

Abstract
Vinculin phosphorylation has been implicated as a potential mechanism for focal adhesion growth and maturation. Four vinculin residues—Y100, S1033, S1045, and Y1065—are phosphorylated by kinases during focal adhesion maturation. In this study, phosphorylation at each of these residues is simulated using molecular dynamics models. The simulations demonstrate that once each phosphorylated vinculin structure is at equilibrium, significant local conformational changes result that may impact either vinculin activation or vinculin binding to actin and PIP2. Simulation of vinculin activation after phosphorylation shows that the added phosphoryl groups can prime vinculin for activation. It remains to be seen if vinculin can be phosphorylated at S1033 in vivo, but these simulations highlight that in the event of a S1033 phophorylation vinculin will likely be primed for activation.
Introduction
To understand how a cell senses and responds to its physical environment, it is imperative to understand the signaling logic of the adhesome, the set of proteins structurally, mechanically, and biochemically associated with integrin-mediated signals and response (1). Adhesion complexes impact a vast and divergent set of cellular and physiological behaviors including such cell migration dependent processes as blood coagulation, leukocyte extravasation, tissue differentiation and repair, bone resorption, and cancer metastasis (2–6). The adhesion complex, initiated through integrin and extracellular matrix (ECM) contact, propagates and matures through mechano-induced scaffold formation, and mechano-induced biochemical signal transduction (2,7,8). Although individual modes of action are understood for a number of these structural and biochemical signals, the integrated mechanism by which the initial integrin-ECM contact develops into a mature focal adhesion remains uncertain. Following integrin-ECM interaction, the scaffolding protein talin links integrin to F-actin filaments, constituting the initial focal contact (9). Continued maturation, however, requires the force-induced recruitment of vinculin to the focal contact (10). Understanding the mechanism and regulation of vinculin recruitment is thus essential to understanding the signaling dynamics by which focal adhesions are formed.
Vinculin, a 1066 residue globular protein, consists of a head region (Vh), composed of four helical bundle domains (D1, D2, D3, and D4), which is connected by a proline-rich flexible linker region to a fifth helical bundle domain known as the tail region (Vt or D5) (11) (Fig. 1). Vinculin's direct role in focal adhesion maturation is a cross-linking event where vinculin acts to reinforce an initial weak talin-actin filament linkage (12). Upon activation, the vinculin binding domain of talin inserts into the hydrophobic core of D1, and Vt binds two subunits of F-actin (13–15). In addition, vinculin interacts with many of the other FA and actin cytoskeleton scaffolding proteins such as α-actinin, paxillin, VASP, and Arp2/3 (11). These interactions compete, however, with a strong autoinhibitory binding (Kd < 10−9 M) of Vt to D1–D4 in a manner that sterically occludes the F-actin and talin- binding interfaces on vinculin, as well as intermolecular binding of vinculin in general (12,16). For vinculin to bind its partners it must undergo a conformational change—vinculin activation—that renders actin-binding sites in Vt accessible to F-actin. Vinculin's role in focal adhesion maturation is therefore highly dependent on a process of vinculin activation.
Figure 1.
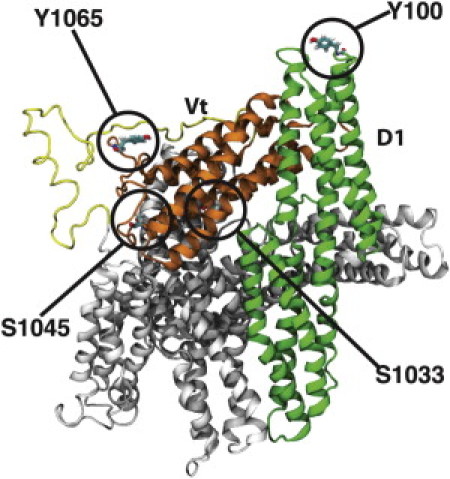
Structure of vinculin and its phosphorylation sites. A cartoon representation of molecular vinculin in the autoinhibited state, with phosphorylation sites circled and labeled. The domain 1 (D1) helix bundle is colored in green, with the remaining helix bundles (D2–4) in the head domain colored in gray. The proline-rich linker region is colored in yellow and the tail domain (Vt) helix bundle is colored in orange. Talin would bind vinculin at D1, whereas Vt is suggested to interact with actin filaments. Movement of D1 away from Vt allows for Vt to potentially interact with actin. Phosphorylation of vinculin at each of the labeled residues has been implicated in regulating the growth and maturation of focal adhesions (20,26). Two of these, pY100 and pS1033, are at the interface between Vt and D1 and likely impact vinculin activation. Two of these, pS1045 and pY1065, are near Vt binding sites for actin or PIP2 and likely impact Vt binding.
Molecular dynamics (MD) modeling offers a valuable approach to understanding the mechanisms of vinculin activation. In previous studies from our group, MD simulations of vinculin activation have investigated the trajectory of vinculin conformational changes leading to activation and the impact of interaction with talin on the activation process (12,17). These simulations suggested a cooperative binding model for vinculin activation, consistent with experimental observations (18). Similarly, talin was shown to bind vinculin only after the vinculin activation process, not before (12). Together these studies illustrate a principle of cooperation between multiple focal adhesion forming molecules in facilitating the focal adhesion maturation process. Furthermore, the MD approach suggests a specific activation mode for vinculin in which D1 is moved away from Vt (17) allowing for binding of both actin and talin to vinculin.
One aspect not considered in previous MD simulations of vinculin activation is the effect of potential phosphorylation modifications of vinculin by kinases downstream of the mechanical stimuli that lead to focal adhesion maturation. Experimental studies have linked phosphorylation of vinculin residues Y1065 and Y100 with Src family kinases (SFKs), and S1033 and S1045 as substrates, at least in vitro, of protein kinase C (PKC) (19–21). Y822 is also implicated as a phosphorylation target, though unlike Y1065 and Y100 this Y822 modification has not been detected in studies using antibodies to phosphorylated tyrosine (19,20,22,23). Though direct effects of mutating either S1033 or S1045 to a constitutive mimic of the phosphorylated or unphosphorylated form remain untested, activation of PKC stimulates cell adhesion and migration; although, downregulation of PKC stymies spreading, migration, and adhesion, and models have been put forward as to how this phosphorylation may affect recruitment (21). Studies downregulating Src kinases or testing mutations of Y100 and Y1065 to constitutive phosphorylation or dephosphorylation mimics have linked their phosphorylation to cell spreading, the force cells apply to surfaces, lamipodial spreading, and wound healing behavior, as well as abolishing interaction with Arp2/3 (19,20). Y1065 phosphorylation has been specifically linked to increased exchange dynamics with nacent focal adhesions, promoting their growth (25,26). On a molecular level, the phosphorylation of Y1065 decreases insertion of the vinculin C-terminal residues into lipid membranes, and both mutation of this residue and deletion of the vinculin C-terminal residues yielded similar decreases in cell traction and force generation (27,28). Thus, each of these phosphorylation events appear to impact the cellular behaviors of focal adhesion maturation, with a specific role in regulating the C-terminal lipid anchor implied for Y1065.
The connection between vinculin phosphorylation and maturing focal adhesions does not elucidate the effect of these modifications on the molecular state of vinculin, nor how such effects lead to observed cellular force generation. Perhaps specific phosphorylation events modify intramolecular affinities sufficiently to cause a conformational change into the active state, a mode of phosphorylation action classically seen in protein kinase activation for instance (29–31). Alternatively, phosphorylation may increase affinity to certain vinculin binding partners, allowing these partners to better compete with the auto-inhibited state. Or perhaps, in the case of vinculin, phosphorylation could impact focal adhesion development via a novel mechanism.
In this study, we develop MD models to determine local and global conformational effects of each vinculin phosphorylation event. Furthermore, we simulate vinculin activation with the inclusion of these phosphorylation events and evaluate directly the impact of vinculin phosphorylation on vinculin activation.
Methods
Phosphorylation of vinculin model
CHARMm topology patches and force field parameters for pSer and pTyr were used to build models of vinculin with Y100, Y1065, S1033, or S1045 modified to their phosphorylated forms (32). The dianionic form of pSer and pTyr were used in each modification, as the pKa for the second deprotonation of each residue typically occurs at pH ∼5.7 to 5.9, well below physiological pH (33,34). Using the crystal structure of unphosphorylated vinculin (PDB ID = 1ST6) (35) a model of the missing proline-rich linker (residues 843–877) was added using a homology model built by SWISSMODEL (36) as previously described (17). Several different structures were created: with Y100 modified to pTyr, with Y1065 modified to pTyr, with S1033 to pSer, with S1045 to pSer, and with both Y100 modified to pTyr and S1033 modified to pSer. Phosphorylation at both Y100 and S1033 was produced as both of these residues are at the interface between Vt and D1, an interface critical to vinculin autoinhibition.
MD of each phosphorylation model
NAMD (37) was used for subsequent simulations of each of the created vinculin models. Each model was immersed in a water box with a minimum 12 Å of padding surrounding the molecule, using more than 60,000 water molecules for each simulation. Additional layers of water were added to regions of vinculin known to undergo conformational change during activation to ensure vinculin remains within the periodic cell. Solvent padding was as high as 35 Å in these regions of vinculin. The water box measured 99 Å × 145 Å × 157 Å (Fig. S1 in the Supporting Material). Each model was minimized for 1000 steps followed by MD simulation of over 50 ns, and the root mean-square deviation of the molecule were calculated (Fig. S2) to ensure equilibration of the phosphorylated structures. CHARMm 27 protein force fields were used (38,39). The particle mesh Ewald method was used for electrostatic interactions, nonbonded interactions were cutoff at 12 Å, and a Langevin damping coefficient of 5/ps was used for temperature control. Rigid bonds between hydrogen atoms and associated larger atoms (40) were used in conjunction with 2 fs timesteps. Simulations were produced in the NPT ensemble using the Langevin piston Nose-Hoover (41) method for pressure control and a Langevin dynamics for temperature control. Temperature was set to 310 K and pressure at 1 Atm.
Simulation of activation
Following equilibration, models of phosphorylated vinculin that were to be investigated for their impact on vinculin activation (pY100 model, pS1033 model, and model with both pY100 and pS1033) were subjected to a conformational change consistent with previous studies of vinculin activation (12,17). pY1065 and pS1045 are not likely to affect the movement of D1 away from Vt as they do not lie near the interface between D1 and Vt (Fig. 1). D1 of vinculin was pulled away from Vt, simulating vinculin activation. The center of mass of Vt (residues 926, 958, 988, and 1031) was fixed while the center of mass of D1 (residues 15, 51, 81, and 115) was pulled away from Vt. The direction of pull was defined as the vector direction from the constrained residues (mass center of Vt) to the pulled residues (mass center of D1). Pulling simulations were carried out using a dummy atom pulled away at a constant velocity of 0.001 Å/ps (0.1 m/s) and connected to the mass center of D1 with a spring constant of 694.79 pN/Å using the SMD package of NAMD. Simulation of vinculin activation here was set up to be similar to previous MD simulations of vinculin activation (12,17).
Results
Phosphorylation-induced local conformational changes
Four residues in vinculin have been implicated as possible sites of phosphorylation by a kinase: Y100, S1033, S1045, and Y1065 (Fig. 1) (20,21,26). To simulate the local conformational changes that would result in vinculin from each of these potential phosphorylation events, a structure of full-length vinculin with each of these residues modified to their phosphorylated form were produced. MD simulations of each phosphorylated vinculin molecule were run to test for structural changes that would occur within the nanosecond timescale. Evaluation of whether these phosphorylation events could activate vinculin directly would require millisecond simulations, beyond feasible computational time if one wants to explore a useful number of conditions. The simulations presented in this study evaluate the initial conformational changes that result from the phosphorylation events.
Phosphorylation of Y100, producing pY100, caused rearrangement of some charged residues at the interface between Vt and D1 (Fig. 2). Phosphorylated Y100 forms an electrostatic interaction with the ammonium group of K35, also in D1. This newly formed interaction is sustained through a large part of the 50 ns simulation as quantified by the reduced distance between pY100 and K35. Y100 is located in the turn between helix 3 and 4 of D1, and K35 between helix 1 and 2. The impact of this new interaction is to remove K35 from a long chain of electrostatic interactions at the Vt D1 interface—R1008, E106, R105, E31, and K35 are linked together through interactions between their charged side-chain moieties; guanidinium, ammonium, and carboxyl groups for arginine, lysine, and glutamate, respectively. Once pY100 is introduced, K35 swings away from the E31 carboxyl group toward the Y100 phosphate moving at least 5 Å away from E31 and disrupting the hydrogen bond network contributing to the Vt-D1 interface. One possible outcome of Y100 phosphorylation, then, is weakening of the Vt-D1 binding. The interaction between Vt and D1 is critical to vinculin autoinhibition (17) and its degradation could affect vinculin activation, these possibilities are explored in the next section.
Figure 2.
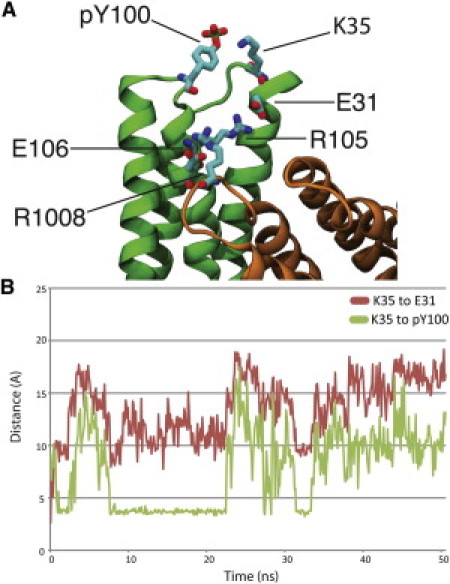
Phosphorylation of vinculin at residue Y100. (A) D1 of vinculin is shown in green, whereas Vt of vinculin is shown in orange. Phosphorylation of Y100 at the top of the D1 helix bundle domain introduces a new electrostatic interaction between K35 and pY100. This new interaction removes K35 from a chain of electrostatic interactions stabilizing the autoinhibited vinculin conformation, potentially reducing the strength of binding between Vt and D1. Other residues involved in the chain of electrostatic interactions include E31, R105, E106, and R1008. (B) As the vinculin with pY100 is simulated for 50 ns K35 moves and reorients itself toward vinculin. The red plot shows the increase in distance between K35 and E31 throughout the 50 ns simulation as a result of the interaction with pY100, and the green plot captures the decrease in distance between pY100 and K35 for nearly 15 ns (between 8 ns of simulation and 23 ns of simulation) due to their association.
Phosphorylation of S1033 also affects the interface between Vt and D1 (Fig. 3). A new electrostatic interaction is formed between the phosphate on pS1033 and the R987 guanidinium group. This breaks a previous interaction between the R987 guanidinium group and the E186 carboxyl group causing the distance between the two residues to increase by 7 Å over the 50 ns simulation. After introduction of the phosphoryl group and rearrangement of the electrostatic interactions, the hydration of E186 and its nearby residues at the interface between Vt and D1–D3 is increased by 15,000 Å2 (Fig. S3). The increased hydration weakens the link between Vt and D1 by both decreasing the surface area of hydrophobic contacts between Vt and D1 in the region, and by shielding the strength of the electrostatic interactions between Vt and D1 in the region. Y100 and S1033 lay on opposite ends of a series of interactions that stabilize the interface between Vt and D1 in the autoinhibited form. Breaking of these interactions is necessary to reach the activated state (17). To test the impact of these phosphorylation events on the transition to the activated state, we simulated vinculin activation in the presence of these phosphoryl groups as discussed in the next section.
Figure 3.
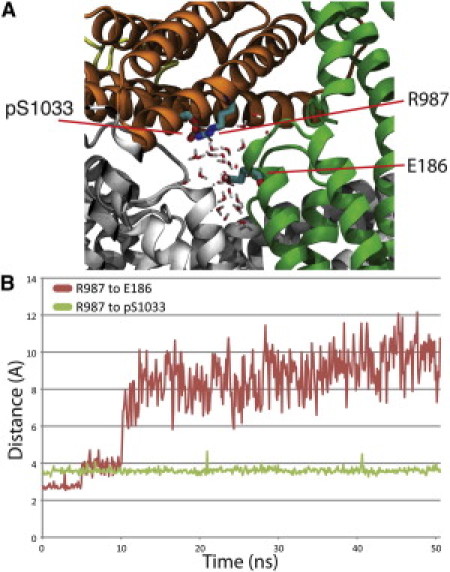
Phosphorylation of vinculin at residue S1033. (A) Phosphorylation of S1033 causes a shift of R987 toward the new phosphoryl group. After this structural rearrangement the hydration of vinculin is increased through penetration of the D1-Vt interface. D1 is shown in green, Vt in orange, and D2–D4 in gray. (B) The separation of R987 from E186 (red plot) and the association of R987 with pS1033 (green plot) is captured throughout the 50 ns of simulation.
The two other phosphorylation sites do not lie at the interface between Vt and D1. Yet, phosphorylation of both S1045 and Y1065 leads to noticeable local conformational changes (Figs. 4 and 5). Phosphorylation of S1045 to pS1045 leads to the formation of a new salt-bridge between the pS1045 phosphate and the guanidinium groups of R1057 and R1060, seen by a decrease in the distance between R1057 and pS1045 of more than 10 Å that is sustained throughout the 50 ns simulation (Fig. 4). This interaction causes the N-terminal of the proline-rich loop region to move away from Vt during simulation. Residue Q851 in the loop region moves more than 20 Å away from R1057; the previous interaction between Q851 and R1057 had stabilized the association of the loop region with this section of Vt. Phosphorylation of Y1065 to pY1065 leads to the formation of a new interaction between the pY1065 phosphate and the ammonium group of K881 in the loop region. The newly formed interaction reduced the distance between K881 and its nearby loop region and pY1065 and its nearby Vt region by 3 Å (Fig. 5). This new interaction links the N-terminal strap of the loop domain to Vt. Although not suggested to play a direct role in vinculin activation, the N-terminal strap has been implicated in both vinculin binding to actin and to recruitment to the cell membrane through PIP2 binding (42). Furthermore, pS1045 lies near an electrostatic actin-binding region of Vt (14) and pY1065 lies on the C-terminal hairpin, a region more likely to be involved in association of vinculin with PIP2. The proximity of pS1045 and pY1065 to actin and PIP2 binding sites, and the impact of both phosphorylated residues on the loop domain suggests phosphorylation of S1045 and Y1065 would impact focal adhesion development not by affecting vinculin activation, but by affecting the intermolecular binding between vinculin and either PIP2 or F-actin. The results from simulation with each vinculin model are summarized in Table S1. The initial conformational changes induced by phosphorylation at each of the sites (Figs. 2–5) and the resulting changes in distances between residues can be compared to the changes between the same residues in simulation of vinculin without phosphorylation (Fig. S4). The changes are not seen in the absence of phosphorylation and are likely due to the phosphorylation event.
Figure 4.

Phosphorylation of vinculin at residue S1045. (A) Phosphorylation of S1045 releases the loop region near D4. After phosphorylation pS1045 interacts with both R1057 and R1060 releasing Q851 and the loop region from association with R1057. The flexible loop region is shown in yellow, the Vt domain in orange, D1 in green, and D2–D4 in gray. (B) Four distances were tracked throughout the 50 ns simulation. The distance between R1057 and Q851 increases early in the simulation (red plot) and remains separated. The R1057 then associates with pS1045 (purple plot) along with R1060 (blue plot). The released Q851 has intermittent association with a nearby loop region residue D841 (green plot).
Figure 5.

Phosphorylation of vinculin at residue Y1065. (A) Phosphorylation of Y1065 introduces a new interaction between Vt and the loop region near Vt. K881 forms an interaction with pY1065. Vt is shown in orange, D2–D4 in gray, and the loop region in yellow. (B) The new linkage between K881 and pY1065 is formed early and is maintained throughout the 50 ns simulation.
A shift in the D4 domain with respect to the D3 domain in the head was observed during simulation of the pY1065 phosphorylation. This initially appeared to be a conformational change triggered by phosphorylation. To control if the domain shift was truly a result of the phosphorylation, a control simulation was produced of nonphosphorylated vinculin at equilibrium. A similar shift was found to occur in the nonphosphorylated vinculin simulation, suggesting the shift to be a result of the molecular flexibility. Indeed, previous normal mode analysis of vinculin (17) found a normal mode of D4 shifting with respect to D3.
Impact of phosphorylation on vinculin activation
The local conformational changes induced by phosphorylation of vinculin at Y100 or S1033 suggest that these events could affect vinculin activation. To evaluate this potential effect, vinculin activation was simulated after residue modification to the phosphorylated form. Vinculin activation was simulated using a constant velocity pull of D1 away from Vt, reflecting the mechanism of vinculin activation explored previously (17). Four simulations were produced using this activation scheme, including: a vinculin model with pY100, a model with pS1033, a model with both modifications, and a model with no phosphorylation.
In these simulations, the maximum level of force needed to achieve the activation is reduced only after phosphorylation at pS1033 (Fig. 6). The conformational changes seen during each set of vinculin activation simulations illuminates the impact of phosphorylation at pS1033 on vinculin activation (Fig. 7). In previously published MD investigation of vinculin activation (17) three sets of electrostatic interactions were described as stabilizing the autoinhibited vinculin conformation. Similarly, in the simulations of activation here, two critical steps toward activation can be described: a), the separation of Vt and D1 in regions near S1033 (Fig. 7 A), and b), the breakage of the electrostatic chain of interactions involving residues R1008, E106, R105, E31, and K35 (Fig. 7 B). These steps directly involve two of the three electrostatic interactions previously described (17). In simulating activation of nonphosphorylated vinculin, 800 pN of force is supplied to D1 within 10 ns of simulation. This force is enough to allow for both a and b to occur without the need for any subsequent peaks in supplied force. In the simulation of vinculin with only pS1033 there is less force needed for a as the phosphorylation has already rearranged the electrostatic interactions and weakened the link in this region. The force needed only increases to ∼700 pN at 30 ns of simulation. After hydration of the residues near pS1033 the separation of D1 from Vt near pS1033 (∼10 ns of simulation) requires ∼600 pN of force. The separation of D1 from Vt in this region makes separation near Y100 with less force possible at 30 ns of simulation. In contrast, simulation of vinculin with only pY100 shows no initial separation of D1 from Vt at the residues near pS1033 and shows an increase in the force peak required to separate charged residues in D1 from R1008. This increased force can be attributed to an interaction between pY100 and R1008 that is formed during the pulling simulation. This interaction was not seen during the equilibrium simulation. Simulation of vinculin with both pY100 and pS1033 shows the same trajectory to activation as phosphorylation with only pS1033 and activation after ∼35 ns of simulation. The additional time is likely required due to the interaction between pY100 and R1008. The peak force required is 700 pN, similar to activation of vinculin with pS1033 only, and less than activation of vinculin without pS1033.
Figure 6.

Effect of phosphorylation on vinculin activation. Four models of vinculin were simulated with an activating force applied to D1: unphosphorylated vinculin (red), vinculin with pY100 (green), vinculin with pS1033 (purple), and vinculin with both pS1033 and pY100 (blue). Activation of unphosphorylated vinculin and vinculin with pY100 required over 800 pN of force, whereas activation of vinculin with pS1033 and with both pS1033 and pY100 required 700 pN of force. The reduced level of force required results from significant local conformational changes introduced by pS1033 that prime vinculin for force-induced activation.
Figure 7.

Trajectory of vinculin activation. Two critical events are necessary for vinculin to activate: (a) D1 regions near residue S1033 need to separate from Vt and (b) the chain of electrostatic interactions involving R1008, E106, R105, E31, and K35 needs to be broken. (A) Simulation of vinculin with pS1033 showed rearrangement of residues near pS1033 allowed for separation of D1 from Vt near pS1033 even before (b) occurs. pS1033 also increased the hydration of the interface between D1 and Vt (arrow). D1 is shown in green, Vt is shown in orange, and D2–D4 is shown in gray. (B) Simulation of vinculin with pY100 showed increased resilience to activation. Part of this increased stability of the autoinhibited conformation resulted from an interaction between pY100 and R1008 of Vt (arrow).
An increased level of force needed for activation in these simulations (more than 700 pN) to the level of force reported in our previous simulations (simulated with a constant force ranging between 50 and 200 pN) (17) arises from viscosity induced by water collisions in the explicit solvent treatment and frictional dampening from the temperature control. In the previous simulations (17) the EEF1 implicit solvent model (43) was used, whereas in these simulations the TIP3 (44) solvent model is used. Even with the higher levels of force needed in the explicit solvent simulations: the trajectory to activation was the same as the previous simulations with the implicit solvent. Even though the forces seen here are much larger and the timescale much shorter than would be seen experimentally, the decrease in force needed to activate vinculin should still hold true in vivo.
Discussion
Vinculin phosphorylation has been repeatedly suggested as one of the mechanisms by which focal adhesions mature; Vinculin phosphorylation has even been linked with vinculin activation and its recruitment to focal adhesions (20,21,26). Yet little evidence exists showing the molecular mechanism by which vinculin phosphorylation would impact these cellular processes. Using MD, this study has illustrated one mechanism by which vinculin phosphorylation could impact vinculin activation: by priming vinculin for activation. Vinculin priming considers the phosphorylation increasing the chances that vinculin would become activated by reducing the stability of the autoinhibition with respect to the pulling trajectory needed for force activation.
Phosphorylation has an impact on the function of a multitude of molecules, and is used as a regulatory mechanism throughout the cell. Primarily, phosphorylation regulates cellular processes either by altering intermolecular binding or by directly causing activating conformational changes by changing intramolecular binding (30,31,45,46). In the case of vinculin, involved in mechanotransduction, we suggest a third mode: priming. A priming phosphorylation would not alter binding affinities to the extent to directly cause domain scale conformation change and activation, but would instead modulate a vinculin molecule's susceptibility to activation by pN-force, specifically the cooperative pulling interaction with talin and actin (17,18). It remains possible that vinculin phosphorylation could activate vinculin in the absence of a cooperative pulling interaction. Evaluating that possibility with MD would require simulation of events on the millisecond timescale.
Two of the phosphorylation sites on vinculin lie at the interface between Vt and D1 and two of the phosphorylation sites lie on Vt near its binding sites for actin and PIP2 (Fig. 1). The equilibrium structure of vinculin with each of its potential phosphoryl-groups suggests that phosphorylation S1033 affects vinculin activation (Figs. 2 and 3), whereas phosphorylation at Y1065 or S1045 could affect Vt binding to actin or PIP2 (Figs. 4 and 5). The impact of phosphorylation on Vt binding to its partners is a topic worthy of consideration in future studies. One possibility is that phosphorylation at multiple sites, for example at Y100 and at Y1065, could enhance vinculin activation by modulating the strength of the autoinhibited conformation and modulating the binding affinity of Vt for PIP2. Any future study considering the impact of phosphorylation on PIP2 binding should also investigate this possible joint mechanism.
Although association of PKC and vinculin has been shown in vivo, evidence for phosphorylation at S1033 and S1045 comes from in vitro kinase assays (21). It could be that phosphorylation at S1033 or S1045 is merely conjecture and would play no role in the physiological process of vinculin activation. Phosphorylation at S1033 and S1045 could also prove to be possible in vivo and an effective part of vinculin activation and focal adhesion formation. The vinculin structure places S1033 away from solvent suggesting phosphorylation at S1033 to be less likely. Studies of vinculin dynamics and vinculin activation point to several interfaces linking Vt to the vinculin head domains, including an interface between Vt and D4 (16), suggesting residues on the Vt surface, such as S1033, could gain solvent exposure as a result of the dynamic movements between vinculin domains. In this study, the separation between Vt and D1 and the role of phosphorylation in priming that separation is considered. Separation of Vt from other vinculin head domains could precede D1 separation. Such domain movements, even if they are temporary and do not lead to vinculin activation themselves, could allow for phosphorylation at S1033 or S1045. Understanding the order of events leading to vinculin activation and focal adhesion formation is critically important. The simulations presented here cannot definitively clarify the order of events leading to vinculin activation, but they do evaluate the possibility of vinculin activation after priming by a phosphoryl group.
The impact of phosphorylation on vinculin activation was considered here, and simulation of vinculin activation with phosphorylation showed that the combined phosphorylation of S1033 decreases the force needed to activate vinculin (Fig. 6). Phosphorylation at Y100 alone had minimal impact on reducing the force needed for vinculin activation, and in fact increased the force needed as a result of its interaction with R1008 formed during the pulling trajectory. Phosphorylation of S1033 then is critical to vinculin priming and one must consider that PKC would need to access S1033 for this priming to occur. Although in a number of other proteins it has been shown that phosphorylation is sufficient to induce activation (30,31,45,46), this study shows that in the case of vinculin a cooperative binding event involving actin and talin is still necessary for vinculin activation.
Although each phosphorylation altered specific side-chain interactions, none of the phosphorylation events caused major domain shifts. Vinculin's resilience to activation from only phosphorylation is consistent with its cellular role as a reinforcing agent (11). Vinculin has been implicated in both reinforcement of the talin-actin linkage (12) and the α-actinin-actin linkage (48). That phosphorylation can prime vinculin for activation but not completely cause vinculin activation means that vinculin can only be activated under a condition in which its binding partners are present, and perhaps there is some activating tension, i.e., vinculin is only activated when reinforcement is needed. The priming reduces the threshold to reinforcement, but maintains its discipline as a reinforcing agent. In a force-induced activation model, vinculin phosphorylation would then modulate sensitivity to tension inputs.
Our simulation of vinculin activation has employed a stretching force between D1 and Vt. Although it is hypothesized this stretch is due to the simultaneous binding of D1 to talin and Vt to actin (12,17) the stretching force could also result from a tension across vinculin when recruited to the cell membrane or to focal adhesions. A recent experimental investigation (49) showed the presence of a mechanical tension across vinculin in vivo. It is unclear whether vinculin was phosphorylated before the measured tension in their experiment; our results suggest this is possible, and the phosphorylation could have reduced the level of tension needed to cause a strain in vinculin. Perhaps an experiment could address this by considering the level of tension across vinculin in cells with PKC knocked-out. One possibility is that the reduced barrier to vinculin activation by vinculin phosphorylation increases the frequency of vinculin activation within the cell in vivo only after priming by phosphorylation.
One phosphorylation target not considered in this study is phosphorylation at Y822. Although some investigations point to phosphorylation at Y822 (22), other studies fail to detect phosphorylation at Y822 by Src kinases (19). Furthermore, investigation of the impact of phosphorylation at Y822 on the growing focal adhesion has demonstrated that the phosphorylation event has no affect on focal adhesion formation (19), whereas the phosphorylation sites investigated in this study have not only been demonstrated to be possible, but are also suggested to impact the mechanotransduction response. Previous experimental studies (20,21,25,26) have demonstrated an overlap between the effects of vinculin and PKC in focal adhesion maturation, potentially caused by phosphorylation of vinculin by PKC. Phosphorylation of vinculin by SFKs has been shown in vivo and our study illustrates one mechanism by which these kinases could be contributing to focal adhesion growth: their phosphorylation of vinculin could be priming vinculin for activation. After phosphorylation by PKC or SFKs, vinculin becomes more susceptible to activation either by simultaneous interaction or by stretch. It is not likely however, that vinculin activation would result directly from phosphorylation in the absence of some other activating interaction or stimuli. Zeigler et al. (11) suggests a model to vinculin activation in which vinculin is recruited to the lipid membrane alongside PKC. Our model suggests that this would allow for S1033 phosphorylation and prime vinculin for later force-induced activation.
This study has specifically explored the impact of phosphorylation on vinculin. However, a number of focal adhesion forming molecules have been implicated in regulation by phosphorylation (2,50). It would be interesting to see an investigation of how phosphorylation may regulate other molecules involved in focal adhesion maturation. Perhaps priming, where chemical modifications increase susceptibility to a specific forcing stimulus, is a phenomenon associated with many molecules involved in mechanotransduction, not just vinculin.
Acknowledgments
The authors are thankful to M. Azimi, R. Moussavi-Baygi, H. Shams, and other members of the Molecular Cell Biomechanics Laboratory for fruitful discussions during the course of this study.
Financial support by National Science Foundation through a CAREER award to M.R.K.M. (CBET 0955291) and a Graduate Research Fellowship to T.J.W. (DGE 1106400) is gratefully acknowledged.
Supporting Material
References
- 1.Zaidel-Bar R., Itzkovitz S., Geiger B. Functional atlas of the integrin adhesome. Nat. Cell Biol. 2007;9:858–867. doi: 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huveneers S., Danen E.H. Adhesion signaling - crosstalk between integrins, Src and Rho. J. Cell Sci. 2009;122:1059–1069. doi: 10.1242/jcs.039446. [DOI] [PubMed] [Google Scholar]
- 3.Moore S.W., Roca-Cusachs P., Sheetz M.P. Stretchy proteins on stretchy substrates: the important elements of integrin-mediated rigidity sensing. Dev. Cell. 2010;19:194–206. doi: 10.1016/j.devcel.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar S., Weaver V.M. Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer Metastasis Rev. 2009;28:113–127. doi: 10.1007/s10555-008-9173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao J., Guan J.L. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28:35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- 6.Gardel M.L., Schneider I.C., Waterman C.M. Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 2010;26:315–333. doi: 10.1146/annurev.cellbio.011209.122036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geiger B., Spatz J.P., Bershadsky A.D. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 2009;10:21–33. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- 8.Vogel V. Mechanotransduction involving multimodular proteins: converting force into biochemical signals. Annu. Rev. Biophys. Biomol. Struct. 2006;35:459–488. doi: 10.1146/annurev.biophys.35.040405.102013. [DOI] [PubMed] [Google Scholar]
- 9.Izzard C.S. A precursor of the focal contact in cultured fibroblasts. Cell Motil. Cytoskeleton. 1988;10:137–142. doi: 10.1002/cm.970100118. [DOI] [PubMed] [Google Scholar]
- 10.Galbraith C.G., Yamada K.M., Sheetz M.P. The relationship between force and focal complex development. J. Cell Biol. 2002;159:695–705. doi: 10.1083/jcb.200204153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziegler W.H., Liddington R.C., Critchley D.R. The structure and regulation of vinculin. Trends Cell Biol. 2006;16:453–460. doi: 10.1016/j.tcb.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 12.Golji J., Lam J., Mofrad M.R. Vinculin activation is necessary for complete talin binding. Biophys. J. 2011;100:332–340. doi: 10.1016/j.bpj.2010.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Humphries J.D., Wang P., Ballestrem C. Vinculin controls focal adhesion formation by direct interactions with talin and actin. J. Cell Biol. 2007;179:1043–1057. doi: 10.1083/jcb.200703036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janssen M.E., Kim E., Hanein D. Three-dimensional structure of vinculin bound to actin filaments. Mol. Cell. 2006;21:271–281. doi: 10.1016/j.molcel.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 15.Izard T., Evans G., Bois P.R. Vinculin activation by talin through helical bundle conversion. Nature. 2004;427:171–175. doi: 10.1038/nature02281. [DOI] [PubMed] [Google Scholar]
- 16.Cohen D.M., Chen H., Craig S.W. Two distinct head-tail interfaces cooperate to suppress activation of vinculin by talin. J. Biol. Chem. 2005;280:17109–17117. doi: 10.1074/jbc.M414704200. [DOI] [PubMed] [Google Scholar]
- 17.Golji J., Mofrad M.R. A molecular dynamics investigation of vinculin activation. Biophys. J. 2010;99:1073–1081. doi: 10.1016/j.bpj.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen H., Choudhury D.M., Craig S.W. Coincidence of actin filaments and talin is required to activate vinculin. J. Biol. Chem. 2006;281:40389–40398. doi: 10.1074/jbc.M607324200. [DOI] [PubMed] [Google Scholar]
- 19.Moese S., Selbach M., Meyer T.F. The Helicobacter pylori CagA protein disrupts matrix adhesion of gastric epithelial cells by dephosphorylation of vinculin. Cell. Microbiol. 2007;9:1148–1161. doi: 10.1111/j.1462-5822.2006.00856.x. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Z., Izaguirre G., Haimovich B. The phosphorylation of vinculin on tyrosine residues 100 and 1065, mediated by SRC kinases, affects cell spreading. Mol. Biol. Cell. 2004;15:4234–4247. doi: 10.1091/mbc.E04-03-0264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ziegler W.H., Tigges U., Jockusch B.M. A lipid-regulated docking site on vinculin for protein kinase C. J. Biol. Chem. 2002;277:7396–7404. doi: 10.1074/jbc.M110008200. [DOI] [PubMed] [Google Scholar]
- 22.Subauste M.C., Pertz O., Hahn K.M. Vinculin modulation of paxillin-FAK interactions regulates ERK to control survival and motility. J. Cell Biol. 2004;165:371–381. doi: 10.1083/jcb.200308011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldmann W.H., Galneder R., Ezzell R.M. Differences in elasticity of vinculin-deficient F9 cells measured by magnetometry and atomic force microscopy. Exp. Cell Res. 1998;239:235–242. doi: 10.1006/excr.1997.3915. [DOI] [PubMed] [Google Scholar]
- 24.Reference deleted in proof.
- 25.Möhl C., Kirchgessner N., Hoffmann B. Becoming stable and strong: the interplay between vinculin exchange dynamics and adhesion strength during adhesion site maturation. Cell Motil. Cytoskeleton. 2009;66:350–364. doi: 10.1002/cm.20375. [DOI] [PubMed] [Google Scholar]
- 26.Küpper K., Lang N., Hoffmann B. Tyrosine phosphorylation of vinculin at position 1065 modifies focal adhesion dynamics and cell tractions. Biochem. Biophys. Res. Commun. 2010;399:560–564. doi: 10.1016/j.bbrc.2010.07.110. [DOI] [PubMed] [Google Scholar]
- 27.Diez G., Kollmannsberger P., Goldmann W.H. Anchorage of vinculin to lipid membranes influences cell mechanical properties. Biophys. J. 2009;97:3105–3112. doi: 10.1016/j.bpj.2009.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wirth V.F., List F., Goldmann W.H. Vinculin's C-terminal region facilitates phospholipid membrane insertion. Biochem. Biophys. Res. Commun. 2010;398:433–437. doi: 10.1016/j.bbrc.2010.06.094. [DOI] [PubMed] [Google Scholar]
- 29.Vuori K., Ruoslahti E. Activation of protein kinase C precedes alpha 5 beta 1 integrin-mediated cell spreading on fibronectin. J. Biol. Chem. 1993;268:21459–21462. [PubMed] [Google Scholar]
- 30.Johnson L.N. Glycogen phosphorylase: control by phosphorylation and allosteric effectors. FASEB J. 1992;6:2274–2282. doi: 10.1096/fasebj.6.6.1544539. [DOI] [PubMed] [Google Scholar]
- 31.Nolen B., Taylor S., Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell. 2004;15:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 32.Feng M., Phillippopoulos M., Lim C. Structural characterization of the phosphotyrosine binding region of a high-affinity SH2 domain-phosphopeptide complex by molecular dynamics simulation and chemical shift calculations. J. Am. Chem. Soc. 1996;118:11265–11277. [Google Scholar]
- 33.Domchek S.M., Auger K.R., Shoelson S.E. Inhibition of SH2 domain/phosphoprotein association by a nonhydrolyzable phosphonopeptide. Biochemistry. 1992;31:9865–9870. doi: 10.1021/bi00156a002. [DOI] [PubMed] [Google Scholar]
- 34.Errington N., Doig A.J. A phosphoserine-lysine salt bridge within an alpha-helical peptide, the strongest alpha-helix side-chain interaction measured to date. Biochemistry. 2005;44:7553–7558. doi: 10.1021/bi050297j. [DOI] [PubMed] [Google Scholar]
- 35.Bakolitsa C., Cohen D.M., Liddington R.C. Structural basis for vinculin activation at sites of cell adhesion. Nature. 2004;430:583–586. doi: 10.1038/nature02610. [DOI] [PubMed] [Google Scholar]
- 36.Arnold K., Bordoli L., Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 37.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 38.MacKerell A., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 39.Mackerell A.D., Jr. Empirical force fields for biological macromolecules: overview and issues. J. Comput. Chem. 2004;25:1584–1604. doi: 10.1002/jcc.20082. [DOI] [PubMed] [Google Scholar]
- 40.Kräutler V., Gunsteren W.F.van, Hünenberger P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001;22:501–508. [Google Scholar]
- 41.Hoover W.G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 42.Palmer S.M., Playford M.P., Campbell S.L. Lipid binding to the tail domain of vinculin: specificity and the role of the N and C termini. J. Biol. Chem. 2009;284:7223–7231. doi: 10.1074/jbc.M807842200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lazaridis T. Effective energy function for proteins in lipid membranes. Proteins. 2003;52:176–192. doi: 10.1002/prot.10410. [DOI] [PubMed] [Google Scholar]
- 44.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 45.Russo A.A., Jeffrey P.D., Pavletich N.P. Structural basis of cyclin-dependent kinase activation by phosphorylation. Nat. Struct. Biol. 1996;3:696–700. doi: 10.1038/nsb0896-696. [DOI] [PubMed] [Google Scholar]
- 46.Machida K., Mayer B.J. The SH2 domain: versatile signaling module and pharmaceutical target. Biochim. Biophys. Acta. 2005;1747:1–25. doi: 10.1016/j.bbapap.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 47.Reference deleted in proof.
- 48.Golji J., Collins R., Mofrad M.R. Molecular mechanics of the alpha-actinin rod domain: bending, torsional, and extensional behavior. PLOS Comput. Biol. 2009;5:e1000389. doi: 10.1371/journal.pcbi.1000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Grashoff C., Hoffman B.D., Schwartz M.A. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 2010;466:263–266. doi: 10.1038/nature09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shattil S.J., Kim C., Ginsberg M.H. The final steps of integrin activation: the end game. Nat. Rev. Mol. Cell Biol. 2010;11:288–300. doi: 10.1038/nrm2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.