000-fold lower half-maximal effective concentration (EC50) of UK432097 compared with adenosine (21,22). In the adenosine case, the binding pocket is well hydrated, and consequently the ligand must compete with water for hydrogen (H)-bonds. In the UK432097 case, bulky aromatic prostheses at the extracellular end of the ligand serve to plug the binding pocket, keeping it relatively dry compared with the adenosine bound receptor.
Methods
The initial structures for A2A+adenosine and A2A+UK432097 were taken from the Protein Data Bank codes 2YDO (10) and 3QAK (9), respectively. The 2YDO structure is missing residues 214–223, and the 3QAK is missing residues 149–157 and has an inserted T4 lysozyme between residues 208 and 221. We excised the T4L and reconstructed these missing regions using the corresponding parts of our previously published well-equilibrated A2A structure (11).
We set up the systems using Maestro version 9.2 (23) as follows: First, we embedded the protein (residues 3–310) in a palmitoyloleyol phosphatidylcholine (POPC) bilayer by aligning the structure to structure ID (3EML) in the Orientations of Proteins in Membranes database (24) with at least 10 Å between the protein and its closest periodic image. Two cholesterol molecules were included in the hypothesized cholesterol-binding site, as described in previous work (11). The protein-membrane system includes ∼90 POPC lipid molecules and ∼9000 TIP3P water molecules in a box ∼80 Å × 60 Å × 100 Å. We modeled the protein using the CHARMM22 force field with cmap correction (25), the POPC lipid and cholesterol using the CHARMM36 parameter set (26), the ligand using the CGenFF small-molecule force field (27), and waters using with CHARMM version of TIP3P. Relaxation and molecular-dynamics calculations were performed with Desmond v. 30110 (28) as follows: We relaxed all of the systems using a lipid-protein equilibration/relaxation protocol distributed with the Schrödinger 2009 suite. We performed the production run under constant pressure of 1 atm and a temperature of 310 K, thermostated and barostated according to the Martyna-Tobias-Klein method (29), with a coupling constant of 0.5 (2.0) ps for the thermostat (barostat). Hydrogen positions were constrained by the M-SHAKE algorithm, allowing a time step of 2 fs. Long-range electrostatics were computed every time step by the particle mesh Ewald (PME) method with a cutoff radius of 10 Å and tail correction. In the PME method, we used 60 Fourier mesh points along each cell axis and the complex-to-complex (c2c) transform option. We obtained two trajectories (∼1 μs each) for each system (A2A+adenosine or A2A+UK432097).
We obtained all of the molecular images using the Visual Molecular Dynamics package (30). In Table 1, the mean-squared deviation (MSD) values for the ligand center of mass (CoM) position are defined by , where ri is the position vector of ligand CoM at a frame i, and ravg is the position vector of ligand CoM averaged over the total frames N in the time periods indicated in the table. MSD ± σ(MSD) values are reported in the table. We obtained σ(MSD) using the blocking method (31) (data shown in Fig. S1 of the Supporting Material). The θ-value is defined as , where ui is the unit vector of the ligand axis at a frame i (at initial, 0 μs), and u is a vector in the plane of the bicyclic core and is chosen so that it points toward the extracellular side. In the table, values are shown, where < > means the average over the indicated time periods. We obtained the H-bonding contacts shown in Table 2 by using a distance cutoff of 4.0 Å (where the distance between donor and acceptor is <4.0 Å) and angle cutoff of 25° (where 180°, the angle of the donor-hydrogen-acceptor, is <25°). We obtained the π-stacking interactions by using a distance cutoff of 4.0 Å (where the CoM distance between the hexagon ring of Phe-168 and the bicyclic adenine core is <4.0 Å) and angle cutoff of 20° (where the angle between the perpendicular axes of the two rings is <20°). In the orientational autocorrelation (see Fig. 3), is calculated by , where ni (nj) is unit vector of the ligand axis at a frame i (j), , and (here, the initial frame 1 is chosen at 0.1 μs and the last frame is at 0.9 μs), and n is the perpendicular of the plane of the bicyclic core. We obtained the number of waters inside a protein using Lightweight Object-Oriented Structure (LOOS) version 1.7.2 (32) (see Fig. 5). Here, the binding pocket was defined as a bounding box for residues 88, 177, 249, 253, 274, and 278.
Table 1.
MSD of CoM position and rotation (θ) for adenosine or UK432097 bound to A2A
| Adenosine | UK432097 | ||||
|---|---|---|---|---|---|
| MSD () | 0.1–0.9 μs | 10.67∗ | 7.91 ± 1.51 | 0.88 ± 0.07 | 2.81∗ |
| 0.1–0.5 μs | 10.92 ± 1.89 | 6.56 ± 1.97 | 0.91 ± 0.11 | 3.41 ± 0.31 | |
| 0.5–0.9 μs | 2.34 ± 0.25 | 7.50 ± 2.31 | 0.76 ± 0.07 | 0.73 ± 0.15 | |
| θ (degree) | 0.1–0.9 μs | 103 ± 56 | 74 ± 48 | 23 ± 4 | 20 ± 7 |
| 0.1–0.5 μs | 65 ± 50 | 85 ± 50 | 23 ± 4 | 19 ± 8 | |
| 0.5–0.9 μs | 140 ± 29 | 63 ± 42 | 23 ± 4 | 21 ± 5 |
Data are reported for two independent trajectories of each ligand. The first trajectory results are reported in the first column and the second trajectory results are reported in the second column. MSD ± σ(MSD) and values (calculated over the indicated time periods) are shown, as defined in Materials and Methods. The values indicated by an asterisk were observed to not converge by the F-P block averaging.
Table 2.
H-bonding and π-stacking interactions of adenosine or UK432097 with A2A
| A2A | +Adenosine | First % | Second % | +UK432097 | First % | Second % | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Glu-13 | O1 | 13 | (22) | 1 | (3) | |||||
| O2 | 6 | (11) | 2 | (6) | ||||||
| Ile-66 | N5 | 3 | (22) | 2 | (11) | |||||
| Ser-67 | N5 | 12 | (68) | 21 | (67) | |||||
| N6 | 31 | (72) | 14 | (62) | ||||||
| Thr-88 | N3 | 7 | (14) | N4 | 24 | (37) | 50 | (70) | ||
| O2 | 10 | (52) | 4 | (34) | ||||||
| Phe-168 | π-stack | 29 | 13 | π-stack | 85 | 34 | ||||
| Glu-169 | N3 | 5 | (10) | 4 | (9) | N5 | 10 | (19) | ||
| O2 | 2 | (3) | N6 | 12 | (19) | |||||
| Asn-253 | N3 | 5 | (14) | 4 | (22) | N3 | 2 | (5) | 17 | (35) |
| His-278 | O1 | 4 | (10) | 3 | (9) | O1 | 22 | (51) | 25 | (49) |
| O2 | 3 | (9) | O2 | 3 | (44) | 7 | (68) |
Criteria used to define H-bonding and π-stacking are defined in Materials and Methods. Data are presented for two independent trajectories for each system. The first trajectory results are shown in the column marked First, and the second trajectory results are shown in the column marked Second. For H-bonds, two measurements are shown: one with an angular restriction and one without (in parentheses). The percentage shows how often the contact is formed, averaged over the last 0.9 μs of simulation time. In some cases an H-bond may switch between two different donors or acceptors on a particular side chain, and in these cases the percentage represents the fraction of time in which any H-bond is formed. The atoms on the ligands that are involved in the contacts (as defined in Fig. 2) are listed.
Figure 3.
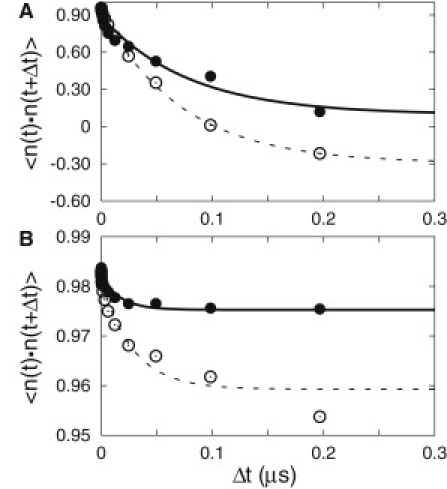
Orientational autocorrelation of (A) adenosine and (B) UK432097. Data for two independent trajectories of each ligand are shown. The first trajectory results are shown in solid circles and solid line, and the second trajectory results are shown by open circles and dashed line. The circles are obtained from , as defined in Materials and Methods. The lines are exponential functions of , fitted to the circles. The fitted decay constants τ for the adenosine case (A) are 78 ns (72 ns) for the solid (dashed) line, and for the UK432097 case (B) are 17 ns (28 ns) for the solid (dashed) line.
Figure 5.
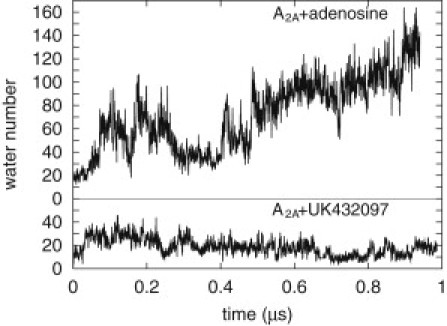
Number of water molecules in the ligand-binding pocket of A2A with adenosine or UK432097 bound. The binding pocket is defined in Materials and Methods. The second trajectory results are shown in Fig. S3.
Results
Ligand dynamics
Fig. 1 compares the motion of the two ligands in the ligand-binding site (ligand structures are shown in Fig. 2). The CoM of adenosine clearly explores a significantly larger volume compared with that of UK432097. To quantify this behavior, we measured the MSD of the CoM of the ligand for four trajectories: two trajectories with adenosine bound, and two trajectories with UK432097 bound (Table 1). The picture suggested in Fig. 1 is confirmed by the overall CoM MSD, averaged over the entire trajectories. More complex behavior was revealed, however, when we sought to compute the statistical error of our MSD estimates. The errors reported in Table 1 were calculated using the blocking approach first published by Flyvbjerg and Petersen (31), known as the F-P block method, which computes the variance of an estimated quantity by considering larger and larger blocks of data (see Fig. S1). When the blocks are independent, the estimate of the variance converges, signaled by a plateau in a plot of the estimated variance as a function of block size. The F-P block averaging indicated convergence of the MSD estimate for the second adenosine trajectory and the first UK432097 trajectory. In the other two cases, the F-P block averaging did not show a strong signal of convergence. A closer examination showed that in each of these two cases, the ligand dynamics are better considered as consisting of two parts, characterized by distinctly different MSDs. This is evident from the same analysis applied separately to the two halves of each trajectory, reported in the second and third rows of Table 1. Although the general conclusion is borne out by this more-detailed analysis, we note that, given less data, it may not be possible to distinguish between the adenosine and UK432097 CoM MSDs (i.e., the second half of adenosine trajectory 1 and the first half of UK432097 trajectory 2).
Figure 1.
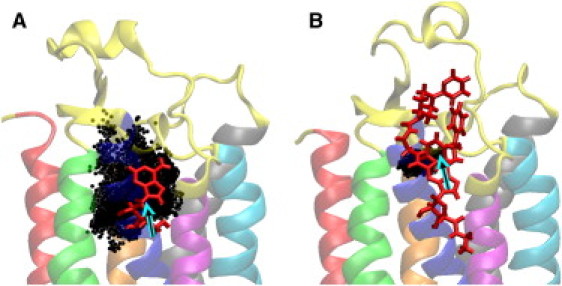
Motion of ligands (A) adenosine and (B) UK432097 when bound to A2A. The instantaneous CoM positions of the ligand are marked by black dots. In each figure, the ligand is shown by red sticks over the black dots and its average CoM position is indicated by an arrow. The transmembrane helices are shown in different colors: red (H1), green (H2), blue (H3), magenta (H4), cyan (H5), gray (H6), and orange (H7).
Figure 2.
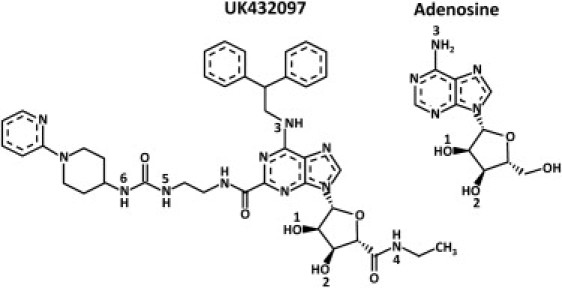
Structures of adenosine and UK432097. Marked atom numbers correspond to H-bonds listed in Table 2.
The orientational dynamics are also quite different in the adenosine and UK432097 cases (see Table 1). This is an interesting point because the orientation of the ligand in the pocket partly determines the binding pose. Attaining the correct pose is believed to be essential for effecting activation, and is therefore an important focus of crystal structure analysis and predictive docking calculations. The gross orientation of the ligand was quantified by a vector in the plane of the bicyclic core (see Fig. 2). The vector was chosen so that it would point toward the extracellular side when the ligands were in their crystallized poses. Taking as a reference the initial orientation, we measured the average angle to this reference and its standard deviation. Consistent with our observation that UK3432097 is relatively stable in the binding pocket, the ligand did not sample multiple orientations. Adenosine, on the other hand, was small enough to rotate within the binding pocket, and frequently sampled poses that were inverted relative to the crystal structure pose. This is reflected by an average angle that differs significantly from the crystal structure orientation, together with a standard deviation that indicates significant fluctuations around the average.
The orientational dynamics of bound adenosine suggests an experimental probe: The decay of polarization of fluorescent light emitted by the bicyclic core is sensitive to the reorientational dynamics of ligand between absorption and emission (33). To investigate whether the orientational dynamics could be observed by time-resolved fluorescence polarization spectroscopy, we considered the autocorrelation of the vector normal to the plane of the bicyclic core. Although the emission dipole is probably not exactly perpendicular to the plane, our choice is a reasonable proxy in the absence of more detailed information. Fig. 3 shows that the autocorrelation of the UK432097 emission dipole decays exponentially with a time constant of 17 ns for the first trajectory (28 ns for the second trajectory), and appears to decay to a nonzero long time limit. The fact that in the long time limit the dipole never fully loses its memory is typical of hindered motion, and thus is expected in this case (33). The autocorrelation of adenosine, on the other hand, is observed to lose its memory exponentially, with a time constant of 78 ns for the first trajectory (72 ns for the second trajectory). The dramatic difference in the long time behavior, together with the timescale of the adenosine orientational correlations (of the same order of the fluorescence lifetime) suggests that the dynamics of the two ligands can be distinguished by time-resolved fluorescence polarization spectroscopy.
Ligand-protein contacts
The dynamic character of adenosine suggests that the contacts between the protein and ligand are dynamic as well. Fig. 4 compares time series of ligand-protein contacts between two trajectories of A2A bound to both ligands (also see Fig. S2). A cursory glance shows that, overall, the contacts that formed in the adenosine case are much more transient than those in the UK432097 case, in accord with the observations of Fig. 1 and Table 1. The only contact that is clearly formed over a significant fraction in both cases is the aromatic stacking between Phe-168 and the bicyclic core of the ligands. This interaction is present in all published structures of A2A cocrystallized with any ligand (agonist (9,10) or antagonist (6)), and also has a profound impact on the affinity of the receptor for both agonists and antagonists (34). However, even this interaction is formed only 29% of the time when A2A is bound to adenosine, compared with 85% of the time for UK432097, as shown in Table 2.
Figure 4.
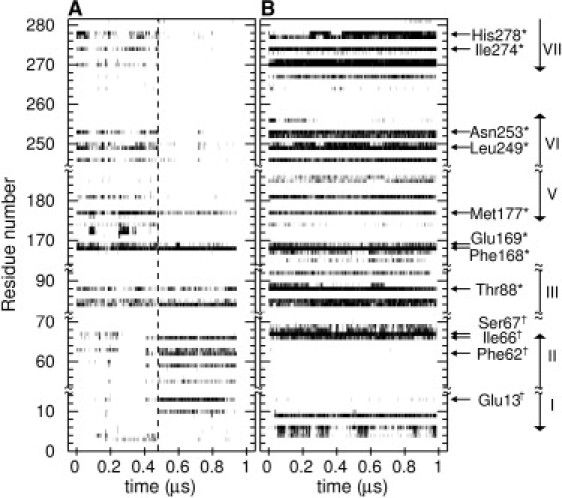
Ligand-residue contact time series for A2A with (A) adenosine or (B) UK432097. A contact (heavy atom distance < 4 Å) is indicated by a thin vertical black line, and stable contacts appear as horizontal solid black bars. The residues shown in Table 2 are indicated, with residues identified by mutagenesis to be important for ligand binding indicated by ∗, and those that form the binding site for our hypothesized inverted pose indicated by †. The vertical arrows indicate the transmembrane helices. The second trajectory results are shown in Fig. S2.
We observe that in comparison with the ring-stacking interaction, H-bonds are much more transient, and considerably more so in the adenosine-bound system. Of particular note are interactions between the ribose hydroxyls (O1 and O2; atom numbering refers to Fig. 2) and Ser-277 and His-278, both of which are important for binding the agonist CGS21680 (35). Because all orthosteric agonists share the ribose moiety, and these hydroxyls are observed in the crystal structures to form H-bonds with key residues that have been determined to be essential to activation, they bear consideration for their role in receptor activation. Remarkably, in the adenosine case, these hydroxyls are in contact with His-278 only ∼10% of the time. Thus, if these H-bonds are indeed essential to activation, our data suggest that, from the single receptor point of view, adenosine only transiently activates A2A. On the other hand, in the UK432097 trajectory these contacts are formed roughly half of the time. If we imagine an ensemble of receptors bound to either adenosine or UK432097, our data together with the mutagenesis data suggest that a significantly larger fraction of the UK432097 ensemble is in an active conformation compared with adenosine. The rest of the tabulated H-bonds are consistent with the trend observed for the O1 and O2 hydroxyls, with the UK432097 H-bonds overall more stably formed. The most stable of all were those in the UK432097 case, formed between Ser-67 and the urea group (atoms N5 and N6), and between Thr-88 and the ribose hydroxyls and 5′-ethyluronamide (atom N4). Thr-88 has also been shown to be important for binding the agonist CGS21680 (35). Remarkably, Thr-88 also interacts with adenosine in our simulations, but with the ligand in an inverted pose, to which we now turn.
At ∼500 ns in the first adenosine trajectory (indicated by the vertical dashed line in Fig. 4), the ligand breaks its crystallographically observed contacts and finds a new inverted pose. The transiently formed H-bonds with Asn-253 and His-278 on H5, H6, and H7 are broken, whereas new H-bonds are formed between the ribose hydroxyls and Glu-13 on H1, and between the exocyclic amide nitrogen (N3) and Thr-88 on H3. Four residues on H2 (Ala-59, Phe-62, Ala-63, and Ile-66) form a hydrophobic pocket for the small hydrophobic moiety on the ribose. Interestingly, the ring-stacking interaction with Phe-168 is re-formed in the inverted pose, making it the only contact that is common to both poses. The same pose is observed in the second adenosine trajectory, though it is less stable because the ligand samples a number of alternate configurations, and therefore does not present a clear signature in the contact time series (see Fig. S2). In both trajectories, the ligand eventually spontaneously unbinds and escapes the binding pocket into the solvent.
Binding-pocket hydration
Considering that the cost of breaking an H-bond is on the order of a few times kBT, how is it that we see such transient and dynamic H-bonds in the adenosine case? And why does it differ so dramatically from the UK432097 case? We suggest that the answer lies in the difference in the solvation of the binding pocket, noting that previous work on rhodopsin (15,16) and the β2 adrenergic receptor (17) showed a connection between binding-pocket hydration and ligand conformation and dynamics. In this case, we expected that the transient nature of H-bonds, especially those formed with the ribose hydroxyls, would be due to competition with water in the binding pocket.
We first simply counted water molecules in the binding pocket, defining the binding pocket by a rectangular volume bounded by residues 88, 177, 249, 253, 274, and 278. (Although the binding pocket is better defined by an irregular volume, incorporating such an irregular shape requires a much more computationally demanding analysis, and we expect the outcome will not qualitatively change the results. Furthermore, we also present a volumetric map of the binding-pocket water, which presents a spatially resolved analysis.) The data clearly indicate that significantly more water is present in the binding pocket in the adenosine case, as shown in Fig. 5. In the adenosine case, the data appear to continue to trend upward, most likely because, as mentioned earlier, the adenosine is in the process of spontaneously unbinding. Nonetheless, we can estimate the number of waters in the binding pocket by considering the portion of the data from 100 ns to 900 ns, where the UK432097 data appear converged and the adenosine is still bound. We find that there are on average 73 waters in the adenosine-binding pocket, and only 18 in the UK432097 case. These data are consistent with the crystal structure data: nine water molecules were resolved in the binding pocket in the adenosine case, compared with only two in the UK432097 case. Data for two independent trajectories are shown in Fig. S3 and are consistent with this analysis, showing 57 waters in the adenosine case and 24 in the UK432097 case.
Where exactly is the water in each case? In particular, is water found deep in the binding pocket where it can compete with the ribose hydroxyls? To answer this question, we built a volumetric map of the water density, essentially a three-dimensional histogram of the binding pocket volume. The histogram is visualized in Fig. 6 and Fig. S4 as water density isosurfaces; in this case, every point contained in the surface has an average water density at least 0.3 of the density of bulk water. From a comparison of the isosurfaces, it is clear that there is significantly more water in the vicinity of the ribose hydroxyls in the adenosine case than in the UK432097 case, supporting the hypothesis that competition with water for H-bonds is responsible for the transient nature of the key ribose contacts in the adenosine case. A comparison of the extracellular surfaces of the protein in each case offers a possible explanation for the difference: In UK432097, the aromatic appendages (see Fig. 2) plug the entrance to the binding pocket, blocking access of water to the interior of the protein. We revisit this observation below.
Figure 6.
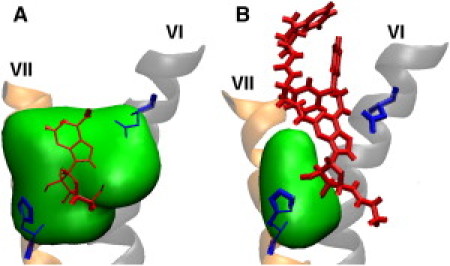
Isosurface of water density in the ligand-binding pocket of A2A with (A) adenosine or (B) UK432097. The water density is averaged over 0.1–0.9 μs, and the isosurface with 0.3 of the bulk water density is drawn in green. In each figure, H6 and H7 are marked by VI and VII, respectively, the ligand is shown in red, and Asn-253 on H6 and His-278 on H7 are shown in blue. The portion of the ligand buried in isosurface is rendered with thinner lines. The second trajectory results are shown in Fig. S4.
Discussion
We have presented molecular-dynamics data for the adenosine A2A receptor bound to either the native ligand adenosine or a highly potent synthetic agonist, UK432097. In two independent trajectories totaling 2 μs of data, adenosine is found to be very dynamic in the binding pocket, as evidenced by the CoM motion and the orientational dynamics. The dynamics of UK432097 as observed in two independent trajectories totaling 2 μs contrast sharply with adenosine: the CoM explores a much smaller volume, and the extended nature of the ligand prevents significant orientational dynamics while it is bound. The dynamics have consequences for the contacts formed with the protein. Of special significance are the H-bonds formed with the ribose hydroxyls, which are thought to be crucial for activation of the receptor. We find that in the adenosine case, the binding pocket is much better hydrated, and that competition with water for H-bonds with the protein results in much more transient H-bonds in the adenosine case and is partly responsible for the observed difference in dynamics. Our observations are corroborated by recent calculations that suggested that caffeine may sample multiple poses when bound to A2A (36), and by earlier simulations of bovine rhodopsin (15), squid rhodopsin (16), and the β2 adrenergic receptor (17).
The dynamic nature of adenosine relative to UK432097 ought to have observable consequences. Our data suggest that the reorientation dynamics of adenosine would not be directly observable by a time-resolved measurement of the loss of polarized fluorescence emission from the ligand. Our simulations indicate that the reorientational dynamics of adenosine occur on ∼100 ns timescales, whereas the fluorescence lifetime of adenosine is on the order of 10 ns. However, the difference between the adenosine and UK432097 cases may be detected because of the difference in the long time limit arising from the hindered nature of the UK432097 reorientation.
Our data also suggest the possibility of an additional binding site located closer to the extracellular entrance to the binding pocket, formed by Glu-13 on H1, Phe-168, and a small hydrophobic pocket on H3. To our knowledge, the role of Glu-13 in ligand binding has yet to be studied by means of mutagenesis. It is possible that the proposed inverted pose is visited as the ligand binds, and thus mutagenesis of Glu-13 might reduce the on-rate for ligand binding. If this notion is confirmed, Glu-13 and the small hydrophobic pocket on H2 might serve as a target for allosteric control of ligand binding, perhaps by incorporating a hydrophobic plug much like the extracellular end of UK432097. Allosteric modulation of GPCR function is an area of very active research (37). Allosteric modulators of adenosine receptor function are known, but, by and large, the mechanisms by which they exert their effects are not (5).
Finally, we argue that the 100- to 1000-fold lower EC50 of UK432097 (21,22) provides evidence in support of our observations, when viewed in the context of conformational selection and its connection to G-protein activation. We suggest that the difference in potency is explained by the fact that UK432097, by forming more robust H-bonds with the hydroxyl moiety, stabilizes a much tighter neighborhood of active conformations, which manifests as an increased likelihood of G-protein activation per unit time. Adenosine, on the other hand, allows the receptor access to a much broader range of conformations. We stress that this hypothesis is based on experimental evidence for the key role of the hydroxyl H-bonds in activation (34,35) and the stark differences observed between the UK432097 and adenosine cases, and not on any inference about the affinity of the two ligands for the receptor. Perhaps the ensemble of conformations that are stabilized by the native ligand is promiscuous enough to be steered down different pathways under different circumstances, for example, by a change in the membrane environment or by dimerization with another receptor. We intend to follow this lead in future work to explore the hypothesis that there is a direct link between conformational promiscuity and functional selectivity.
Acknowledgments
The authors thank Anne Robinson and the members of her laboratory for their thoughts and discussions.
This work was supported by grants from the National Center for Research Resources (5P30RR031160-03) and the National Institute of General Medical Sciences, National Institutes of Health (8 P30 GM103519-03). This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (grant OCI-1053575).
Supporting Material
References
- 1.Gilchrist A. John Wiley & Sons; Hoboken, NJ: 2010. GPCR Molecular Pharmacology and Drug Targeting. [Google Scholar]
- 2.Congreve M., Marshall F.H. The impact of GPCR structures on pharmacology and structure-based drug design. Br. J. Pharmacol. 2010;159:986–996. doi: 10.1111/j.1476-5381.2009.00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Congreve M., Langmead C.J., Marshall F.H. Progress in structure based drug design for G protein-coupled receptors. J. Med. Chem. 2011;54:4283–4311. doi: 10.1021/jm200371q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaakola V.-P., Ijzerman A.P. The crystallographic structure of the human adenosine A2A receptor in a high-affinity antagonist-bound state: implications for GPCR drug screening and design. Curr. Opin. Struct. Biol. 2010;20:401–414. doi: 10.1016/j.sbi.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Fredholm B.B., IJzerman A.P., Müller C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol. Rev. 2011;63:1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaakola V.-P., Griffith M.T., Stevens R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hino T., Arakawa T., Murata T. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature. 2012;482:237–240. doi: 10.1038/nature10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doré A.S., Robertson N., Marshall F.H. Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure. 2011;19:1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu F., Wu H., Stevens R.C. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lebon G., Warne T., Tate C.G. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lyman E., Higgs C., Voth G.A. A role for a specific cholesterol interaction in stabilizing the apo configuration of the human A2A adenosine receptor. Structure. 2009;17 doi: 10.1016/j.str.2009.10.010. 1660–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Malley Michelle A., Helgeson Matthew E., Wagner Norman J., Robinson Anne S. Biophys. J. 2011;101:1938–1948. doi: 10.1016/j.bpj.2011.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodríguez D., Piñeiro Á., Gutiérrez-de-Terán H. Molecular dynamics simulations reveal insights into key structural elements of adenosine receptors. Biochemistry. 2011;50:4194–4208. doi: 10.1021/bi200100t. [DOI] [PubMed] [Google Scholar]
- 14.Martínez-Mayorga K., Pitman M.C., Brown M.F. Retinal counterion switch mechanism in vision evaluated by molecular simulations. J. Am. Chem. Soc. 2006;128:16502–16503. doi: 10.1021/ja0671971. [DOI] [PubMed] [Google Scholar]
- 15.Grossfield A., Pitman M.C., Gawrisch K. Internal hydration increases during activation of the G-protein-coupled receptor rhodopsin. J. Mol. Biol. 2008;381:478–486. doi: 10.1016/j.jmb.2008.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jardón-Valadez E., Bondar A.-N., Tobias D.J. Coupling of retinal, protein, and water dynamics in squid rhodopsin. Biophys. J. 2010;99:2200–2207. doi: 10.1016/j.bpj.2010.06.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romo T.D., Grossfield A., Pitman M.C. Concerted interconversion between ionic lock substates of the β(2) adrenergic receptor revealed by microsecond timescale molecular dynamics. Biophys. J. 2010;98:76–84. doi: 10.1016/j.bpj.2009.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dror R.O., Pan A.C., Shaw D.E. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA. 2011;108:13118–13123. doi: 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dror R.O., Arlow D.H., Shaw D.E. Identification of two distinct inactive conformations of the β2-adrenergic receptor reconciles structural and biochemical observations. Proc. Natl. Acad. Sci. USA. 2009;106:4689–4694. doi: 10.1073/pnas.0811065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hurst D.P., Grossfield A., Reggio P.H. A lipid pathway for ligand binding is necessary for a cannabinoid G protein-coupled receptor. J. Biol. Chem. 2010;285:17954–17964. doi: 10.1074/jbc.M109.041590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daly J.W. Adenosine receptors: targets for future drugs. J. Med. Chem. 1982;25:197–207. doi: 10.1021/jm00345a001. [DOI] [PubMed] [Google Scholar]
- 22.Poucher S.M., Keddie J.R., Coll M.G. The in vitro pharmacology of ZM 241385, a potent, non-xanthine A2a selective adenosine receptor antagonist. Br. J. Pharmacol. 1995;115:1096–1102. doi: 10.1111/j.1476-5381.1995.tb15923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maestro, version 9.2. 2011. Schrödinger, LLC, New York.
- 24.Lomize M.A., Lomize A.L., Mosberg H.I. OPM: orientations of proteins in membranes database. Bioinformatics. 2006;22:623–625. doi: 10.1093/bioinformatics/btk023. [DOI] [PubMed] [Google Scholar]
- 25.MacKerell A.D., Jr., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 26.Klauda J.B., Venable R.M., Pastor R.W. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vanommeslaeghe K., Hatcher E., Mackerell A.D., Jr. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bowers K.J., Chow E., Shaw D.E. Scalable algorithms for molecular dynamics simulations on commodity clusters. Proc. ACM/IEEE SC2006 Conf., Tampa, FL. 2006 [Google Scholar]
- 29.Martyna G.J., Tobias D.J., Klein M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 30.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 31.Flyvbjerg H., Petersen H.G. Error estimates on averages of correlated data. J. Chem. Phys. 1989;91:461–466. [Google Scholar]
- 32.Romo T.D., Grossfield A. LOOS: an extensible platform for the structural analysis of simulations. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2009;2009:2332–2335. doi: 10.1109/IEMBS.2009.5335065. [DOI] [PubMed] [Google Scholar]
- 33.Lakowicz J.R. Plenum Press; New York: 1983. Principles of Fluorescence Spectroscopy. [Google Scholar]
- 34.Jaakola V.-P., Lane J.R., Stevens R.C. Ligand binding and subtype selectivity of the human A(2A) adenosine receptor: identification and characterization of essential amino acid residues. J. Biol. Chem. 2010;285:13032–13044. doi: 10.1074/jbc.M109.096974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim J., Wess J., Jacobson K.A. Site-directed mutagenesis identifies residues involved in ligand recognition in the human A2a adenosine receptor. J. Biol. Chem. 1995;270:13987–13997. doi: 10.1074/jbc.270.23.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y., Burger S.K., Vöhringer-Martinez E. Computational study of the binding modes of caffeine to the adenosine A2A receptor. J. Phys. Chem. B. 2011;115:13880–13890. doi: 10.1021/jp2022049. [DOI] [PubMed] [Google Scholar]
- 37.Conn P.J., Christopoulos A., Lindsley C.W. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.