

Abstract
There is increasing evidence for the presence of an alternative code imprinted in the genome that might contribute to gene expression regulation through an indirect reading mechanism. In mammals, components of this coarse-grained regulatory mechanism include chromatin structure and epigenetic signatures, where d(CpG) nucleotide steps are key players. We report a comprehensive experimental and theoretical study of d(CpG) steps that provides a detailed description of their physical characteristics and the impact of cytosine methylation on these properties. We observed that methylation changes the physical properties of d(CpG) steps, having a dramatic effect on enriched CpG segments, such as CpG islands. We demonstrate that methylation reduces the affinity of DNA to assemble into nucleosomes, and can affect nucleosome positioning around transcription start sites. Overall, our results suggest a mechanism by which the basic physical properties of the DNA fiber can explain parts of the cellular epigenetic regulatory mechanisms.
Introduction
Determining the mechanisms that regulate gene expression in complex organisms is the next frontier of genomics research (1). In the traditional paradigm, specific proteins regulate gene expression through the recognition of certain sequence signals (by means of specific hydrogen-bond interactions) upstream of the transcription start sites (TSSs) (2). Nevertheless, there is increasing evidence about the presence of an alternative code that may contribute to a rough regulation of gene expression through an indirect reading mechanism, probably signaled by chromatin structure and epigenetic marks (3,4). This mechanism is unlikely (even in a synergistic manner) to achieve the fine-tuning and specificity of the direct protein-DNA readout. Conversely, it probably plays a pivotal role in basal gene expression, which requires less regulation and for which the extreme cost of developing a highly specific protein regulation infrastructure seems unjustified. Key players in this regulatory mechanism may be d(CpG) steps, which despite being largely underrepresented in the genome of complex organisms are enriched in nearly 60% of human promoters, where they often define ultrarich CpG segments, the so-called CpG islands (5,6). Even if CpG islands do not contain specific transcription binding motifs, they clearly favor the downstream binding of the transcription machinery (7), particularly for those genes that are usually active. The molecular basis of the d(CpG) effect on gene regulation remains unclear, although it has been suggested to be related to the definition of DNA fiber properties (8).
One of the most intriguing features of the d(CpG) step is its ability to undergo nonmutagenic chemical modifications such as cytosine methylation (9). In mammalian genomes, DNA methyltransferases (DNMTs) can transfer a methyl group from S-adenosylmethionine to cytosine at CpG dinucleotides (10). The bulk of the methylation takes place during DNA replication in the S-phase of the cell cycle (11), and is the most abundant form of post-replicative DNA modification observed in eukaryotic organisms (12). During this process, the cytosine is flipped 180° out of the DNA backbone into an active-site pocket of the enzyme (13) where methylation of cytosine takes place.
Intriguingly, methylation of cytosines seems to be an erroneous decision of evolution because it dramatically increases the chances of C→T mutation, but this seeming disadvantage is compensated for by the gain in regulatory possibilities offered by methylation. Indeed, highly methylated DNA is typically associated with inactive genes, whereas methylation depletion is observed for active genes (14,15). Furthermore, most cytosines in CpG steps, except those in CpG islands, are methylated in vertebrate somatic cells (16,17). The first step of methylation occurs early in mammalian development as a result of de novo DNMTs (Dnmt3a and Dnmt3b) (18) that methylate CpG steps in both DNA strands. The methylation profile is conserved by maintenance DNA methyltransferase (Dnmt1) throughout cell divisions. During replication, daughter strands are nonmethylated, resulting in hemimethylated DNA. Dnmt1 recognizes hemimethylated CpG steps and methylates the daughter strand (19). Recent studies have demonstrated that changes in methylation patterns along CpG islands and CpG shores (methylation hotspots on the outskirts of CpG islands (20)) correlate with tissue differentiation and cancer, proving the role of methylation in cellular reprogramming (20,21,23,24). The regulatory function of methylated cytosines (hereafter referred to as MeC) was traditionally explained by their interaction with methyl-CpG binding domain proteins (MBDs) (18), but considering the prevalence of cytosine methylation, MBDs alone cannot entirely account for the role of d(CpG) methylation in the cell. An increased level of complexity is provided by the almost nonexistent sequence specificity of the DNMTs, precluding the mechanism underlying the methylation reaction (26).
Here we present a comprehensive theoretical analysis of the d(CpG) properties along with an experimental validation of key theoretical findings. We show that d(CpG) steps display unique physical properties, especially in the context of CpG islands, that severely change upon methylation. Our calculations suggested, and experiments confirmed, that DNA segments containing MeC are very stiff and hard to bend, and display a lower tendency to circularize or form nucleosomes by wrapping around histones. The latter effect has striking consequences for the organization of nucleosome arrays near TSSs, which in turn modifies the accessibility of regulatory proteins, leading to alterations in the pattern of DNA expression. Overall, without diminishing the role of specific regulatory proteins, basic descriptors of DNA physical properties can help us rationalize several seemingly disconnected pieces of the puzzle of DNA regulation.
Methods
Molecular-dynamics simulations
We performed molecular-dynamics (MD) simulations on an array of different oligomers containing CpG steps, in both their methylated and nonmethylated forms (see Table S1 in the Supporting Material). We also included in our analysis trajectories of unmethylated CpG step data from the Ascona B-DNA Consortium (ABC) (27) database in all tetramer environments to enrich the dynamics database. All simulations were carried out in duplicates for 100 ns (after equilibration) using explicit solvent, the parmbsc0 refinement of the Amber force field (28), and state-of-the-art simulation conditions (Supporting Material).
Mesoscopic model of DNA flexibility
We derived a flexibility model from different MD equilibrium trajectories using a harmonic model (29–31). Accordingly, we projected the MD trajectories onto a helical reference system to obtain equilibrium values and derive the covariance matrix, which we then inverted to recover the stiffness matrices for each basepair step, from which a mesoscopic estimate of the energy associated to a given deformation can be easily computed (29,32) as
where ΔXi is the perturbation from equilibrium geometry (Fig. 1), and fij are elements of Θ, where Θ is the 6 × 6 stiffness matrix expressing the stiffness of a given step to deformation in roll, tilt, twist, slide, shift, and rise (see Supporting Material, Fig. 2, and Pérez et al. (33)). Alternatively, global deformation parameters can be derived using a similar approach, but considering global instead of local geometric descriptors (see Fig. S1, Supporting Material, and Lankas et al. (34)). Note that elastic parameters derived from protein-DNA crystal complexes (29,31) or simulation data are in good agreement (33), supporting their use to describe DNA flexibility. Here, we favored the use of MD-derived values for consistency with the newly derived parameters describing MeCpG steps (which cannot be derived from analysis of crystal structures).
Figure 1.
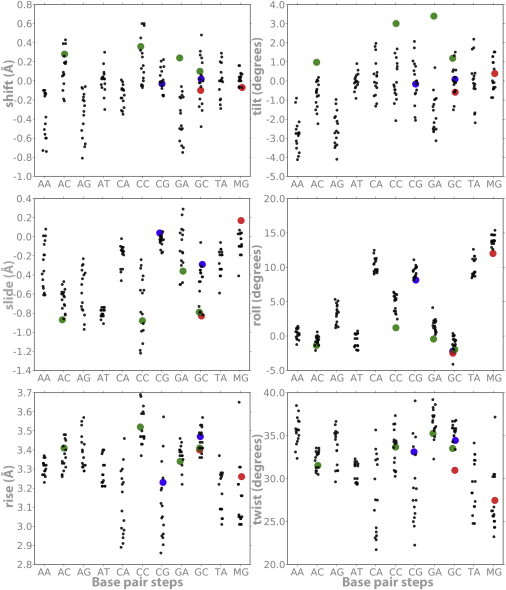
Average helical parameters (translations in angstroms and rotations in degrees) derived from MD simulations of the usual 10 dinucleotides plus d(MeCpG; referred to as MG in the figure). The black dots correspond to control simulations (those performed for this work and those obtained from the ABC database (27)) for each central basepair step representing the different tetramer environments; notice the slight displacement from the vertical to avoid stacking of data. Blue dots stand for d(CpG) and d(GpC) steps when embedded in a poly d(CpG) track. Green dots refer to neighboring methylated steps (XpMeC): d(ApMeC), d(TpMeC)=d(GpA), d(CpMeC), and d(GpMeC). Finally, red dots stand for d(MeCpG) and d(GpMeC) in the context of a poly d(CpG) track.
Figure 2.
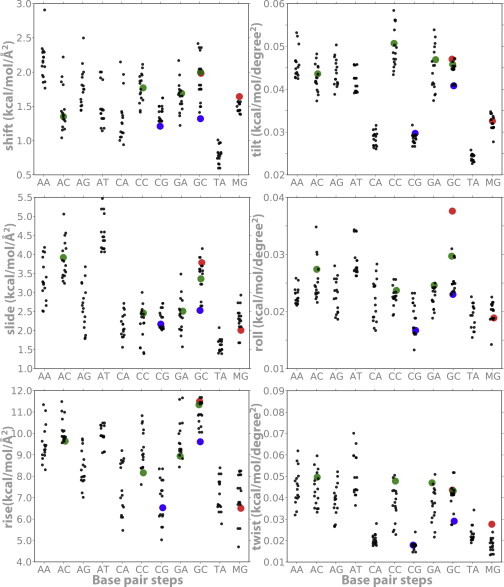
Average stiffness helical parameters (translations in kcal/mol Å2 and rotations in kcal/mol deg2) derived from MD simulations of the usual 10 dinucleotides plus d(MeCpG; referred to as MG in the figure). Same notation as in Fig. 1.
In this work, we used a mesoscopic method to estimate deformation energy related to nucleosome formation and circularization assays. This implies that indirect readout mechanisms prevail over the direct readout for the description of sequence preferences in nucleosome binding. A second implication is that these deformations follow a harmonic behavior. Both of these assumptions represent simplifications, and thus the validity of the method is not always guaranteed (32,35,36).
Circularization assays and modeling
We carried out DNA circularization experiments to validate our theoretical estimations about the impact of methylated cytosines on DNA physical properties. For this purpose, we designed a short polymerizable oligonucleotide (d(GAAAAAACGGGCGAAAAACGG)·d(TCCCGTTTTTCGCCCGTTTTT)) based on a reported sequence favoring the formation of minicircles (37), with the incorporation of a central CpG dinucleotide subject to be methylated and 5′-sticky ends to enable the formation of multimers. Thus, under favorable ligation conditions, the multimers form circles that are as short as allowed by the geometry and flexibility of the DNA (Fig. 3). As a negative control, we selected a previously reported nonbendable oligonucleotide (d(GCAAATATTGAAAAC)·d(GCGTTTTCAATATTT; see Supporting Material for details). The ligation products were analyzed by atomic force microscopy (AFM) and by two-dimensional gel electrophoresis (Fig. 3, Fig. S2, Fig. S3, and Supporting Material).
Figure 3.
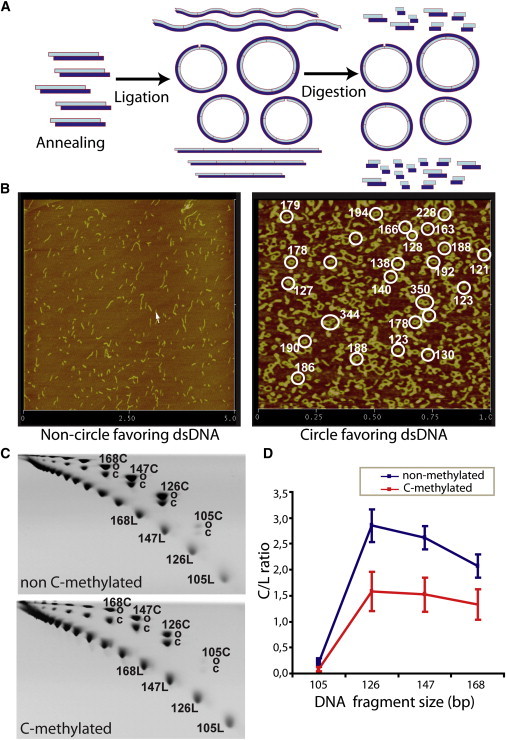
Overview of circularization assays. (A) Schematic diagram of the underlying principle of circularization assays. A DNA oligonucleotide is first annealed to form duplexes and subsequently is multimerized-circularized by a ligation reaction. Under favorable ligation conditions, DNA forms circles as short as allowed by the geometry and flexibility of the DNA. Only circularized DNA is resistant to an exonuclease digestion; linear multimers will be degraded. (B) AFM images of ligation products for 15-bp nonfavoring (left) and 21-bp favoring (right) circularization oligonucleotides; circle size estimations are highlighted in white. (C) 2D polyacrylamide native gels showing different migrations of linear (L) and circular (C) DNA species (which can be either covalently closed (cC) or nicked open (oC)) for nonmethylated and MeC oligomers of 21 bp, respectively. Linear DNA molecules are positioned on the lower diagonal, and circular DNA molecules are positioned on the upper diagonal. (D) The circularization efficiency is expressed as the ratio between C and L molecules of the same size.
Circularization efficiency was determined based on the J-factor, which defines the ratio of circular and linear DNA species for a given sequence length (37). Experimental J-factors were extracted from the linear and circle DNA signal intensities as detected on two-dimensional gels (see Supporting Material and Fig. S3). Although the absolute J-factors depend on the experimental setup, the ratio of J-factors of methylated versus nonmethylated oligos provide a reliable measure of the impact of methylation on DNA circularization. Accordingly, experimental J-factors can validate whether theoretical suggestions regarding physical changes induced by methylation are correct. We derived theoretical J-factors from Monte Carlo simulations (Supporting Material) using mesoscopic descriptors derived from MD simulations (see above).
Mesoscopic model of nucleosome deformation energy
We theoretically determined the ability of a 147-mer DNA sequence to wrap around a nucleosome using harmonic deformation energy as described above considering mesoscopic descriptors. To reduce the noise, we determined the deformation vector (ΔX above) using the target geometry obtained by Fourier-averaging all available crystal structures of nucleosome particles. As noted above, our mesoscopic model is useful in so far as histone-bound DNA deforms harmonically and the indirect readout has an important contribution in directing nucleosome formation. Regarding the first point, the rotational degrees of motion in nucleosomes (twist, roll, and tilt) clearly fall within the normal fluctuations of DNA (38,39), and only the translational parameter slide shows slightly more positive values than expected. Clearly, evolution has optimized nucleosome positioning sequences to have flexible steps such as d(CpA) and d(TpA) in crucial positions to accommodate the deformations required for nucleosome binding (see results of SELEX experiments in Thåström et al. (40)). Note also that by using a single conformation to model all nucleosomes, Tolstorukov et al. (38) and Balasubramanian et al. (39) found a high degree of correlation between the predictions arising from harmonic deformation energies and nucleosome positioning sequences, a result that was consistent with nucleosome location experiments performed by our group (41). Thus, without ignoring its limitations, we believe the model can be useful to rationalize nucleosome positioning.
In vitro nucleosome reconstitution
To assess the effect of methylated DNA on nucleosome assembly, we selected a nucleosome positioning sequence (DNA construct 601.2 in Anderson and Widom (42)) to reconstitute nucleosomes in vitro after incubation with histones, before and after extensive DNA methylation (see Supporting Material). Methylated states were verified by DNA sequencing. The reconstituted nucleosomes were subsequently analyzed by gel shift assays (see Fig. 4).
Figure 4.
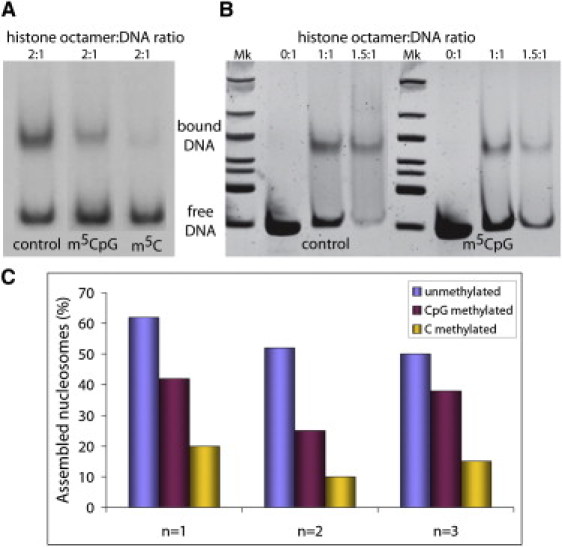
In vitro nucleosome core particle reconstitution. Results of gel mobility shift assays of nucleosomes reconstituted in vitro with a 147-bp 601.2 DNA fragment containing either C or MeC at different histone octamer concentrations are shown. The upper bands correspond to histone core-bound DNA, and lower bands correspond to unbound (or free) DNA. Mk: DNA ladder for size band estimation. (A) Radiolabeled DNA bands. (B) DNA bands stained with SyBr Safe (Invitrogen) and visualized by ultraviolet light. (C) Histograms displaying the percentage of in vitro assembled nucleosomes using the same sequence in different methylation conditions coming from triplicate experiments.
Results
Physical properties of CpG steps and CpG islands
MD simulations performed here in conjunction with those retrieved from the ABC database (27) revealed that sequence is crucial for defining the DNA equilibrium geometries (Fig. 1). In general, Pyr-Pur steps show a lower rise and twist, as well as higher roll values, than the rest of the dinucleotide steps. Additionally, they display an unusually large dispersion in certain key equilibrium helical parameters (e.g., twist), arising from different tetramer environments. Focusing on the different tetramer environments, we can see that the d(CpG) steps are peculiar in presenting bimodal distributions of some parameters (e.g., twist; see Fig. S4), which confirms previous ABC findings (27). Such bimodality is not an artifact that arises from incomplete sampling, because it is also present in multi-microsecond trajectories (43), and suggests that the d(CpG) step is specially flexible, as confirmed by a stiffness analysis (Fig. 2). It is worth noting that the large deformability in twist and roll, combined with large roll values, suggests that protein-induced curvature may be favored in DNA with CpG steps. Fig. S5 shows that CpG, despite the neighboring basepair, is more curved than most basepair steps (only TA and CA are comparable) and is directed preferentially toward the major groove.
One might expect the unique physical properties of d(CpG) steps to be amplified in long d(CpG)n tracks (i.e., CpG islands). Surprisingly, this is not the case, and the global geometrical properties of long d(CpG) segments are different from those expected by extrapolating the individual characteristics of the d(CpG) steps (Fig. S1). Thus, the high flexibility of the d(CpG) steps suggests that poly d(CpG) should be extremely flexible. Conversely, the d(CpG)9 segment studied here is hardly distinguishable from other 18-mer duplexes in terms of global unwinding and isotropic bending. This is hardly surprising when one considers the differences between an individual d(CpG) step and a poly d(CpG). The former has the properties of an individual d(CpG) step, whereas the d(CpG)9 segment has properties due to the alternation of d(CpG) and d(GpC) steps. Thus, the lower flexibility of d(GpC) steps (Fig. S1) makes the whole sequence overall stiffer than a simple extrapolation of d(CpG) properties. Furthermore, whereas the large roll of individual d(CpG) steps would imply a strong curvature of the entire oligonucleotide, the d(CpG)9 curvature is actually very moderate due to the low roll values of d(GpC) steps. In conclusion, the properties of long d(CpG) segments are distinct from the extrapolation of properties of isolated d(CpG) steps, warning against the use of oversimplified rules of DNA flexibility.
Effect of CpG methylation
Early structural experiments suggested that cytosine methylation (Fig. S5) might induce helical transitions from B- to Z-DNA (44). However, a secondary structure analysis of CpG methylated oligonucleotides by circular dichroism spectroscopy (Supporting Material and Fig. S6) revealed that the transition only occurred at nonphysiological salt concentrations (from 1 to 2 M NaCl). This evidence is in agreement with our MD results and previously reported Fourier transform infrared (FTIR) spectroscopy data (45), demonstrating that in vivo DNA remains in the B-form upon methylation, and accordingly, the transition to the Z-form is not the underlying determinant for the physiological role of CpG methylation.
The results of the MD simulations suggest that when methylated, d(CpG) steps increase their average roll value and reduce their twist (Fig. 1), leading to an increase in local curvature. Furthermore, methylation makes d(CpG) steps stiffer, especially in terms of roll and tilt deformations: the MeCpG step has larger tilt and roll force constants on average than the CpG step (MG and CG, respectively, in Fig. 2). Methylation also alters the geometric properties of the basepair step previous to the d(CpG) site, here denoted as d(XpC), where X = A, C, G, or T (Figs. 1 and 2). In canonical DNA, d(XpC) steps tend to compensate for the geometry and relative stiffness of d(CpG) in twist, tilt, and roll. However, upon methylation, we observed an increase of force constants for rotational parameters in both the MeCpG and d(XpMeC) steps (green dots in Fig. 2). Hence, the additive effect of methylation leads to significant alterations in the global physical properties of DNA, especially for CpG islands (Figs. 1 and 2, and Fig. S1).
The higher stiffness of the d(MeCpG) steps should lead to a decrease in the DNA circularization efficiency, which must be especially visible for the smallest circles. Indeed, Monte Carlo calculations using the MD-derived stiffness parameters (see Materials and Methods, and Supporting Material) suggested that circularization of 126- to 189-bp-long methylated oligos (one MeCpG every 21 bp) is more difficult than circularization of unmethylated ones (with a relative J-factor of 0.05–0.2). This result was confirmed by circularization experiments on the same sequence with a relative J-factor of ∼0.5 (see Fig. 3, Materials and Methods, and Supporting Material).
Potential impact on chromatin structure
The sequence-dependent physical properties of DNA play a crucial role in determining nucleosome positioning (46–52) and thus are instrumental in genome regulation (53,54). An analysis of nucleosome distribution in Saccharomyces cerevisiae (55,56) revealed that d(CpG)s are enriched in nucleosome-bound regions but depleted in internucleosomal segments, supporting the idea that d(CpG) steps are easily fitted into the nucleosome structure. Protein-DNA interactions are governed by direct and indirect readouts. The former arises from protein-DNA direct interactions, whereas the latter is based on the ability of a DNA sequence to deform into a conformation that makes the interaction happen. Without underestimating the importance of direct readout mechanisms in nucleosome binding, we note that indirect readout models seem to capture well the global positioning profile of nucleosomes (41). Thus, changes in the physical properties of the DNA fiber related to methylation should have a direct impact on nucleosome affinity and positioning. Our models prompted us to hypothesize that in the absence of external factors (e.g., MBD proteins or chromatin remodelers), the increased stiffness due to d(CpG) methylation leads to a higher deformation energy required to wrap DNA around a nucleosome (Fig. 5). We tested this hypothesis by conducting in vitro nucleosome reconstitution experiments (see Materials and Methods, and Supporting Material) with normal and methylated DNAs. The results confirm that the d(MeCpG) DNA has a lower ability to form nucleosomes than the nonmethylated sequence (Fig. 4).
Figure 5.
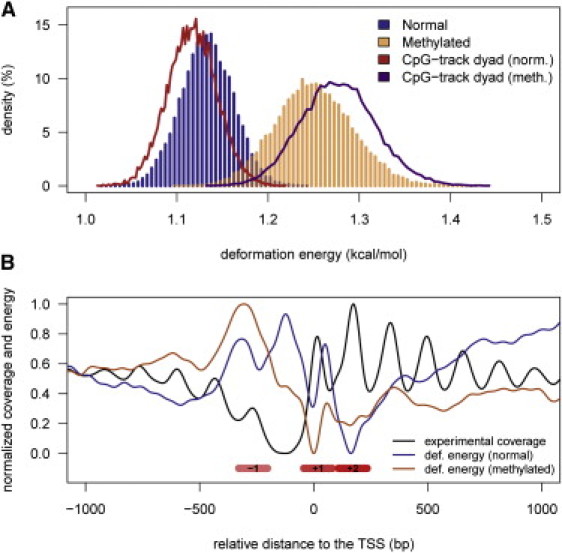
Impact of methylation on nucleosome positioning. (A) The distribution of predicted energies (per base step) for 147-bp-long random DNAs in normal (blue histogram) and methylated (orange histogram) forms, respectively. The curves correspond to the predicted energy (per base step) when random oligos contain a poly d(CpG) track at the dyad, in normal (red) and methylated (magenta) forms. (B) The nucleosome distribution surrounding the TSSs of yeast genes determined from MNase digestion experiments (black line), compared with the predicted distortion energy to wrap a nucleosome in those sites when genomic DNA is normal (blue line) or methylated (orange line). All values were normalized to facilitate the interpretation of the plots. Nucleosome positions −1, +1, and +2 are indicated by red boxes for clarity.
Interestingly, nucleosome formation was further decreased when all cytosines in the DNA were methylated, which confirms that a reduced flexibility is mainly responsible for the lower affinity of methylated DNA for the histones. Other nonmammalian organisms that have alternative cytosine methylation patterns besides the methylation of CpG steps could use this strategy for gene regulation. Tillo and Hughes (57) established that increasing C+G contents correlate well with higher nucleosome formation. Therefore, it is not surprising that a mechanism in which more cytosines are methylated would further rigidify the sequence, further changing the nucleosome positioning preferences (57).
Discussion
d(CpG) steps are statistically underrepresented in the genome, but they appear concentrated in regulatory regions, which suggests that d(CpG) steps provide the suitable physical properties that enable the DNA to efficiently interact with regulatory proteins (58,59) and help define the correct nucleosome positioning. Indeed, our MD results suggest that isolated d(CpG) steps are curved (see Fig. S5) and particularly flexible, making d(CpG) steps appropriate for those regions in which DNA needs to be locally distorted to facilitate protein binding, in agreement with early NMR measures (60). This hypothesis is supported by an analysis of whole-genome nucleosome positioning in yeast, which revealed a d(CpG) step depletion in internucleosomal segments (41,55,56).
Mesoscopic calculations (Fig. 5 A) indicate that a d(CpG)5 segment located at the dyad axis may favor nucleosome formation, and hence very long d(CpG) tracks (CpG islands) are very likely to assemble into nucleosomes. Nucleosomes can easily move along the d(CpG) track, presumably leading to a nucleosome-depleted region at the external borders of the island, and thereby imprinting a distinct nucleosome array organization that would define the accessibility to regulatory regions downstream of the CpG islands, where many promoters are located. This also accords with the fact that high C+G content correlates with nucleosome positioning (57)
Methylation increases the curvature of d(CpG) steps (Fig. S5), although this local geometric effect tends to be compensated for by neighboring steps. In fact, all tested 18-mer methylated oligos (except CpG islands) were less curved and less flexible than their unmethylated counterparts. On the other hand, as previously suggested (61,62), methylation increases d(CpG) stiffness, and this effect propagates to neighboring steps, leading to a global increase in the rigidity of DNA. Our results suggest that this effect alone explains (within the indirect readout model) the limited ability of methylated DNA to interact with certain proteins, such as transcription factors (63–65). Our in silico simulations and in vitro nucleosome reconstitution experiments showed that methylation reduces DNA affinity for nucleosomes, probably due to the increased rigidity of the DNA fiber. Our results are in full agreement with previous findings (66,67), with recent data about the anticorrelation between nucleosome formation and methylation (68), and with physical intuition that suggests that a more flexible fiber should wrap more easily than a rigid one. Additionally, the rigidifying effect of MeCpG is also observed in recent fluorescence resonance energy transfer (FRET)-derived data (69,70) showing that methylating nucleosome-bound DNA results in nucleosome compaction and rigidity. Taken together, these results indicate that DNA methylation may regulate nucleosome dynamics by increasing the rigidity of DNA either before or during nucleosome assembly.
Our combined theoretical and experimental results demonstrate that methylation decreases nucleosome formation. Nucleosome depletion is usually considered as a signal of gene activity (71,72). At the same time, gene activity correlates with low levels of methylation (73). We postulate that the presence of almost 105 MeCpG steps present in the genome could significantly modify the nucleosome positioning landscape. This hypothesis would explain how the gene expression pattern can change while the number of nucleosomes (but not positions) is kept constant in either methylated or canonical genomes. Hence, we analyzed the nucleosome organization around TSSs on the unmethylated yeast genome and subsequently compared the in silico effects of methylation on nucleosome positioning (Fig. 5 B). TSSs are typically characterized by a nucleosome-depleted region and well-positioned −1, +1, and +2 nucleosomes (55,74). As expected, these positions are clearly marked in the energy profiles: regions with high deformation energy signal nucleosome-depleted areas and vice versa (Fig. 5). These profiles support recent claims about particular nucleosome positioning sites (signaled by large in vitro propensities for nucleosome assembly) anchoring the formation of nucleosomal arrays in vivo (75,76). Methylation of d(CpG) steps modifies the deformation energy profile associated with nucleosome wrapping around the TSS, which ultimately may be reflected by a change in the nucleosome array (Fig. 5 B; note that this does not necessarily make the nucleosomes more diffuse) and, accordingly, in gene expression (77). In particular, it seems that upon methylation, the nucleosome-free region is less defined and the nucleosome −1 is moved downstream. Furthermore, our calculations suggest that when CpG islands are methylated, nucleosomes are concentrated at the CpG island edges, leading to a completely different configuration of the nucleosome array around the TSS and consequently to a change in gene activity. Hence, we can partially rationalize the striking effect of methylated CpG islands on the activity of several genes, particularly those involved in cancer (78), by considering the highly unfavorable impact that methylation has on the ability of poly CpG tracks to wrap around nucleosomes (Fig. 5 A). Further work is required to shed more light on this interesting hypothesis.
Taken together, our studies show how an apparently minor covalent change such as methylation can alter the physical properties of DNA, and how such a change can modify the ability of DNA to organize the chromatin fiber, which may be reflected by significant alterations in gene regulation, even in the absence of specific MBDs.
Conclusions
In summary, simple physical properties of DNA (described from calculations based on first principles) can provide a rationale for the seemingly chaotic diversity of gene regulatory signals in developed organisms, particularly epigenetic signatures such as cytosine methylation. Our results support the hypothesis that physical properties define a basal regulatory code that is superposed onto more elaborated mechanisms involving the action of specific proteins when fine-tuning of gene function is required. Furthermore, our findings suggest that simply by varying the physical properties of some distant regions to a particular gene while keeping the specific protein-binding boxes unaltered, we may be able to modulate that gene's biological functionality. This raises interesting possibilities in the emerging field of synthetic biology. From the results of this study, it follows that methylated DNA is not as likely to form nucleosomes. However, the complete picture is even more complex when one considers that DNMTs have a greater preference to target nucleosome-bound DNA, slightly enriching it (1%) in MeCpG steps (79).
Acknowledgments
We thank Anna Aviñó for help with the experiments. The authors declare that they have no competing interests and acknowledge no conflict of interest.
This work was supported by the Spanish Ministry of Science and Innovation (BIO2009-10964 and Consolider E-Science, INB-Genoma España, and COMBIOMED RETICS), the European Research Council, and the Fundación Marcelino Botín. A.P. received support from a Juan de la Cierva postdoctoral fellowship.
Supporting Material
References
- 1.Collins F.S., Green E.D., Guyer M.S., US National Human Genome Research Institute A vision for the future of genomics research. Nature. 2003;422:835–847. doi: 10.1038/nature01626. [DOI] [PubMed] [Google Scholar]
- 2.Hélène C. Recognition of base sequences by regulatory proteins in procaryotes and eucaryotes. Biosci. Rep. 1981;1:477–483. doi: 10.1007/BF01121581. [DOI] [PubMed] [Google Scholar]
- 3.Schueler M.G., Sullivan B.A. Structural and functional dynamics of human centromeric chromatin. Annu. Rev. Genomics Hum. Genet. 2006;7:301–313. doi: 10.1146/annurev.genom.7.080505.115613. [DOI] [PubMed] [Google Scholar]
- 4.Butcher L.M., Beck S. Future impact of integrated high-throughput methylome analyses on human health and disease. J. Genet. Genomics. 2008;35:391–401. doi: 10.1016/S1673-8527(08)60057-0. [DOI] [PubMed] [Google Scholar]
- 5.Bird A.P. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 6.McClelland M., Ivarie R. Asymmetrical distribution of CpG in an ‘average’ mammalian gene. Nucleic Acids Res. 1982;10:7865–7877. doi: 10.1093/nar/10.23.7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elango N., Yi S.V. Functional relevance of CpG island length for regulation of gene expression. Genetics. 2011;187:1077–1083. doi: 10.1534/genetics.110.126094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kundu T.K., Rao M.R. CpG islands in chromatin organization and gene expression. J. Biochem. 1999;125:217–222. doi: 10.1093/oxfordjournals.jbchem.a022276. [DOI] [PubMed] [Google Scholar]
- 9.Doerfler W. DNA methylation and gene activity. Annu. Rev. Biochem. 1983;52:93–124. doi: 10.1146/annurev.bi.52.070183.000521. [DOI] [PubMed] [Google Scholar]
- 10.Goll M.G., Bestor T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005;74:481–514. doi: 10.1146/annurev.biochem.74.010904.153721. [DOI] [PubMed] [Google Scholar]
- 11.Leonhardt H., Page A.W., Bestor T.H. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell. 1992;71:865–873. doi: 10.1016/0092-8674(92)90561-p. [DOI] [PubMed] [Google Scholar]
- 12.Clark T.A., Murray I.A., Korlach J. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 2012;40:e29. doi: 10.1093/nar/gkr1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klimasauskas S., Kumar S., Cheng X. HhaI methyltransferase flips its target base out of the DNA helix. Cell. 1994;76:357–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 14.Chandler L.A., Jones P.A. Hypomethylation of DNA in the regulation of gene expression. Dev. Biol. 1988;5(N Y 1985):335–349. doi: 10.1007/978-1-4615-6817-9_12. [DOI] [PubMed] [Google Scholar]
- 15.Cedar H., Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- 16.Southern E.M. DNA sequences and chromosome structure. J. Cell Sci. Suppl. 1984;1:31–41. doi: 10.1242/jcs.1984.supplement_1.3. [DOI] [PubMed] [Google Scholar]
- 17.Suzuki M.M., Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 2008;9:465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 18.Hermann A., Gowher H., Jeltsch A. Biochemistry and biology of mammalian DNA methyltransferases. Cell. Mol. Life Sci. 2004;61:2571–2587. doi: 10.1007/s00018-004-4201-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hermann A., Goyal R., Jeltsch A. The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J. Biol. Chem. 2004;279:48350–48359. doi: 10.1074/jbc.M403427200. [DOI] [PubMed] [Google Scholar]
- 20.Irizarry R.A., Ladd-Acosta C., Feinberg A.P. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doi A., Park I.H., Feinberg A.P. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat. Genet. 2009;41:1350–1353. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reference deleted in proof.
- 23.Lister R., Pelizzola M., Ecker J.R. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhutani N., Brady J.J., Blau H.M. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463:1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reference deleted in proof.
- 26.Choi S.H., Heo K., Yang A.S. Identification of preferential target sites for human DNA methyltransferases. Nucleic Acids Res. 2011;39:104–118. doi: 10.1093/nar/gkq774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lavery R., Zakrzewska K., Sponer J. A systematic molecular dynamics study of nearest-neighbor effects on base pair and base pair step conformations and fluctuations in B-DNA. Nucleic Acids Res. 2010;38:299–313. doi: 10.1093/nar/gkp834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pérez A., Marchán I., Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys. J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olson W.K., Gorin A.A., Zhurkin V.B. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc. Natl. Acad. Sci. USA. 1998;95:11163–11168. doi: 10.1073/pnas.95.19.11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lankas F., Sponer J., Cheatham T.E., 3rd DNA basepair step deformability inferred from molecular dynamics simulations. Biophys. J. 2003;85:2872–2883. doi: 10.1016/S0006-3495(03)74710-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morozov A.V., Fortney K., Siggia E.D. Using DNA mechanics to predict in vitro nucleosome positions and formation energies. Nucleic Acids Res. 2009;37:4707–4722. doi: 10.1093/nar/gkp475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Orozco M., Noy A., Pérez A. Recent advances in the study of nucleic acid flexibility by molecular dynamics. Curr. Opin. Struct. Biol. 2008;18:185–193. doi: 10.1016/j.sbi.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Pérez A., Lankas F., Orozco M. Towards a molecular dynamics consensus view of B-DNA flexibility. Nucleic Acids Res. 2008;36:2379–2394. doi: 10.1093/nar/gkn082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lankas F., Sponer J., Langowski J. Sequence-dependent elastic properties of DNA. J. Mol. Biol. 2000;299:695–709. doi: 10.1006/jmbi.2000.3781. [DOI] [PubMed] [Google Scholar]
- 35.Paillard G., Lavery R. Analyzing protein-DNA recognition mechanisms. Structure. 2004;12:113–122. doi: 10.1016/j.str.2003.11.022. [DOI] [PubMed] [Google Scholar]
- 36.Pérez A., Luque F.J., Orozco M. Frontiers in molecular dynamics simulations of DNA. Acc. Chem. Res. 2012;45:196–205. doi: 10.1021/ar2001217. [DOI] [PubMed] [Google Scholar]
- 37.Podtelezhnikov A.A., Mao C., Vologodskii A. Multimerization-cyclization of DNA fragments as a method of conformational analysis. Biophys. J. 2000;79:2692–2704. doi: 10.1016/S0006-3495(00)76507-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tolstorukov M.Y., Colasanti A.V., Zhurkin V.B. A novel roll-and-slide mechanism of DNA folding in chromatin: implications for nucleosome positioning. J. Mol. Biol. 2007;371:725–738. doi: 10.1016/j.jmb.2007.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balasubramanian S., Xu F., Olson W.K. DNA sequence-directed organization of chromatin: structure-based computational analysis of nucleosome-binding sequences. Biophys. J. 2009;96:2245–2260. doi: 10.1016/j.bpj.2008.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thåström A., Lowary P.T., Widom J. Sequence motifs and free energies of selected natural and non-natural nucleosome positioning DNA sequences. J. Mol. Biol. 1999;288:213–229. doi: 10.1006/jmbi.1999.2686. [DOI] [PubMed] [Google Scholar]
- 41.Deniz O., Flores O., Orozco M. Physical properties of naked DNA influence nucleosome positioning and correlate with transcription start and termination sites in yeast. BMC Genomics. 2011;12:489. doi: 10.1186/1471-2164-12-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson J.D., Widom J. Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol. 2000;296:979–987. doi: 10.1006/jmbi.2000.3531. [DOI] [PubMed] [Google Scholar]
- 43.Pérez A., Luque F.J., Orozco M. Dynamics of B-DNA on the microsecond time scale. J. Am. Chem. Soc. 2007;129:14739–14745. doi: 10.1021/ja0753546. [DOI] [PubMed] [Google Scholar]
- 44.Behe M., Felsenfeld G. Effects of methylation on a synthetic polynucleotide: the B—Z transition in poly(dG-m5dC).poly(dG-m5dC) Proc. Natl. Acad. Sci. USA. 1981;78:1619–1623. doi: 10.1073/pnas.78.3.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banyay M., Gräslund A. Structural effects of cytosine methylation on DNA sugar pucker studied by FTIR. J. Mol. Biol. 2002;324:667–676. doi: 10.1016/s0022-2836(02)01104-x. [DOI] [PubMed] [Google Scholar]
- 46.Schones D.E., Cui K., Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaplan N., Moore I.K., Segal E. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009;458:362–366. doi: 10.1038/nature07667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chung H.R., Vingron M. Sequence-dependent nucleosome positioning. J. Mol. Biol. 2009;386:1411–1422. doi: 10.1016/j.jmb.2008.11.049. [DOI] [PubMed] [Google Scholar]
- 49.Cui F., Zhurkin V.B. Structure-based analysis of DNA sequence patterns guiding nucleosome positioning in vitro. J. Biomol. Struct. Dyn. 2010;27:821–841. doi: 10.1080/073911010010524947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Travers A., Hiriart E., Di Mauro E. The DNA sequence-dependence of nucleosome positioning in vivo and in vitro. J. Biomol. Struct. Dyn. 2010;27:713–724. doi: 10.1080/073911010010524942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trifonov E.N. Nucleosome positioning by sequence, state of the art and apparent finale. J. Biomol. Struct. Dyn. 2010;27:741–746. doi: 10.1080/073911010010524944. [DOI] [PubMed] [Google Scholar]
- 52.Olson W.K., Zhurkin V.B. Working the kinks out of nucleosomal DNA. Curr. Opin. Struct. Biol. 2011;21:348–357. doi: 10.1016/j.sbi.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Field Y., Kaplan N., Segal E. Distinct modes of regulation by chromatin encoded through nucleosome positioning signals. PLOS Comput. Biol. 2008;4:e1000216. doi: 10.1371/journal.pcbi.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bai L., Morozov A.V. Gene regulation by nucleosome positioning. Trends Genet. 2010;26:476–483. doi: 10.1016/j.tig.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 55.Segal E., Fondufe-Mittendorf Y., Widom J. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee W., Tillo D., Nislow C. A high-resolution atlas of nucleosome occupancy in yeast. Nat. Genet. 2007;39:1235–1244. doi: 10.1038/ng2117. [DOI] [PubMed] [Google Scholar]
- 57.Tillo D., Hughes T.R. G+C content dominates intrinsic nucleosome occupancy. BMC Bioinformatics. 2009;10:442. doi: 10.1186/1471-2105-10-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goñi J.R., Fenollosa C., Orozco M. DNAlive: a tool for the physical analysis of DNA at the genomic scale. Bioinformatics. 2008;24:1731–1732. doi: 10.1093/bioinformatics/btn259. [DOI] [PubMed] [Google Scholar]
- 59.Goñi J.R., Pérez A., Orozco M. Determining promoter location based on DNA structure first-principles calculations. Genome Biol. 2007;8:R263. doi: 10.1186/gb-2007-8-12-r263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bertrand H., Ha-Duong T., Hartmann B. Flexibility of the B-DNA backbone: effects of local and neighbouring sequences on pyrimidine-purine steps. Nucleic Acids Res. 1998;26:1261–1267. doi: 10.1093/nar/26.5.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nathan D., Crothers D.M. Bending and flexibility of methylated and unmethylated EcoRI DNA. J. Mol. Biol. 2002;316:7–17. doi: 10.1006/jmbi.2001.5247. [DOI] [PubMed] [Google Scholar]
- 62.Mirsaidov U., Timp W., Timp G. Nanoelectromechanics of methylated DNA in a synthetic nanopore. Biophys. J. 2009;96:L32–L34. doi: 10.1016/j.bpj.2008.12.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bell A.C., Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- 64.Clark S.J., Harrison J., Molloy P.L. Sp1 binding is inhibited by (m)Cp(m)CpG methylation. Gene. 1997;195:67–71. doi: 10.1016/s0378-1119(97)00164-9. [DOI] [PubMed] [Google Scholar]
- 65.Hark A.T., Schoenherr C.J., Tilghman S.M. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- 66.Davey C.S., Pennings S., Allan J. A determining influence for CpG dinucleotides on nucleosome positioning in vitro. Nucleic Acids Res. 2004;32:4322–4331. doi: 10.1093/nar/gkh749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pennings S., Allan J., Davey C.S. DNA methylation, nucleosome formation and positioning. Brief. Funct. Genomics Proteomics. 2005;3:351–361. doi: 10.1093/bfgp/3.4.351. [DOI] [PubMed] [Google Scholar]
- 68.Felle M., Hoffmeister H., Längst G. Nucleosomes protect DNA from DNA methylation in vivo and in vitro. Nucleic Acids Res. 2011;39:6956–6969. doi: 10.1093/nar/gkr263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choy J.S., Wei S., Lee T.H. DNA methylation increases nucleosome compaction and rigidity. J. Am. Chem. Soc. 2010;132:1782–1783. doi: 10.1021/ja910264z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee J.Y., Lee T.H. Effects of DNA methylation on the structure of nucleosomes. J. Am. Chem. Soc. 2012;134:173–175. doi: 10.1021/ja210273w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee C.K., Shibata Y., Lieb J.D. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat. Genet. 2004;36:900–905. doi: 10.1038/ng1400. [DOI] [PubMed] [Google Scholar]
- 72.Field Y., Fondufe-Mittendorf Y., Segal E. Gene expression divergence in yeast is coupled to evolution of DNA-encoded nucleosome organization. Nat. Genet. 2009;41:438–445. doi: 10.1038/ng.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Choi J.K., Bae J.B., Kim Y.J. Nucleosome deposition and DNA methylation at coding region boundaries. Genome Biol. 2009;10:R89. doi: 10.1186/gb-2009-10-9-r89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Feng J., Dai X., He C. New insights into two distinct nucleosome distributions: comparison of cross-platform positioning datasets in the yeast genome. BMC Genomics. 2010;11:33. doi: 10.1186/1471-2164-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaplan N., Moore I., Segal E. Nucleosome sequence preferences influence in vivo nucleosome organization. Nat. Struct. Mol. Biol. 2010;17:918–920. doi: 10.1038/nsmb0810-918. author reply 920–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Valouev A., Ichikawa J., Johnson S.M. A high-resolution, nucleosome position map of C. elegans reveals a lack of universal sequence-dictated positioning. Genome Res. 2008;18:1051–1063. doi: 10.1101/gr.076463.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang X., Bryant G.O., Ptashne M. An effect of DNA sequence on nucleosome occupancy and removal. Nat. Struct. Mol. Biol. 2011;18:507–509. doi: 10.1038/nsmb.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Esteller M. Epigenetics provides a new generation of oncogenes and tumour-suppressor genes. Br. J. Cancer. 2006;94:179–183. doi: 10.1038/sj.bjc.6602918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chodavarapu R.K., Feng S., Pellegrini M. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466:388–392. doi: 10.1038/nature09147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.