

Abstract
Crystallographic structure and deuterium accessibility comparisons of CcO in different redox states have suggested conformational changes of mechanistic significance. To predict the intrinsic flexibility and low energy motions in CcO, this work has analyzed available high-resolution crystallographic structures with ProFlex and elNémo computational methods. The results identify flexible regions and potential conformational changes in CcO that correlate well with published structural and biochemical data and provide mechanistic insights. CcO is predicted to undergo rotational motions on the interior and exterior of the membrane, driven by transmembrane helical tilting and bending, coupled with rocking of the β-sheet domain. Consequently, the proton K-pathway becomes sufficiently flexible for internal water molecules to alternately occupy upper and lower parts of the pathway, associated with conserved Thr-359 and Lys-362 residues. The D-pathway helices are found to be relatively rigid, with a highly flexible entrance region involving the subunit I C-terminus, potentially regulating the uptake of protons. Constriction and dilation of hydrophobic channels in RsCcO suggest regulation of the oxygen supply to the binuclear center. This analysis points to coupled conformational changes in CcO and their potential to influence both proton and oxygen access.
Abbreviations used: Protein Data Bank, (PDB); cytochrome c oxidase, (CcO); Rhodobacter sphaeroides CcO, (RsCcO); Paracoccus denitrificans CcO, (PdCcO); Thermus thermophilus CcO, (TtCcO); Bos taurus CcO, (BtCcO)
Introduction
CcO, the fourth complex of the electron transport chain in mitochondria, is responsible for accepting electrons from cytochrome c and passing them through bimetallic copper A and heme a to the active site, composed of heme a3 and copper B (Fig. 1). Molecular oxygen is reduced at the active site, releasing energy for CcO to pump protons across the membrane. This proton pumping is responsible for generating an electrochemical gradient used for ATP synthesis (1,2). Deficiencies in CcO, as a result of mutations within CcO subunits or assembly proteins, can cause severe metabolic disease, especially in tissues with high energy requirements (3).
Figure 1.
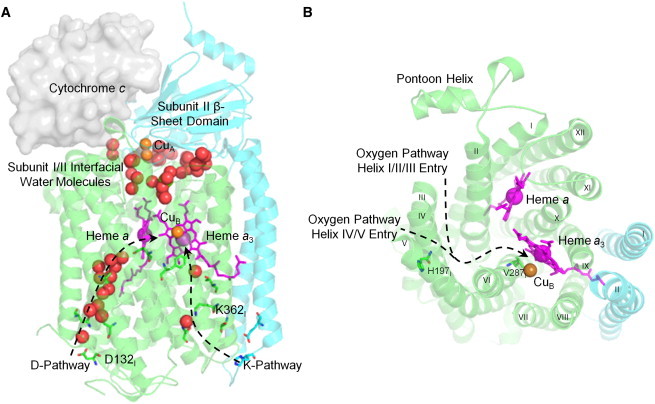
Architecture of the 2-subunit structure essential for RsCcO activity. Subunit I, in green ribbons, includes 12 transmembrane α-helices, heme, and metal cofactors, and the proton and oxygen pathways. Subunit II, in blue ribbons, is composed of two transmembrane α-helices, the β-sheet domain, and the copper A (CuA) site (PDB ID 2GSM (7)). (A) Electrons are sequentially passed from cytochrome c, positioned based on (54), through the bimetallic CuA and heme a to the active site heme a3 and copper B (CuB). The D-proton pathway contains a chain of well resolved water molecules shown in red spheres, whereas the K-pathway contains two bound water molecules. (B) A hydrophobic channel, shown as dashed lines, leads from the oxygen-rich membrane environment to the heme a3/CuB center. Helix numbers appear in Roman numerals.
Structural and mutational studies of RsCcO have defined two conserved proton pathways and a putative oxygen channel. These pathways allow protons to be taken up from the internal, negative side of the membrane and directed to the active site or pumped to the external, positive side. The K-pathway, named after conserved Lys-362, takes up protons for oxygen reduction. The D-pathway, named after conserved Asp-132, takes up the remaining protons consumed in oxygen reduction and all protons pumped across the membrane (4–9). (Note that helices and amino acid residues are located in subunit I unless otherwise indicated.) A bifurcated hydrophobic channel leading from the oxygen-rich membrane environment to the active site in bacterial and mammalian CcO has been defined based on the absence of water molecules in the interior channel and cocrystallization with high-pressure xenon gas (Fig. 1 B) (5,10,11).
There is little agreement on the detailed mechanism of coupling of the oxygen reduction and proton pumping functions of CcO. Hydrogen/deuterium exchange and x-ray crystal structures of CcO in distinct redox states have identified regions that undergo conformational change during the reaction cycle, including rotation of the heme a3 porphyrin ring and farnesyl tail and movement of helix VIII, which contains critical K-pathway residues (12). The redox state of RsCcO proteins during x-ray beam exposure has been monitored microspectrophotometrically. Results suggest that the initial protein conformation (whether oxidized or reduced) is apparently stabilized by crystal contacts and maintained despite reduction of the CcO metal centers during data collection (13). Structural and solvent accessibility changes support alternating proton pathway accessibility to the active site, with the K-pathway open during metal reduction only and the D-pathway open in all the other reaction intermediates (12–15).
However, crystallography provides structural snapshots constrained by intermolecular crystal contacts, rather than a continuum of conformations, whereas deuterium exchange mass spectrometry provides data for a limited subset of the protein fragments. Alternative experimental methods to study conformational change are not readily applicable to CcO due to its size and hydrophobicity. Computational methods have also been used to investigate the CcO mechanism, evaluating local motions on the nanosecond timescale. Molecular dynamics (MD), quantum mechanics/molecular mechanics, and electrostatic analyses have predicted continuous hydrogen bonding chains within the K-pathway, identified potential proton exit pathways, and evaluated the effects of hydration and redox state on the protonation of critical residues (16–23).
In this study, main-chain conformational change, coupling between mobile regions, and the functional implications of such motions are studied by computational analysis of CcO flexibility and low-energy vibrational motions using ProFlex and elNémo. The focus is on aspects of CcO conformation change shared by related structures, for instance, CcO structures in different redox states, resolved by different research groups, and derived from related organisms. ProFlex has successfully predicted flexibility and protein coupling for ligand-bound and ligand-free protein structures (24,25), whereas elNémo normal modes analysis has described the main-chain motions of a number of integral membrane proteins (26–29). The conformational changes predicted in CcO, and their influence on function, are considered in the context of available data.
Materials and Methods
Identification of flexible regions using ProFlex
In ProFlex (24), the input protein structure is represented as a three-dimensional molecular framework with atoms represented as vertices and bonds as edges. Graph-theoretic analysis, based on constraint counting from structural engineering (30), decomposes a protein structure into rigid, isostatic, and flexible regions based on the number of internal degrees of freedom, offset by the number of covalent and noncovalent bonds and bond-coordination angle constraints (24). Protein regions linked by more bonds than needed to form a stable structure are considered rigid. Regions with the same number of bond constraints and bond-rotational degrees of freedom are defined as isostatic (barely rigid), whereas regions with fewer constraints than bond-rotational degrees of freedom are identified as flexible. Details of how ProFlex calculates an energy-dependent hydrogen-bond dilution profile (25) are presented in the Supporting Material, including Fig. S1.
For ProFlex, the three-dimensional atomic coordinates corresponding to one biological unit were selected from the highest resolution bacterial structures (Table S1): 2- and 4-subunit RsCcO, 2-subunit PdCcO, and the 3-subunit TtCcO structure, which is structurally equivalent to the 2-subunit RsCcO. Oxidized and reduced 2-subunit RsCcO structures were produced by the same research group, with only the addition of dithionite for protein reduction (Table S2). ProFlex analysis predicts intrinsic protein flexibility and has shown little sensitivity to small changes in analyzed structures (24), including the redox state. This is desirable for the current analysis, which focuses on global conformational change and coupling. Analyzed structures of CcO include two heme groups, three copper ions, and calcium and magnesium, where appropriate. Bound water molecules that were buried or interfacial between subunits were included in the ProFlex analysis, as recommended, based on identification by PRO_ACT (34) and Consolv (35). Protons and partial charges were assigned for salt bridge identification using WhatIf (version 6 (36)) for protein and water atoms and InsightII with CVFF parameters for heme groups (Accelrys, San Diego, CA).
Identification of low energy motions by normal mode analysis using ElNémo
Collective protein motions have been studied since the early 1980s using normal mode analysis (37). This approach decouples protein motions about an initial, low energy conformation into oscillating orthogonal modes that range from low-frequency, global motions (low numbered modes) to high-frequency, localized motions (high numbered modes). ElNémo normal mode analysis (38) (http://www.igs.cnrs-mrs.fr/elnemo/) was applied to sample the low-frequency global motions in CcO. ElNémo places a harmonic potential, or spring, between each pair of nonhydrogen atoms within 8 Å, and then solves for the frequency and direction of maximal motion in each mode. CcO, with over 6,300 protein and cofactor atoms, requires a rotational-translational-block representation. Two consecutive amino acid residues form a block, and blocks are then coupled by springs. This representation has been shown to have little effect on the resulting low-frequency motions (39). High-resolution bacterial CcO structures were analyzed by elNémo (Table S1). For proper interpretation by elNémo, the heme, copper, and magnesium PDB coordinates were renamed as ATOM records, and heme groups were assigned unique chain identifiers. Bound water molecules were not included, as they do not affect the accuracy of results (40,41).
The modes output by elNémo include six trivial zero frequency modes, 1–6, corresponding to rigid-body protein rotation and translation. This work is focused on the internal motions, starting with mode 7. The displacement in the direction of maximal motion was identified by elNémo, and 11 structural snapshots (centered on the input crystal structure) were output to represent the dynamics trajectory for each mode. The normalized mean-squared displacement value for each Cα, 〈R2〉, was graphed as a function of residue number (Fig. S2). Significantly mobile and immobile regions were defined as three or more consecutive Cαs with 〈R2〉 values at least two standard deviations above or below the mean for all residues, respectively.
Evolutionary conservation
RsCcO amino acid conservation was measured and mapped onto the structure using ConSurf (42) (http://consurf.tau.ac.il). Multiple sequence alignments were constructed by searching the SwissProt database (43) for all protein sequences with 30–95% sequence identity to RsCcO subunits I and II, using five iterations of PSI-BLAST with an E-value cutoff of 0.001. The resulting sequences (254 subunit I and 148 subunit II sequences) were aligned using CLUSTALW (44) (http://www.clustal.org). The ConSurf neighbor-joining method was used to construct a phylogenetic tree of the sequences such that only one representative of each clade was included, to keep clusters of similar sequences from dominating the conservation scores.
Cavity identification
RsCcO internal protein cavities were identified by HOLLOW (45), which places probe atoms in the protein interior on a 0.5 Å grid at points not occupied by protein atoms. A molecular surface tangent to the probe atom van der Waals spheres was used to visualize the cavities. PyMOL (http://www.pymol.org; Schrödinger) was used to render all structural figures.
Results and Discussion
Global motions in CcO
Unlike computational methods previously applied to CcO, ProFlex and elNémo probe regions of global flexibility, intraprotein coupling, and main-chain motions; the elNémo analysis simulates low energy main-chain protein motions, whereas ProFlex identifies underconstrained (coupled flexible) regions and rigid regions connected by hinges that allow motion relative to other parts of the protein. Two-subunit RsCcO structures were selected for analysis, as these structures are of higher isotropic resolution. The loss of two accessory subunits had little effect on the flexibility of the CcOs catalytic core (Fig. S3).
The degree of flexibility and coupling within CcO is highlighted by ProFlex analysis (Fig. 2) of oxidized and reduced RsCcO (PDB entries 2GSM (7) and 3FYE (12)) and the closest homolog PdCcO (PDB entry 1AR1 (46)). At the initial hydrogen-bond energy (see Methods), all three structures have a stable core consisting of helices I–VI, X–XII, and the subunit II β-sheet domain. The middle of helices VII–IX and helix II of subunit II, which surround the K-pathway, are flexible (Fig. 2), as are the exterior surface where cytochrome c interacts and the C-terminus of subunit I. At a higher but still moderate energy with a less-dense hydrogen-bond network (hydrogen bonds at least −4.2 kcal/mol in strength for RsCcO), all helices except those packed against the heme groups (II, VI, and X) attained significant flexibility while remaining coupled (Fig. S4). At significantly higher energy (H-bonds stronger than −7.6 kcal/mol in RsCcO; Fig. S4), RsCcO became a coupled flexible structure surrounding the heme groups. These results indicate that most regions of CcO, except for the helices surrounding the K-pathway (VII and VIII) and residues coordinating heme groups, behave as a coupled structure that is either mutually flexible or rigid, depending on the energy.
Figure 2.
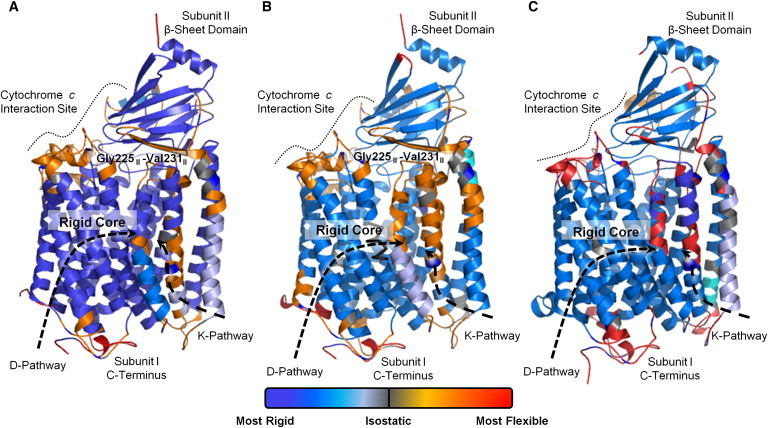
ProFlex prediction of main-chain flexibility and stability in RsCcO. (A) Oxidized RsCcO (7), (B) reduced RsCcO (12), and (C) PdCcO (46) were analyzed. Deep blue indicates regions of greatest stability, with a gradient to light blue (somewhat rigid) to gray (isostatic) to orange (flexible) to red (most flexible). The stable proteins core consists of transmembrane helices I–VI, including the D-pathway and helices X–XII. Most of the subunit II β-sheet domain is also internally rigid. The greatest intramembrane flexibility occurs within helices VII–VIII and subunit II helix II surrounding the K-pathway, whereas the cytochrome c interaction site and subunit I C-terminus exhibit the highest overall flexibility.
ElNémo was used to predict low energy motions about the equilibrium conformation of CcO (Table S1). The three lowest frequency protein motions, modes 7–9, were selected as representing global motions because at least 30% of the residues experienced significant displacement (PDB entry 2GSM (7)). Mode 7 exhibited counter-rotational twisting on the interior and exterior of the membrane (Fig. 3 A; Movie S1) involving helical tilting and bending. The protein located near the inside of the membrane rotated about an axis perpendicular to the membrane plane, whereas on the outside of the membrane the protein rotated about an axis tilted by ∼30°, parallel to the subunit II β-strands. Mode 8 involved counter-rotational motions of CcO on each side of the membrane about an axis perpendicular to the membrane plane (Fig. 3 B; Movie S2). Mode 9 showed a C-clamp-like motion of subunits I and II, with the pontoon helix approaching the interior C-terminus of subunit I near helices I, III, and XII, and then receding (Fig. 3 C; Movie S3). Mechanical twisting motions of integral membrane proteins on either side of the membrane previously have been described in potassium, mechanosensitive, and nicotinic acetylcholine receptor proteins (26–29,48).
Figure 3.
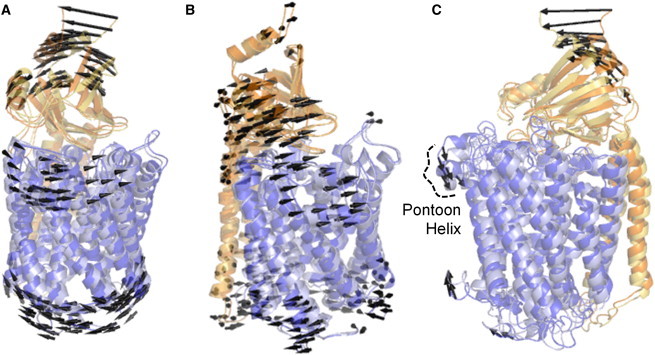
ElNémo simulation of low-frequency motions in RsCcO. Low-energy modes of internal RsCcO motion (PDB entry 2GSM (7)), are shown by superposition of the two extreme conformations in each mode, with subunit I in blue and subunit II in orange. Arrows indicate motion of Cαs undergoing a displacement of at least 4 Å. (A) Counterrotational motions on the membrane interior relative to the exterior were seen in mode 7. On the inside of the membrane, RsCcO helices rotated about an axis perpendicular to the membrane, whereas on the outside, the rotation axis was 30° from the membrane plane, parallel to the β-strands. (B) Mode 8 involved counterrotational motion about an axis perpendicular to the membrane. (C) Mode 9 showed clamp-like compression and expansion, drawing the protein on the interior and exterior of the membrane towards the midpoint of helices I, III, and XII. All three modes showed major rocking of the β-sheet domain while maintaining the subunit I-II interface. Results on PdCcO (PDB entry 1AR1 (46)) and TtCcO (PDB entry 1XME (47)) were substantially similar.
The internal flexibility and low energy motions of CcO were compared to other integral membrane proteins. The β2 adrenergic receptor, KcsA potassium channel, and voltage-dependent anion channel were selected as representing diverse membrane proteins with similar structural resolution to the analyzed CcO proteins and experimental characterization of conformational change (Table S3). ElNémo results indicate that low energy motions of CcO have a similar immobile protein core, mobile secondary structural ends, and a similar percentage of displaced residues relative to the receptor and channel proteins, though β2 adrenergic receptor shows greater mobility at the lowest energy (Table S4; Fig. S5). ProFlex results suggest that CcO and the β2 adrenergic receptor are more rigid than the transporter/channel proteins (Fig. S6), perhaps due to tighter packing within their helical bundles relative to the pore-forming membrane proteins.
Coupled local motions
Local motions underlying the global conformational changes include an interdomain hinge between subunits I and II and coupled helical deformation. ProFlex results for the oxidized and reduced RsCcO structures (Fig. 2) indicate the greatest transmembrane flexibility is located in the central regions of helices VII–VIII. Additionally, the secondary structures at the interface of subunit I and the subunit II β-sheet domain are flexibly coupled. This region includes the helix VII–VIII loop, the ends of helix IX and subunit II helices I–II, and the interfacial subunit II β-strand (residues Gly-225-Val-231) (Fig. 1). This flexibly coupled region acts like an interdomain hinge, which allows the coordinated counter rotational movements of subunit I together with subunit II β-sheet domain rocking as predicted by elNémo (Fig. 3 and Movies S1–S3). In elNémo mode 7, helices I, IX, and X maintain an interface with the β-sheet domain during conformational change (Table S5 and Movie S1). In modes 8 and 9, subunit II helices I–II bend and straighten significantly in concert with the β-sheet domain rocking (Table S5 and Movies S2 and S3). Because the cytochrome c binding site involves one face of this domain (Fig. 1), these motions could couple with cytochrome c binding or affect its affinity for the site. Interdomain hinging while maintaining an interface has been observed previously in normal mode analysis of nicotinic receptors (see (48)).
Within the lipid bilayer, a relatively stationary set of RsCcO residues is found near the midplane in elNémo modes 7 and 8 (Fig. S7), whereas in mode 9, a second set of stationary residues is found near the inside face of the membrane. This is similar to mechanosensitive channels, in which a pivot plane runs through the plane of the membrane and key channel gating residues (26). One potential consequence of the tilting and internal conformational change of the RsCcO helices about these pivots is a change in distance across the membrane, as observed for mechanosensitive channels hypothesized to use a twist-to-open function (26).
The subunit II β-sheet domain is identified as a single, internally rigid domain by the ProFlex method, and low energy motions of this region support that its rocking behavior is essentially rigid-body, based on the ∼1 Å main-chain root mean-square deviation following least squares superposition of this domain between the two extreme conformations in each mode. Experimental analysis of movement for the subunit II β-sheet domain is limited. Crystal structure comparisons provide little information, because this domain is involved in strong crystal contacts in most RsCcO structures (Fig. 4 C) (7,12). However, deuterium exchange analysis supports the flexibility of the subunit I/β-sheet domain interface, with the subunit II Gly-225-Val-231 β-strand found to be differentially solvent accessible (14). Two-dimensional surface-enhanced infrared absorption spectroscopy experiments on membrane bound RsCcO indicate a possible change in association of α-helices and β-strands upon protein activation (49). To better understand the mechanistic implications of the movements that Proflex and elNémo predict, these global motions are considered in the context of the functional regions of CcO.
Figure 4.
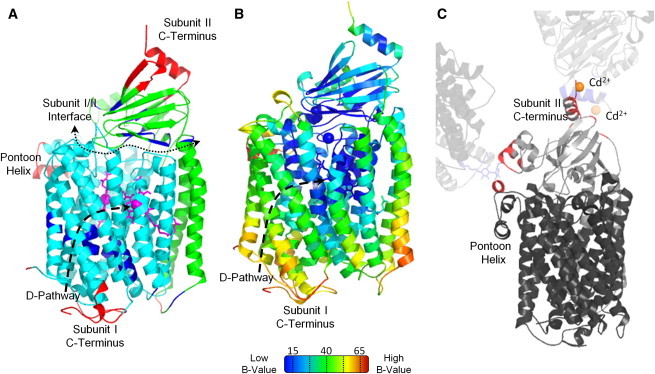
Comparison of RsCcO normal modes and crystallographic temperature factors. (A) Segments undergoing the most (red) or least (dark blue) displacement in modes 7–9, based on having three consecutive Cαs with a displacement, 〈R2〉, two standard deviations above or below the mean value from all three modes. The least mobile regions are in transmembrane helices surrounding the D-pathway and at the subunits' interface, whereas the most mobile regions are the pontoon helix and the C-termini of each subunit. (B) Crystallographic temperature factors (B-values) of reduced RsCcO (PDB entry 3FYE (12)), measuring fluctuation in atomic position via spread in electron density. Blue indicates the least mobile residues and red the most mobile ones. The core of subunit I, including the interior of the D-pathway, has the lowest B-values, whereas the pontoon helix, subunit I C-terminus, and inside of subunit II helix II have the highest. (C) Crystal contacts of subunit II with neighboring molecules. Based on the elNémo analysis, the β-sheet domain would be expected to have high B-values, especially for the C-terminal helix of subunit II. However, the β-sheet domain is stabilized by crystal contacts (shown in red) with neighboring molecules (7,12,13). One molecule of RsCcO is represented in dark gray (subunit I) and medium gray (subunit II). Neighboring molecules appear in light gray.
Protein flexibility along the K-pathway suggests a proton conducting mechanism involving water molecule movement
The CcO K-pathway is responsible for supplying protons to the active site during metal center reduction where they are consumed to make water. Mutagenesis and x-ray crystallographic studies have identified functionally important residues and two stable water molecule binding sites along the K-pathway (5,7,12,13,50). These water molecule sites (HOH6516 and HOH6533 in PDB entry 2GSM (7)), are located between conserved Lys-362 and Ser-299, and Tyr-288 and Thr-359, respectively (Fig. 1). A continuous hydrogen-bond network for proton transfer from the K-pathway entry to the active site is not apparent in most crystal structures, though it is predicted by MD calculations, which show concurrent motion of Thr-359 (18).
According to ProFlex, the most flexible intramembrane region in oxidized and reduced RsCcO and in PdCcO involves the helices composing the K-pathway, VII–VIII and subunit II helix II, which could move independently of the rigid core of subunit I (Fig. 2). Interestingly, in the same region, the low energy motions in elNémo mode 8 suggest the repositioning of water molecules along the K-pathway. These motions alternately produce and occlude two small cavities (Fig. 5), which correspond to the well-resolved K-pathway water molecule positions, even though this method does not consider water molecules in the input structure.
Figure 5.
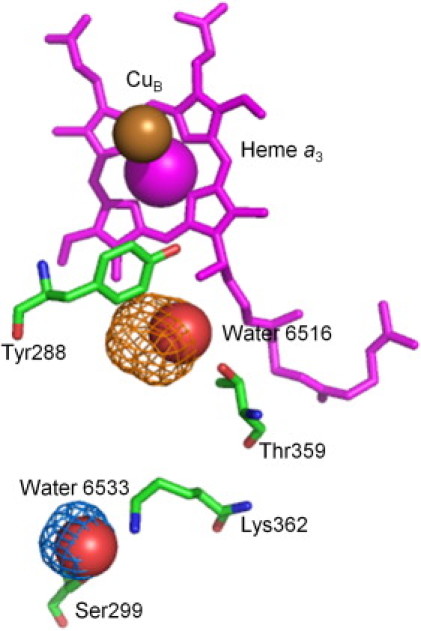
Water molecule movement in the K-pathway. K-pathway residues, in green tubes, and associated water molecules, in red spheres, lead to the active site heme a3, in magenta tubes and CuB depicted as a gold sphere (PDB entry 2GSM (7)). Two molecular surfaces are shown in gold and blue mesh for the two extreme conformations of elNémo mode 8. The blue cavity (coincident with crystallographic Wat-6516) appears in one extreme conformation from elNémo, whereas the gold cavity (coincident with Wat-6533) appears in the other, suggesting that conformational change is associated with water molecule repositioning.
ProFlex and elNémo analyses of intrinsic protein flexibility and low-energy protein motions are consistent with the crystallographic and deuterium accessibility data involving the K-pathway. Specifically, crystallographic analysis shows movement of heme a3 in the reduced form, affecting the conformation of the central region of helix VIII and allowing new water molecules to be resolved in the active site entrance (12). Deuterium accessibility studies also support conformational change in helix VIII upon reduction, as residues Val-354-Trp-366 rapidly incorporate deuterium during metal reduction (14).
The D-pathway remains rigid but may be regulated by the flexible subunit I C-terminus
The D-pathway of CcO conducts protons from the inside of the membrane to both the active site for oxygen reduction chemistry and the exterior for pumping. In contrast to the K-pathway, the D-pathway contains a conserved hydrogen-bonded chain of water molecules (5,7,12,13). Both ProFlex and elNémo analyses indicate relative rigidity along helices II–V composing the D-pathway (Figs. 2 and 3). Only energies at which the enzyme would partly unfold did these regions become flexible or flexibly linked to the rest of subunit I in ProFlex (Fig. S4). Regions surrounding the D-pathway were substantially immobile in elNémo normal modes, based on at least three consecutive residues having displacements at least two standard deviations below the average for all residues (Fig. 4 A). Rigidity of the D-pathway residues and associated water molecules is supported by low crystallographic temperature factor (B) values (Fig. 4 B) (7,12), absence of structural changes between the oxidized and reduced structures (12,13), and lack of deuterium exchange in D-pathway peptide fragments (14).
In contrast to the intramembrane portion of the D-pathway, the solvent-exposed subunit I C-terminus was determined to be highly flexible, based on bond-rotational degrees of freedom identified by ProFlex (Fig. 2) and displacement of C-terminal residues Thr-520–Thr-550 by at least two standard deviations above the average residue in elNémo normal modes (Fig. 4 A). Crystallographic B-values also identify this as one of the most mobile regions of RsCcO (Fig. 4 B), with residues beyond Thr-550 not usually resolved (7,12,13). Although C-terminal mobility is anticipated due to its irregular structure, this region could be essential for regulating the D-pathway. In reaction intermediates with the D-pathway hypothesized to be fully functional (oxidized, peroxy, and ferryl), the peptide fragment from this region incorporates ∼80% deuterium, whereas incorporation is reduced to ∼50% in intermediates with an inactive D-pathway (14). This suggests that C-terminus flexibility is related to D-pathway activity, possibly by recruiting protons via its acidic residues (Glu-552, Asp-558, Asp-559, and Glu-561).
Oxygen supply to the CcO active site may be gated
In RsCcO, a branching hydrophobic channel leading from the membrane environment to the protein's active site has been identified based on solvent accessible surfaces within crystal structures and cocrystallization with xenon gas, which has similar hydrophobicity and size as oxygen (5,10,11). This bifurcated channel has two entries (5), one between helices I, II, and III and another between helices IV and V, with the ceiling formed by the helix III–IV loop (residues Pro-160-Tyr-175) (Fig. 1 B). The channel has been hypothesized to be responsible for oxygen supply to the active site but remains a topic of debate. MD of oxygen diffusion from the helix IV–V entry to the hydrophobic channel and active site suggest that this branch of the channel may supply oxygen for enzyme turnover (10). Furthermore, mutagenesis of hydrophobic residues lining the channel indicated that channel occlusion results in reduced oxygen binding to the active site (51).
Motions of RsCcO sampled by elNémo in low-energy modes 7–9 resulted in alternation between open and constricted states in the oxygen pathway (PDB entry 2GSM (7)) (Fig. 6). Gating occurs in both branches of the channel between the entrance and their junction (Fig. 6, B and C). The helix I-II-III entrance is also periodically closed by the motions of helix III coupled to large amplitude motions of the pontoon helix over the entrance (Fig. 3 C). The importance of this oxygen pathway and its constriction points can be inferred from the conserved residues lining and constricting the channel (Fig. 6, B and C). Additionally, the hydrophobic channel of TtCcO, which naturally functions under significantly reduced oxygen tension, was analyzed with elNémo. TtCcO contains substantially longer hydrophobic channels with more uniform diameter. These channels did not experience constriction, but instead remained continuously dilated.
Figure 6.
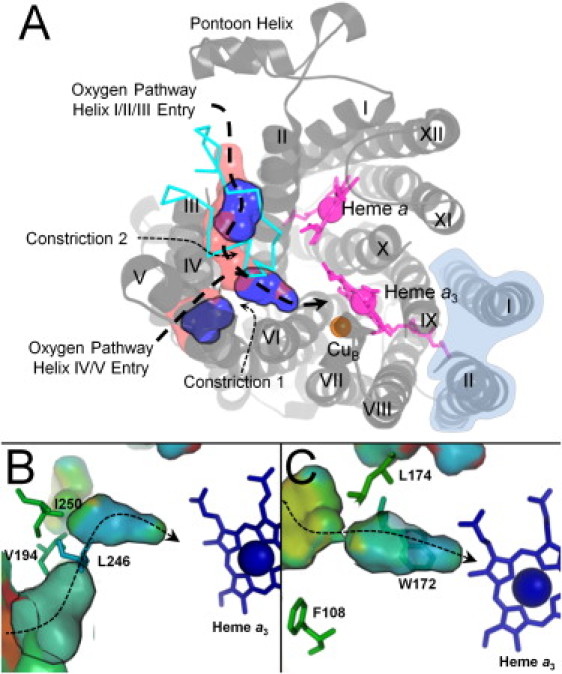
Conformational gating in the RsCcO oxygen channels. (A) Converging, hydrophobic channels for oxygen, defined by thick dashed lines, are formed by residues in helices I–VI, in gray ribbons, and the external loop joining helices III and IV, in cyan. Motions sampled in all three normal modes result in alternating constriction, shown as discontinuous blue cavities, and dilation, shown as a virtually continuous Y-shaped channel in pink. Constriction occurs at two points. Subunit 2 is shaded in light blue. (B) In the channel starting near helices IV–V, ConSurf indicates moderate conservation, colored in green surface and side-chain tubes, of channel-forming residues and increasingly high conservation, colored in blue, between the constriction (Val-194, Leu-246, and Ile-250) and the active site. (C) The channel starting at helices I–III is also increasingly conserved between its constriction (Phe-108, Trp-172, and Leu-174) and the active site.
Previous comparison of BtCcO, RsCcO, PdCcO, and TtCcO structures defined hydrophobic channels in the same region but with differing lengths and diameters (11). BtCcO, RsCcO, and PdCcO channels were shorter than the long, continuous channel of TtCcO. On the other hand, RsCcO channels are of considerably larger volume while being lined with large hydrophobic residues that produce constriction points along the channel (11). Based on crystallographic comparisons in the presence and absence of xenon gas (11) and the low energy motion simulations of CcO presented here, we propose that variation in the pathway diameter of RsCcO may regulate oxygen occupancy next to the active site, whereas the larger, unconstricted channel in TtCcO enhances oxygen flow to the active site.
In addition to providing an oxygen channel ceiling in RsCcO, the helix III–IV loop has also been suggested to be involved in proton pumping. Mutations in this loop (Gly-171-Asp, Trp-172-Phe, Trp-172-Gln, and Tyr-175-Ala) show reduced proton pumping and increased proton backflow (52,53). MD simulations suggest this loop can move deeper into the membrane, allowing transient water molecule chains to form between the top of the D-pathway and the hypothesized proton exit regions including the Mg2+ site (53). ProFlex analysis supports the flexibility of this loop (RsCcO Gly-161-Trp-172; Fig. 2, A and B), and is in agreement with the changes of solvent accessibility between reaction intermediates (Gly-169-Tyr-175) (14). Flexibility, conservation, mutational analysis, and differential solvent accessibility in reaction intermediates all support a role of the helix III–IV loop in CcO pumping.
Conclusions
Normal modes and flexibility analysis indicate that the core of CcO undergoes global counterrotational and compressional motions on the interior and exterior of the membrane, coupled with rocking of the β-sheet domain. These motions are a consequence of helix tilting, bending, and compression, potentially regulating oxygen flux to the active site while the D-pathway integrity is maintained. The subunit I C-terminus is predicted to be the most mobile region in the protein, suggesting conformational regulation of proton uptake at the D-pathway entrance. The K-pathway helices support water molecule movement in the pathway and are the most flexible intramembrane region, consistent with conformational changes in crystal structures. This work predicts global motions and underlying internal flexibility of CcO that is consistent with experimental observations, while providing a different perspective on K-pathway proton conductance, regulation of proton uptake in the D-pathway, and gating of oxygen flux to the active site.
Acknowledgments
The authors thank Drs. Yves-Henri Sanejouand and Karsten Suhre for their helpful discussions and suggestions involving elNémo, Dr. Denise Mills for discussions on cytochrome c oxidase, and Dr. Sean Law for the script (http://www.pymolwiki.org/index.php/Modevectors) used to create Fig. 3.
This work was supported by National Institutes of Health GM26916 (to S.F.-M.) and Michigan State University College of Natural Science and Quantitative Biology Distinguished Graduate Student Fellowships (to L.B.).
Supporting Material
References
- 1.Hosler J.P., Ferguson-Miller S., Mills D.A. Energy transduction: proton transfer through the respiratory complexes. Annu. Rev. Biochem. 2006;75:165–187. doi: 10.1146/annurev.biochem.75.062003.101730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brzezinski P., Gennis R.B. Cytochrome c oxidase: exciting progress and remaining mysteries. J. Bioenerg. Biomembr. 2008;40:521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diaz F. Cytochrome c oxidase deficiency: patients and animal models. Biochim. Biophys. Acta. 2010;1802:100–110. doi: 10.1016/j.bbadis.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 4.Fetter J.R., Qian J., Ferguson-Miller S. Possible proton relay pathways in cytochrome c oxidase. Proc. Natl. Acad. Sci. USA. 1995;92:1604–1608. doi: 10.1073/pnas.92.5.1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Svensson-Ek M., Abramson J., Iwata S. The x-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J. Mol. Biol. 2002;321:329–339. doi: 10.1016/s0022-2836(02)00619-8. [DOI] [PubMed] [Google Scholar]
- 6.Brändén M., Tomson F., Brzezinski P. The entry point of the K-proton-transfer pathway in cytochrome c oxidase. Biochemistry. 2002;41:10794–10798. doi: 10.1021/bi026093+. [DOI] [PubMed] [Google Scholar]
- 7.Qin L., Hiser C., Ferguson-Miller S. Identification of conserved lipid/detergent-binding sites in a high-resolution structure of the membrane protein cytochrome c oxidase. Proc. Natl. Acad. Sci. USA. 2006;103:16117–16122. doi: 10.1073/pnas.0606149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lepp H., Salomonsson L., Brzezinski P. Impaired proton pumping in cytochrome c oxidase upon structural alteration of the D pathway. Biochim. Biophys. Acta. 2008;1777:897–903. doi: 10.1016/j.bbabio.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu J., Han H., Gennis R.B. Decoupling mutations in the D-channel of the aa(3)-type cytochrome c oxidase from Rhodobacter sphaeroides suggest that a continuous hydrogen-bonded chain of waters is essential for proton pumping. Biochemistry. 2010;49:4476–4482. doi: 10.1021/bi100344x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hofacker I., Schulten K. Oxygen and proton pathways in cytochrome c oxidase. Proteins. 1998;30:100–107. [PubMed] [Google Scholar]
- 11.Luna V.M., Chen Y., Stout C.D. Crystallographic studies of Xe and Kr binding within the large internal cavity of cytochrome ba3 from Thermus thermophilus: structural analysis and role of oxygen transport channels in the heme-Cu oxidases. Biochemistry. 2008;47:4657–4665. doi: 10.1021/bi800045y. [DOI] [PubMed] [Google Scholar]
- 12.Qin L., Liu J., Ferguson-Miller S. Redox-dependent conformational changes in cytochrome c oxidase suggest a gating mechanism for proton uptake. Biochemistry. 2009;48:5121–5130. doi: 10.1021/bi9001387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu J., Qin L., Ferguson-Miller S. Crystallographic and online spectral evidence for role of conformational change and conserved water in cytochrome oxidase proton pump. Proc. Natl. Acad. Sci. USA. 2011;108:1284–1289. doi: 10.1073/pnas.1012846108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Busenlehner L.S., Salomonsson L., Armstrong R.N. Mapping protein dynamics in catalytic intermediates of the redox-driven proton pump cytochrome c oxidase. Proc. Natl. Acad. Sci. USA. 2006;103:15398–15403. doi: 10.1073/pnas.0601451103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharpe M.A., Ferguson-Miller S. A chemically explicit model for the mechanism of proton pumping in heme-copper oxidases. J. Bioenerg. Biomembr. 2008;40:541–549. doi: 10.1007/s10863-008-9182-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pomès R., Hummer G., Wikström M. Structure and dynamics of a proton shuttle in cytochrome c oxidase. Biochim. Biophys. Acta. 1998;1365:255–260. [Google Scholar]
- 17.Wikström M., Verkhovsky M.I., Hummer G. Water-gated mechanism of proton translocation by cytochrome c oxidase. Biochim. Biophys. Acta. 2003;1604:61–65. doi: 10.1016/s0005-2728(03)00041-0. [DOI] [PubMed] [Google Scholar]
- 18.Cukier R.I. A molecular dynamics study of water chain formation in the proton-conducting K channel of cytochrome c oxidase. Biochim. Biophys. Acta. 2005;1706:134–146. doi: 10.1016/j.bbabio.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 19.Song Y., Michonova-Alexova E., Gunner M.R. Calculated proton uptake on anaerobic reduction of cytochrome C oxidase: is the reaction electroneutral? Biochemistry. 2006;45:7959–7975. doi: 10.1021/bi052183d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henry R.M., Yu C.-H., Pomès R. Functional hydration and conformational gating of proton uptake in cytochrome c oxidase. J. Mol. Biol. 2009;387:1165–1185. doi: 10.1016/j.jmb.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 21.Sugitani R., Stuchebrukhov A.A. Molecular dynamics simulation of water in cytochrome c oxidase reveals two water exit pathways and the mechanism of transport. Biochim. Biophys. Acta. 2009;1787:1140–1150. doi: 10.1016/j.bbabio.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh N., Prat-Resina X., Cui Q. Microscopic pKa analysis of Glu-286 in cytochrome c oxidase (Rhodobacter sphaeroides): toward a calibrated molecular model. Biochemistry. 2009;48:2468–2485. doi: 10.1021/bi8021284. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J., Gunner M.R. Multiconformation continuum electrostatics analysis of the effects of a buried Asp introduced near heme a in Rhodobacter sphaeroides cytochrome c oxidase. Biochemistry. 2010;49:8043–8052. doi: 10.1021/bi100663u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobs D.J., Rader A.J., Thorpe M.F. Protein flexibility predictions using graph theory. Proteins. 2001;44:150–165. doi: 10.1002/prot.1081. [DOI] [PubMed] [Google Scholar]
- 25.Hespenheide B.M., Rader A.J., Kuhn L.A. Identifying protein folding cores from the evolution of flexible regions during unfolding. J. Mol. Graph. Model. 2002;21:195–207. doi: 10.1016/s1093-3263(02)00146-8. [DOI] [PubMed] [Google Scholar]
- 26.Valadié H., Lacapcre J.J., Etchebest C. Dynamical properties of the MscL of Escherichia coli: a normal mode analysis. J. Mol. Biol. 2003;332:657–674. doi: 10.1016/s0022-2836(03)00851-9. [DOI] [PubMed] [Google Scholar]
- 27.Shrivastava I.H., Bahar I. Common mechanism of pore opening shared by five different potassium channels. Biophys. J. 2006;90:3929–3940. doi: 10.1529/biophysj.105.080093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miloshevsky G.V., Jordan P.C. The open state gating mechanism of gramicidin a requires relative opposed monomer rotation and simultaneous lateral displacement. Structure. 2006;14:1241–1249. doi: 10.1016/j.str.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 29.Szarecka A., Xu Y., Tang P. Dynamics of heteropentameric nicotinic acetylcholine receptor: implications of the gating mechanism. Proteins. 2007;68:948–960. doi: 10.1002/prot.21462. [DOI] [PubMed] [Google Scholar]
- 30.Maxwell J. On the calculation of the equilibrium and stiffness of frames. Philos. Mag. 1864;27:294–299. [Google Scholar]
- 31.Reference deleted in proof.
- 32.Reference deleted in proof.
- 33.Reference deleted in proof.
- 34.Williams M.A., Goodfellow J.M., Thornton J.M. Buried waters and internal cavities in monomeric proteins. Protein Sci. 1994;3:1224–1235. doi: 10.1002/pro.5560030808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raymer M.L., Sanschagrin P.C., Kuhn L.A. Predicting conserved water-mediated and polar ligand interactions in proteins using a K-nearest-neighbors genetic algorithm. J. Mol. Biol. 1997;265:445–464. doi: 10.1006/jmbi.1996.0746. [DOI] [PubMed] [Google Scholar]
- 36.Vriend G. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-v. 29. [DOI] [PubMed] [Google Scholar]
- 37.Brooks B., Karplus M. Harmonic dynamics of proteins: normal modes and fluctuations in bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. USA. 1983;80:6571–6575. doi: 10.1073/pnas.80.21.6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suhre K., Sanejouand Y.-H. ElNémo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32(Suppl. 2) doi: 10.1093/nar/gkh368. W610-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tama F., Gadea F.X., Sanejouand Y.H. Building-block approach for determining low-frequency normal modes of macromolecules. Proteins. 2000;41:1–7. doi: 10.1002/1097-0134(20001001)41:1<1::aid-prot10>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 40.Roux B., Karplus M. The normal modes of the gramicidin-A dimer channel. Biophys. J. 1988;53:297–309. doi: 10.1016/S0006-3495(88)83107-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Essiz S.G., Coalson R.D. Langevin dynamics of molecules with internal rigid fragments in the harmonic regime. J. Chem. Phys. 2007;127:104109. doi: 10.1063/1.2756044. [DOI] [PubMed] [Google Scholar]
- 42.Landau M., Mayrose I., Ben-Tal N. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33(Suppl. 2) doi: 10.1093/nar/gki370. W299-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bairoch A., Boeckmann B. The SWISS-PROT protein sequence data bank. Nucleic Acids Res. 1991;19(Suppl):2247–2249. doi: 10.1093/nar/19.suppl.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignments. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ho B.K., Gruswitz F. HOLLOW: generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct. Biol. 2008;8:49. doi: 10.1186/1472-6807-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ostermeier C., Harrenga A., Michel H. Structure at 2.7 A resolution of the Paracoccus denitrificans two-subunit cytochrome c oxidase complexed with an antibody FV fragment. Proc. Natl. Acad. Sci. USA. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hunsicker-Wang L.M., Pacoma R.L., Stout C.D. A novel cryoprotection scheme for enhancing the diffraction of crystals of recombinant cytochrome ba3 oxidase from Thermus thermophilus. Acta Crystallogr. D Biol. Crystallogr. 2005;61:340–343. doi: 10.1107/S0907444904033906. [DOI] [PubMed] [Google Scholar]
- 48.Taly A., Delarue M., Changeux J.P. Normal mode analysis suggests a quaternary twist model for the nicotinic receptor gating mechanism. Biophys. J. 2005;88:3954–3965. doi: 10.1529/biophysj.104.050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nowak C., Laredo T., Naumann R.L.C. 2D-SEIRA spectroscopy to highlight conformational changes of the cytochrome c oxidase induced by direct electron transfer. Soft Matter. 2010;6:5523–5532. doi: 10.1039/c0mt00083c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koepke J., Olkhova E., Michel H. High resolution crystal structure of Paracoccus denitrificans cytochrome c oxidase: new insights into the active site and the proton transfer pathways. Biochim. Biophys. Acta. 2009;1787:635–645. doi: 10.1016/j.bbabio.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 51.Riistama S., Puustinen A., Wikström M. Binding of O(2) and its reduction are both retarded by replacement of valine 279 by isoleucine in cytochrome c oxidase from Paracoccus denitrificans. Biochemistry. 2000;39:6365–6372. doi: 10.1021/bi000123w. [DOI] [PubMed] [Google Scholar]
- 52.Namslauer I., Brzezinski P. A mitochondrial DNA mutation linked to colon cancer results in proton leaks in cytochrome c oxidase. Proc. Natl. Acad. Sci. USA. 2009;106:3402–3407. doi: 10.1073/pnas.0811450106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seibold S.A., Mills D.A., Cukier R.I. Water chain formation and possible proton pumping routes in Rhodobacter sphaeroides cytochrome c oxidase: a molecular dynamics comparison of the wild type and R481K mutant. Biochemistry. 2005;44:10475–10485. doi: 10.1021/bi0502902. [DOI] [PubMed] [Google Scholar]
- 54.Roberts V.A., Pique M.E. Definition of the interaction domain for cytochrome c on cytochrome c oxidase. III. Prediction of the docked complex by a complete, systematic search. J. Biol. Chem. 1999;274:38051–38060. doi: 10.1074/jbc.274.53.38051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.