

Abstract
Paragangliomas are rare tumors of neural crest origin. They are benign in the majority of cases and are characterized by a strong vascularisation.
In the head and neck region they most commonly occur as carotid body tumors. Jugulotympanic and especially vagal paragangliomas are seen less frequently. Complete surgical resection represents the only curative treatment option even though resection of locally advanced tumors regularly results in lesions of the lower cranial nerves and major vessels.
Appoximately 30% of all head and neck paragangliomas (HNPs) are hereditary and associated with different tumor syndromes. The paraganglioma syndromes 1, 3 and 4 (PGL 1, 3 and 4) make up the majority of those familial cases. PGL 1 is associated with mutations of the succinate dehydrogenase subunit D (SDHD) gene, PGL 3 is caused by SDHC and PGL 4 by SDHB gene mutations. Multiple HNPs and the occurance of HNPs together with pheochromocytomas are seen in SDHD as well as SDHB mutation carriers. In patients with SDHB mutations the risk for the development of malignant paraganglial tumors is significantly higher compared to SDHC and SDHD patients as well as patients with sporadic tumors. SDHC mutation carriers almost exclusively present with benign HNP that are unifocal in the majority of cases. The role of transmission is autosomal dominant for all three symptoms. Interestingly, there is a “parent-of-origin-dependent-inheritance” in subjects with SDHD gene mutations. This means that the disease phenotype may only become present if the mutation is inherited through the paternal line. We recommend screening for mutations of the genes SDHB, SDHC and SDHD in patients with HNPs. Certain clinical parameters can help to set up the order in which the three genes should be tested.
Keywords: paraganglioma, glomus tumor, pheochromocytoma, paraganglioma syndrome, rare diseases
1. Introduction
Head and neck paragangliomas (HNPs) represent rare tumors of neural crest origin [1], [2]. They are highly vascular neoplasms that are benign in the majority of cases [1], [2], [3]. They may occur along the paraganglia's pathway of embryologic migration extending from the skull base to the pelvic floor [1], [2], [4], [5], [6]. Paraganglia play an importatnt role in organismic homeostasis by acting as chemoreceptors or by secretion of catecholamines in response to stress [4]. Paraganglial tumors in the thorax and the abdomen are referred to as pheochromocytomas [1], [7], [8].
Terminology
Terminology in science and clinical practice regarding tumors of the paraganglial system is divergent [9], [10], [11]. According to the suggestions of Neumann and coworkers we will use the term pheochromocytoma for all tumors in the thorax and the abdomen whereas the term head and neck paraganglioma (HNP) is only used for tumors in the head and neck region [9]. The vast majority of all pheochromocytomas are endocrinologically active. HNP on the other hand are almost exclusively nonfunctioning [9].
The terms glomus tumor and chemodectoma are often used for HNP [12], [13]. The term glomus tumor is somewhat confusing though, since it may easily be mixed up with a benign tumor of the subcutaneous skin, the so called glomangioma [2], [14], [15]. The term chemodectoma has first been used by Mulligan in 1950 [16]. Strictly speaking it is not correct either, since regarding paraganglia in the head and neck only the carotid body paraganglia act as chemoreceptors [17]. The terms glomus tumor and chemodectoma should therefore not be used any more [17], [18].
Incidence and localisation
HNP are rare neoplasms comprising about 0.03% of all human tumors [2], [6]. The yearly incidence is estimated to be at around 0.001% [2].
The site of origin defines the name given to paragangliomas in the head and neck. The CBT represents the most common tumor type [1], [19], [20], [21]. Other paragangliomas (PGs) that are frequently detected in the head and neck include jugular PGs, tympanic PGs and, although less common vagal PG [2], [22], [23]. PGs in the nose [24], the paranasal sinuses [25], parotid gland [26], cervical sympathatic chain [27], [28], larynx [29], [30], [31], thyroid gland [32], parathyroid gland [33], esophagus [34] or the orbit [35] are exceedingly rare.
Function of paraganglia in the head and neck
Paraganglia in the carotid body work as chemoreceptors measuring hypoxia, hypercapnia and ph value in arterial blood. Via the glossopharyngeal nerve they are directly connected to the respiratory centers in the central nervous system. Carotid body paraganglia are involved in regulation of respiration and blood pH value, blood pressure and heart rate [36], [37], [38], [39]. The function of the other head and neck paraganglia remains less clear [40].
Etiology
Many etiologic aspects of HNP development remain unclear [36], [41]. Sporadic CBTs have been demonstrated to be more frequent in people living at high altitudes [42] and in patients with chronic obstructive lung disease [42]. Therefore, a chronic hypoxic stimulation probably plays an important role in the occurance of those unusual neoplasms [36], [42], [43].
Age at diagnosis and sex distribution
HNPs may occur at any age [36], [44]. Most patients become symptomatic between their 30th and their 60th birthday [36], [45]. Concerning sex distribution, there is a clear female predominance by a factor of approximately 3:1 until 4:1 [1], [10], [17], [46], [47], [48], [49].
2. Discussion
2.1 Symptoms
CBTs often remain clinically silent before they present as a painless, slowly enlarging mass in the lateral neck [1], [4], [21], [36], [45], [48]. In later stages of tumor development dysphagia, deficits of the cranial nerves VII, IX, X, XI and XII as well as Horner's syndrome may be seen [4], [48].
Physical examination typically reveals a rubbery, non-tender mass in the lateral neck. The lesion is more freely moveable in a horizontal plane than vertically, which is referred to as a positive Fontaine's sign. The finding of a carotid bruit or a pulsatile character of the tumor strenthens the tentative diagnosis of a CBT. A careful examination for deficits of the lower cranial nerves as well as the cervical sympathetic chain is mandatory [4], [48].
Tympanic PGs usually become symptomatic as a pulsatile tinnitus that might be accompanied by a conductive hearing loss. In patients with jugular PGs there may be lower cranial nerve deficits in addition [2], [3], [46], [49], [50]. Tympanic as well as jugular PGs usually present to the otorhinolaryngologist by means of a bluish, pulsating mass behind the ear drum [2].
Vagal PGs most commonly arise from the inferior nodose ganglion but they may occur at any point along the course of the cervical vagal nerve [4]. Therefore, there is a wide variety of possible symptoms ranging from a painless mass in the lateral neck to dysphagia, cranial nerve deficits and Horner´s syndrome [17].
The rate of functioning HNPs is low. About 1-3% of HNPs present with elevated catecholamines and subsequent tachycardia, tremor and hypertension [18], [48], [51], [52]. Secretion of dopamine with resulting hypotension is very infrequently seen in HNP patients [52].
2.2 Diagnosis
2.2.1 Radiology
After taking the patient´s history and a thorough examination of the head and neck region an evaluation by imaging procedures is absolutely necessary to establish the diagnosis of a HNP.
In cervical HNPs, B-mode sonography with color-coded Doppler sonography represents an inexpensive, non-invasive diagnostic tool frequently used as the first imaging step [1], [2], [53], [54]. A CBT will typically present as a solid, well-defined, hypoechoic tumor with a splaying of the carotid bifurcation [1], [54]. The external carotid artery (ECA) is usually displaced anteriorly and medially whereas the internal carotid artery (ICA) is typically displaced posteriorly and laterally [54], [55].
Vagal PGs often displace both the ECA and the ICA anteriorly. The internal jugular vein is typically compressed and displaced posteriorly [55], [56]. In large vagal PGs there may also be a splaying of the carotid bifurcation [56].
In color-coded duplex sonography, cervical HNPs typically present as hypervascular masses. It is important to mention though that the absence of hypervascularity does not exclude a cervical HNP [54].
Magnetic resonance imaging (MRI) today plays the most important role in the pretherapeutic evaluation of HNPs. HNPs typically show a hyperintense signal on T2-weighted images and a distinct contrast enhancement on the T1-weighted images [57]. MRI also presents the exact extension of the tumor and its exact relationship to the carotid vessels. The classification of CBTs is according to criteria set forth by Shamblin [58]. Class I tumors are localized tumors with splaying of the carotid bifurcation but little attachment to the carotid vessels. Class II tumors partially surround the carotid vessels and Shamblin class III CBTs intimately surround the carotids [58]. Figure 1 (Fig. 1) demonstrates those three classes of CBTs.
Figure 1. Figure 1a: CBT (class I): The axial T1-weighted MRI with contrast medium shows a CBT in typical location. Splaying of the left carotid bifurcation without any surrounding of the carotid vessels.

Figure 1b: CBT (class II): Axial T1-weighted MRI with contrast showing a right-sided CBT partially surrounding the internal and external carotid artery.
Figure 1c: CBT (class III): A large left-sided CBT intimately surrounding the carotid vessels (axial T1 weighted MRI with contrast).
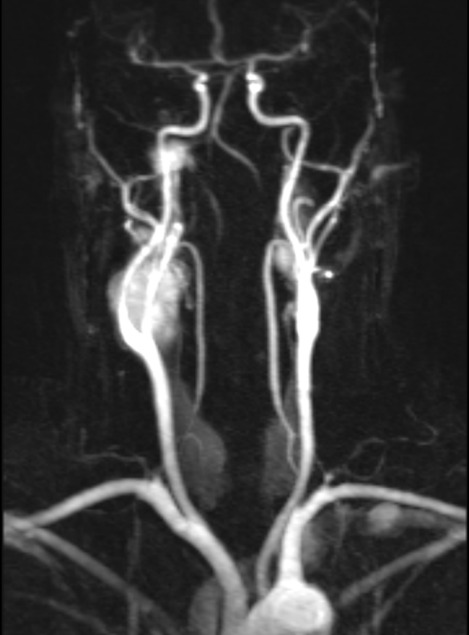
In jugulotympanic HNPs the MRI is helpful to detect dura infiltration and intradural tumor growth. It is also needed to exclude other tumor entities occuring in the area such as neurinomas and endolymphatic sac tumors (ELST) [59]. Today, the MRI will be performed in combination with a dynamic contrast-enhanced magnetic resonance angiography (MRA). The MRA helps to diagnose or rule out additional paragangliomas in the head and neck (Figure 2 (Fig. 2)) [59].
Figure 2. Dynamic contrast-enhanced magnetic resonance angiography (MRA) of a 44 year old male SDHD mutation carrier. Note bilateral CBTs and a jugular paraganglioma on the right in the early arterial phase.
Computed tomography (CT) is needed to evaluate jugulotympanic HNPs. The extend of temporal bone destruction is important to classify those tumors according to the Fisch classification (Table 1 (Tab. 1)) [60]. This preoperative classification is essential, since the surgeon will choose the operative approach depending on tumor stage [59].
Table 1. Classification of jugulotympanic paragangliomas according to Fisch & Mattox [60].

Digital substraction angiography (DSA) provides an arterial „map“ and identifies the blood supply and flow dynamics of the tumor. It also provides endovascular access for tumor embolisation [1], [2], [3], [48], [54]. As an invasive imaging procedure, DSA is not free of side affects. Today, it should only be performed when there is the need for pre-operative tumor embilisation. In all other cases, an MRA should be performed [59], [61].
2.2.2 Nuclear medicine
The role of metabolic imaging procedures in the diagnosis of HNPs as well as pheochromocytomas has greatly increased within the last 10 years [13], [57], [62], [63], [64], [65].
The 123I-Metaiodobenzylguanidin-szintigraphy (MIBG szintigraphy) has often been used as a metabolic imaging tool in the diagnosis of neuroendocrine tumors [62], [63], [64]. The tracer needed, [123I]MIBG, is readily availeable in all nuclear medicine centers in Germany. MIBG szintigraphy also has some clear disadvantages though. The images are made two and 24 hours after intraveneous application of the tracer which means that patients do have to come in twice. The tracer also interferes with many common drugs such as heart medication, blood pressure medication and antidepressants. Moreover, [123I]MIBG is often accumulated in the salivary glands which complicates proper diagnosis [57]. In addition, sensitivity is not very high [57].
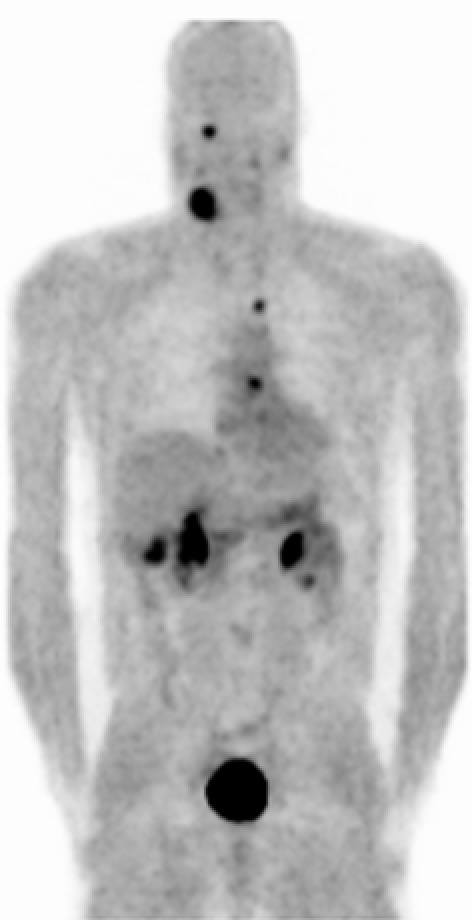
At the University of Freiburg Medical Center the 18F-Fluorodihydroxyphenylalanin positron emission tomography (18F-DOPA-PET) has been used for more than ten years in the diagnosis of paraganglial tumors. Hoegerle et al. could demonstrate as early as 2002 that the 18F-DOPA-PET is an excellent tool for the detection of pheochromocytomas which is superior to MIBG szintigraphy [13]. The following year, the same could be shown for HNPs [57]. 18F-DOPA-PET is capable to detect even very small paraganglial tumors and metastatic disease. The tracer is not accumulated in the head and neck. With a single procedure, paraganglial tumors in the head and neck, the thorax as well as the abdomen may be detected (Figure 3 (Fig. 3)). On the downhand side, the tracer is only available at selected nuclear medicine centers [57].
Figure 3. 18F-DOPA-PET presenting a right CBT, a right jugular paraganglioma, a remnant of a left CBT as well as pheochromocytomas in the left atrium and in projection to the aortic arch. This 41 year old male SDHD patient had already undergone surgery for bilateral abdominal pheochromocytomas. Physiologic tracer uptake is seen in the gallbladder, the renal pelvis and the urinary bladder.
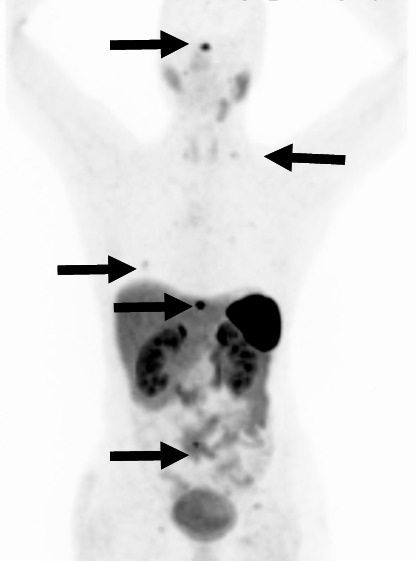
Other nuclear medicine procedures frequently used for the diagnosis of paraganglial tumors include 18F-fluorodopamin-PET (18F-FDA-PET), 18F-fluoro-2-deoxy-D-glucose-PET (18F-FDG-PET) [64] and the 68Ga-DOTA-D-Phe1-Tyr3-octreotate-PET (68Ga-DOTATATE-PET) (Figure 4 (Fig. 4)) [66], [67].
Figure 4. 68Ga-DOTATATE-PET with multiple bone metastases in the spine, the rips and at the skull base (marked by arrows). Physiologic tracer uptake in the liver, the kidneys, the adrenals, the stomach, pancreas, spleen and the small bowel. This 41 year old female SDHB mutation carrier had first undergone resection of a malignant carotid body tumor with regional lymph node metastases seven years earlier.
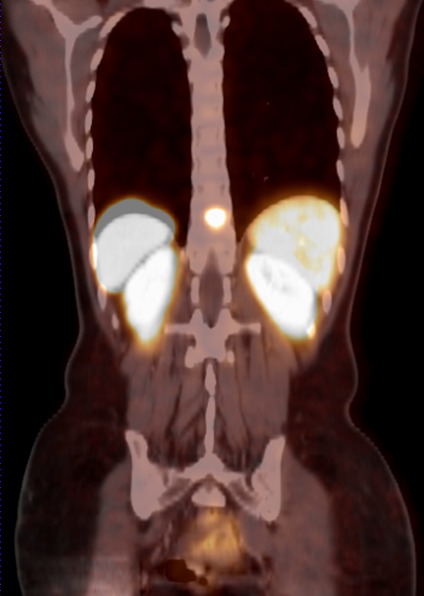
The PET examinations mentioned are often combined with a CT scan. Those fusions between PET and CT (Figure 5 (Fig. 5)) lead to an increased specifity and sensitifity [65]. Luster et al. recently examined 25 paraganglioma and pheochromocytoma patients with 18F-DOPA-PET/CT fusions. Sensitivity in this study was 100%, specifity was 88% with a positive predictive value of 100% and a negative predictive value of 88% [65].
Figure 5. Frontal fusion between computed tomography and PET affirms the spinal metastasis in the SDHB mutation carrier from Figure 4.
2.2.3 Histopathology and immunhistochemistry:
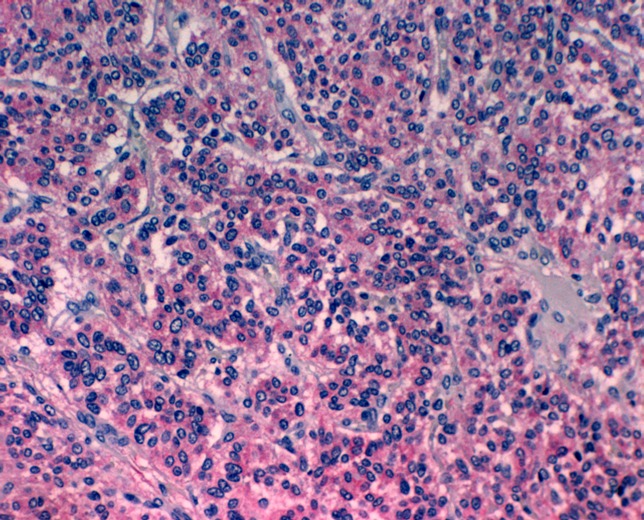
HNPs as well as pheochromocytomas are basically composed of two cell types: chief cells and sustentacular cells [6], [68], [69], [70], [71]. The histological aspect of the tumor is the typical „Zellballen“ pattern where nests or cords of chief cells are surrounded by sustentacular cells (Figure 6 (Fig. 6)). Paraganglial tumors derive from the chief cells and are benign neoplasms in the majority of cases [4], [20], [72], [73]. Overall less than 10% of HNPs are cited to be malignant [1], [3], [6], [48]. Interestingly, there are no histopathological or immunohistochemical criteria for the diagnosis of a malignant paraganglial tumor. Therefore, a malignant HNP or pheochromocytoma may only be diagnosed when there is metastasis to non neuroendocrine tissue [1], [3], [4], [6], [18], [48]. In HNPs, metastases are most frequently found in cervical lymph nodes. Distant metastases are most common in bone, liver and lung [44], [74], [75], [76], [77], [78], [79].
Figure 6. The hematoxiline-eosine staining reveals chief cells in a classical “Zellballen” pattern surrounded by ectatic blood vessels and sustentacular cells (original magnification x100).
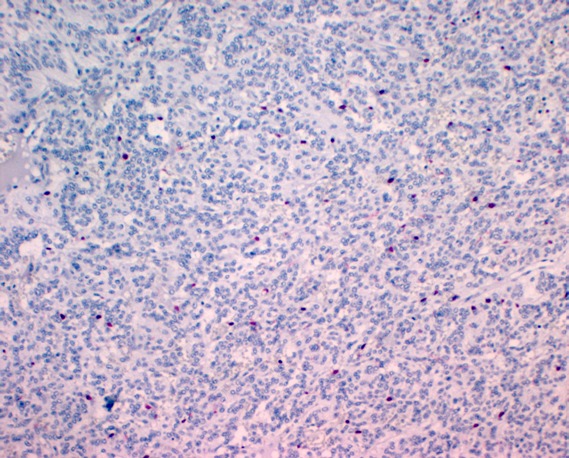
Immunohistochemically, chief cells express neuroendocrine markers such as CD56, synaptophsin, chromogranin (Figure 7 (Fig. 7)), and neuron specific enolase (Figure 8 (Fig. 8)) [68], [69], [74], [80]. Sustentacular cells are positive for the S-100 protein (Figure 9 (Fig. 9)) [68], [69], [74], [81].
Figure 7. On immunhistochemistry the chief cells are positive for chromogranin (original magnification x200).
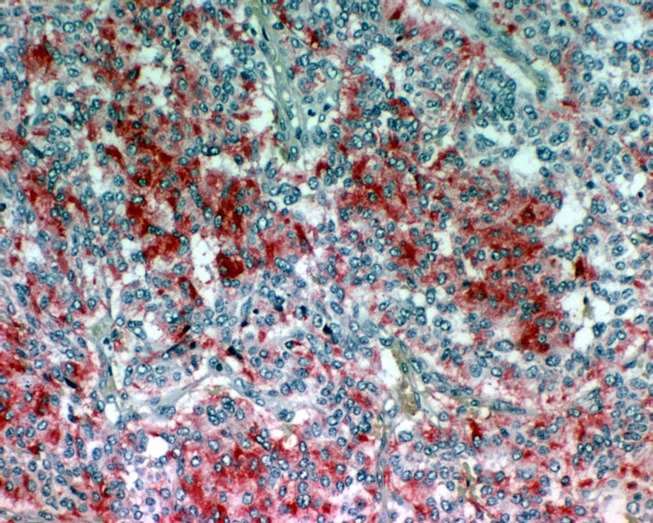
Figure 8. Positive staining of the chief cells for NSE (original magnification x100).
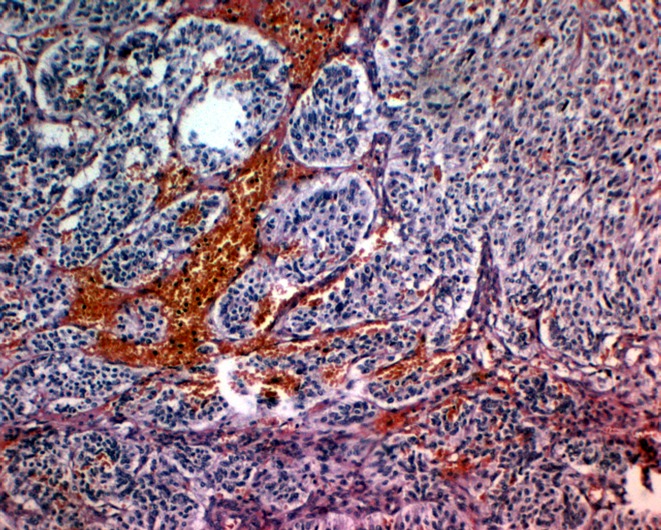
Figure 9. S-100 positive sustentacular cells surrounding the chief cells (original magnification x100).
Whether fine needle aspiration biopsy (FNA) should be used as a means of confirming a clinical diagnosis of HNP is controversial. Monatabi and coworkers report on their results regarding FNA in 13 patients with CBT [82]. The authors state that the correct diagnosis of a CBT with FNA is „extremely difficult“. Nevertheless they regard FNA as an important tool for the pre-therapeutic diagnosis of HNPs. On the other hand, aspirates from CBTs have been mistaken for many different neoplasms including neurofibroma, neurofibrosarcoma, malignant melanoma, different forms of thyroid cancer as well as metastases from a carcinoma [68]. In addition to the questionable diagnostic value of a FNA in HNPs one has to consider potential side effects such as heavy bleeding [68].
We think, that FNA does not deserve any role in pre-operative diagnosis of HNPs. It is a potentially dangerous examination with a very low sensitivity and specifity.
2.3 Differential diagnosis
Cervical HNPs often become symptomatic by means of a painless, slowly enlarging lateral neck mass [1], [4], [21], [36], [45], [48]. The differential diagnosis of a non-tender lateral neck mass includes lymphadenopathies, branchial cleft cysts, salivary gland tumors, neurogenic tumors, aneurysms of the carotid artery as well as HNPs [36], [45]. Differentiation between a HNP and the other masses mentioned can usually easily be made by means of the radiologic as well as functional imaging modalities mentioned above.
Jugulotympanic HNPs often present with pulsatile tinnitus [2], [3], [46], [49], [50]. Gehrking et al. reviewed possible causes of pulse synchronous tinnitus [50]. Jahnke and Klingbeil reported on a patient with pulsatile tinnitus caused by an intratympanic encephalocele [83]. Schikschneit and coworkers added an amputation neuroma of the middle ear as another unusual cause for pulsatile tinnitus [84].
Table 2 (Tab. 2) features possible causes for the symptom „pulsatile tinnitus“.
Table 2. Differential diagnosis of pulsatile tinnitus (modified according to Gehrking [50]).

Differentiation between a jugulotympanic PG and the other causes mentioned can be obtained by a thorough examination of the ear in combination with the radiologic examinations above.
2.4 Therapy
The management of HNPs remains controversial [1], [3], [4], [37], [48], [85], [86], [87]. The current treatment options include surgical resection [1], [2], [3], [19], [48], [59], [88], [89], conventional radiation therapy [47], [90], [91], [92], stereotactic radiosurgery (for jugulotympanic PGs only) [85], [86], [93], permanent embolisation [20], [37] and a combination of those modalities.
Due to the fact that HNPs are usually slow-growing tumors it may be appropriate under certain circumstances to withhold any kind of invasive therapy and to observe tumor growth with serial MRI studies („wait and scan“) [1], [2], [3], [45].
The tentative diagnosis of a HNP has to be made pre-operatively in any case, so that the surgeon does not face the tumor unprepared. Matthews wrote all the way back in the year 1915: “...this rare tumor presents with unusual difficulties to the surgeon, and should one encounter it without having suspected the diagnosis, the experience will not soon be forgotten“ [94].
2.4.1 Therapy of cervical paragangliomas
Therapeutic options for the treatment of cervical HNPs include complete surgical resection [1], [2], [3], [4], [19], [48], [88], [95], conventional radiotherapy [47], [90], [91], [92] and permanent embolisation [20], [37]. Due to generally slow tumor growth, a “wait and scan” policy might be justified in certain cases [1], [2], [3], [45].
In every case, benefits and potential risks of all the treatment options mentioned have to be taken into consideration for every individual patient. Factors that play an important role in the search for optimal therapy include size, classification and exact localisation of the tumor(s), age and general health of the patient as well as pre-therapeutic cranial nerve deficits [1], [3], [48].
Surgical resection
Complete surgical resection represents the only curative treatment option for HNPs [1], [4], [19], [49], [88]. The first successful resection of a CBT dates back to 1889 (cited in [36]). Before the development of vascular surgical reconstructive techniques in the 1960s and 1970s the results of sugical resection were miserable [37]. Mortality rates of 5-30 % have been reported in those days (cited in [37]). Today, complete surgical resection of cervical HNPs is considered the treatment of choice for the vast majority of patients by most authors around the world [1], [2], [3], [4], [19], [48], [56], [88], [89], [95], [96], [97], [98].
The resection of CBTs is performed via a transcervical approach [36], [59], [95], [98]. Special care is taken to avoid injury to the cranial nerves. Proximal and distal control of the large vessels is then optained [36], [59]. Especially in Shamblin class III tumors, there might be injury to the common or internal carotid artery. Therefore the surgeon should be familiar with the use of intraluminal vascular shunts as well as vascular reconstructive techniques [59].
When we resect a cervical HNP, we also perform a selective neck dissection in the regions IIA, IIB and III [99] to exclude or varify lymph node metastases [1], [59].
Cure rates after complete surgical resection of CBTs are generally reported to be as high as 89-100% [1], [19], [47], [88], [95]. The major morbidity associated with surgery is related to post-operative cranial nerve dysfunction [1], [3], [17], [48], [100]. Patetsios et al. described their experiences with the resection of CBTs. Permanent post-operative cranial nerve deficits were seen in five out of 29 patients (17.2%) [48]. Wang and coworkers detected cranial nerve deficits in seven out of 29 patients (24.1%) after tumor resection [36]. Anand and coworkers reviewed the post-surgical results of 1181 patients with CBTs. Permanent cranial nerve deficits were seen in 21.8% of patients [100].
In patients with vagal PG, the affected vagal nerve usually has to be sacrificed during surgery [3], [19].
Primary radiation
The aim of conventional radiotherapy in HNPs is to achieve long term tumor control. One has to keep in mind that radiotherapy of HNPs is not a curative treatment option [47], [49].
Histopathologic studies could demonstrate that radiation therapy does not have any effect on the chief cells but leads to structural changes of the vascular connective tissue [19], [48], [91].
Radiation of cervical HNPs is reported to achieve long term tumor control in up to 96% of cases [47]. The dose commonly recommended is 45 to 56 Gy. When doses of less than 40 Gy are applied, failure rates have been shown to be as high as 22% [101].
Permanent cranial nerve deficits seem to be less common after radiation therapy of cervical HNPs when compared to surgical resection [3], [47], [86]. Nevertheless, the potential risk for radiation induced malignancies has to be taken into serious account not only in young patients [86], [102]. This potential risk has been estimated to be as high as 3-5% [102].
In our opinion, radiation therapy of cervical HNPs should be considered as a treatment option when a tumor is clearly not resectable without reasonable risk, when the patient is in poor health or does not want any surgical procedure or in the setting of multiple HNPs [1], [3].
Alternative therapeutic options
Due to slow tumor growth, a “wait and scan” policy may be an option in selected cases [1], [3], [45], [59]. Tumor doubling time has been estimated to be between 4.2 and 13.8 years for HNPs [3], [86].
Persky and coworkers suggest a “wait and scan” policy in old patients with multiple medical problems [3]. Westerband et al. report on an asymptomatic 77 year old patient with a small CBT (diameter of 0.9 x 0.5 cm). Due to the very small size, the authors decided to follow up the patient in yearly intervals.
A so called permanent embolisation may have the potential to slow down tumor growth for a short period of time [20], [37]. In our opinion, this option should only be considered in a clearly palliative setting.
2.4.2 Therapy of jugulotympanic paragangliomas
The different treatment options have been a matter of discussion in international medical literature when it comes to treatment of jugulotympanic PGs. The current treatment options include surgical resection [2], [19], [46], [59], [103], [104], [105], [106], [107], conventional radiation therapy [47], [90], [91], [92], stereotactic radiosurgery [85], [86], [93], permanent embolisation [20], [37] and a “wait and scan” policy [3], [59].
As in patients with cervical HNPs, benefits and potential risks of all the treatment options mentioned have to be taken into account for every individual patient. Factors that play an important role in the search for optimal therapy include size, classification and exact localisation of the tumor(s), age and general health of the patient and, especially in patients with jugular PGs, pre-therapeutic cranial nerve deficits [1], [3], [48].
Surgical resection
The first (partial) resection of a jugular paraganglioma was described by Rosenwasser in 1945 [cited in 49]. Until the development of microsurgical techniques, new approaches to the skull base as well as modern interventional neuroradiology is was not possible to completely resect jugular PGs [49]. Potentially life-threatening vascular complications were frequently seen, the rate of post-operative cranial nerve deficits was extremely high and, due to incomplete resection, patients faced recurrent tumor growth in the vast majority of cases [49].
Today local tumor control is achieved even in extensive jugular PGs in 80-90% in most surgical series [3], [17], [108], [109], [110], [111].
Compared to all other treatment options, surgical resection bears two major advantages. First of all, it is the only curative treatment option [1], [3], [59]. In addition, it is the only way to obtain material for histopathologic examination, since taking an open biopsy as well as FNA is obsolete in those neoplasms [59].
For patients with tympanic PGs (Fisch class A and B) complete surgical resection clearly represents the therapy of choice whenever there are no contraindications against a general anesthesia [3], [17], [46], [49], [59], [112]. Those tumors become symptomatic by means of pulsatile tinnitus and conductive hearing loss at an early stage in the majority of cases. Complete surgical resection can be optained in almost every case with a low risk for damaging of the lower cranial nerves [46], [49], [59], [112]. Schick and coworkers report on their results in surgical resection of tympanic PGs [49]. Ten tympanic PGs were resected in that study. There were no post-operative cranial nerve deficits and the rate of tumor recurrance was 0% [49]. Gstoettner et al. did not detect any cranial nerve deficits after resection of six class B tympanic PGs [46].
When it comes to optimal treatment of jugular PGs the discussion in international medical literature is much more controversial [3], [4], [47], [49], [90]. Fisch class C and D jugular PGs are resected via an infratemporal approach in the majority of cases. In classes C1, C2 und De,i1/2 the juxtacondylar approach represents an alternative. An intra-operative neuromonitoring of the cranial nerves VII, IX, X, XI and XII has been shown to be of benefit in patients with class C and D jugular PGs [59]. When it comes to class Di3 tumors, the attempt of a surgical resection does not make any sense. Those patients should be send for palliative radiation therapy [59].
The downside of surgical resection of jugular PGs is the large percentage of post-operative cranial nerve deficits. As jugular PGs are intimately involved with the lower cranial nerves, patients frequently suffer from post-operative facial palsy as well as voice, swallowing, articulation and shoulder weakness problems [3], [19], [49], [108], [109], [110].
Persky et al. detected post-operative cranial nerve deficits in 10 out of 14 patients with jugular PGs (71.4%) [3]. Schick et al. described new cranial nerve deficits after resection of 32 class C and D jugular PGs as often as: N. VII in 37.9%, N. IX in 31%, N. X in 45.8%, N. XI in 7.1% and N. XII in 36% of cases [49]. Additional surgical procedures such as vocal cord medialization and others were required in 15 out of 32 patients in this study (46.9%) [49].
Due to the fact that older patients find it more difficult to cope with post-operative cranial nerve deficits, Glasscock and coworkers suggested a maximum age of 60 years for the surgical resection of jugular PGs [113]. In our opinion though, the age of a patient alone should not be the only reason whether surgical resection should be suggested to the patient or not.
Primary radiation
One advantage of conventional radiation therapy has been said to avoid the morbidity of surgery while offering a high probability of local tumor control [47], [90], [91], [92]. Powell and coworkers achieved a local tumor control rate of 90% after radiation of 46 patients with jugulotympanic PGs. Median follow-up in this study was nine years [92]. Chino et al. reported on long-term follow-up in a group of 31 patients with jugulotympanic PGs. Local control rates 5 years, 10 years and 15 years after surgery where 96%, 90% and 90% respectively [114]. Boyle even reported a tumor control rate of 100%. Unfortunately, there are only nine patients in this study and follow-up time was very heterogenous, ranging from one to 12 years [90].
Lower cranial nerve deficits after radiation therapy do occur less frequently after radiation compared to surgery [3], [47], [86]. In the study by Powell et al. a facial nerve palsy was detected in 2/46 patients (4.7%) after radiation of a jugulotympanic PG [92]. In a group of 53 patients reported by Hinerman and coworkers, there was a facial palsy in just one patient (1.9%) [47].
Nevertheless, a number of serious complications have been reported after radiation of jugular PGs including temporal bone osteomyelitis, necrosis of the temporal lobe as well as insufficiency of the pituitary gland [47], [49], [86]. More frequently seen but less dramatic side affects of temporal bone radiation include chronic otitis media, chronic stenosis of the external ear canal as well as chronic problems in the maxillary joint. Those kind of „minor“ complications were seen in 5/53 patients (9.4%) reported by Hinerman [47].
In addition to the fact that radiation therapy of jugulotympanic PGs represents no curative but only a palliative treatment option, the potential risk for radiation induced malignancies has to be taken into serious account not only in young patients [86], [102].
In addition, surgical resection is more difficult in cases where local tumor control is not achieved by radiation therapy [49].
Stereotactic radiosurgery
Stereotactic radiosurgery („gamma knife“) represents a rather new treatment option for patients with jugulotympanic PGs [85], [86], [93]. In contrast to conventional radiotherapy, the whole radiation dose of 12-18 Gy may be applied within a single day [85]. Foote et al. report on their experiences with stereotactic radiosurgery in the treatment of 25 patients with jugulotympanic PGs [86]. During a median follow-up time of 37 months the authors did not detect any tumor growth in those patients. Addiotionally, there were no cranial nerve deficits due to therapy. Feigenberg and coworkers on the other hand report on two out of five patients that presented with growing jugulotympanic paragangliomas after stereotactic radiosurgery [85]. One patient in that study also developed a deficit of the trigeminal nerve.
Concerning stereotactic radiosurgery, we agree with Hinerman that larger studies with a longer follow up are needed to judge the oppertunities of this treatment option in the therapy of jugulotympanic paragangliomas [93].
Alternative therapeutic options
As in patients with cervical PGs, tumor growth is also slow in the majority of jugulotympanic PGs [49], [59]. There are reports of patients in the literature that survived without any kind of treatment for more than 40 years [115].
A „wait and scan“ policy is always to be considered in elderly patients with underlying medical conditions or very small tumors as well as in patients with small recurrent tumors after therapy [59].
A permanent embolisation only has a palliative character [20], [37].
In the last few years, there have been promising results of heavy ion therapy in the treatment of benign and malignant neoplasms of the skull base [116], [117], [118]. Heavy ion therapy is a treatment option that may play a future role in patients with recurrent and extensive jugular PGs.
2.4.3 Recommendations for therapy
Our therapy recommendations for patients with HNPs are listed in Table 3 (Tab. 3).
Table 3. Recommendations for therapy of head and neck paragangliomas.

In our opinion, the majority of patients with HNPs should undergo complete tumor resection. One has to keep in mind, that complete resection represent the only curative treatment option for those unusual neuroendocrine neoplasms.
The recommendations in Table 3 (Tab. 3) are only valid for patients that may be operated in general anesthesia without any major risks. Especially in large vagal PGs, class III CBTs as well as class C and D jugular PGs a number of additional factors have to be considered when therapy is planned on an individual basis for every patient. Those factors include the age and general health of the patient, size, class and localisation of the tumor(s) as well as pre-operative cranial nerve deficits.
2.4.4 Therapy of multifocal paragangliomas
Patients with multifocal HNPs represent a special challenge [1], [18], [20], [59], [119], [120]. A one step surgery in subjects with bilateral HNPs is usually obsolete since it is bearing the risk of bilateral cranial nerve palsies resulting in severe disabilities [1], [18], [20], [59], [119], [120]. Velegrakis et al. recommend to always resect the largest tumor first. The remaining HNP(s) can be observed or, if needed, treated by radiation therapy or permanent embolisation [119]. Sobol and coworkers think that a one step surgical procedure may be an option in patients with multiple unilateral HNPs. They report on a 27 year old patient with a CBT and a vagal PG on the right side. During the resection of the vagal PG, the vagal nerve had to be sacrificed. The class I ipsilateral CBT could be resected without problems during the same surgery [18].
Based on our experiences in the treatment of multifocal HNPs we recommend to resect the largest tumor first whenever possible [20], [59], [120]. Depending on the post-operative cranial nerve status, therapy of the remaining tumor(s) may be individually planned. In the case of a 45 year old patient with bilateral jugular PGs, we could completely resect both tumors without the occurance of cranial nerve deficits within one year [120]. The same was true for a 34 year old patient with bilateral CBTs [1]. A 38 year old patient presented with a palsy of the vagal and hypoglossal nerve after resection of a class III CBT. The two remaining contralateral HNPs therefore underwent radiation therapy [20].
Patients with bilateral vagal PGs are somewhat of a special issue. Since it is usually not possible to resect those tumors without sacrificing the vagal nerve, we suggest resection of the larger tumor followed be radiation of the remaining paraganglioma.
2.4.5 Follow-up
No matter what kind of therapy has been applied, patients with HNPs need long time follow-up. Forrest et al. recommend yearly intervals within the first five years after therapy. Thereafter intervals may be stretched to three to five years [112]. At our department, patients are seen every six months during the first five years. After that, patients come in once a year. All patients undergo a thorough examination of the head and neck during those follow-ups. In patients with cervical HNPs be perform a bilateral B-mode sonography with color-coded duplex sonography. Whenever needed, an MRA is performed in addition. Patients with jugulotympanic HNPs undergo an MRI and an MRA.
Especially patients with jugular PGs need life long follow-up. Recurrent tumors have been seen in those patients decades after initial treatment.
2.5 Preoperative embolisation
The primary goals of pre-operative embolisation of HNPs are to reduce blood loss in the surgical field, minimize the risk of intraoperative complications, prevent recurrence by contributing to complete resection as well as minimizing the need for blood transfusions [4], [37], [121].
Since embolisation is an invasive and potentially dangerous procedure, the risk of potential complications from embolisation always have to be weighed against the advantages. An experienced vascular neuroradiology team has to be absolutely familiar with the complexities and possible variations in head and neck vascular anatomy. At the Department of Neuroradiology of Freiburg Medical School different polyvinyl alcohol (PVA) particles with diameters between 50 and 600 µm are routinely used for embolisation of HNPs. Occasionally, coils may be useful in addition. Proper timing of pre-operative embolisation is needed. In our opinion, embolisation should be performed between two and four days before surgery [1], [4].
Possible minor side effects of embolisation include fever as well as local pain and hematomas in the groin. Severe side effects include skin necrosis, blindness, cranial nerve deficits and a large number of neurological deficits including unilateral palsy and death [3], [20], [36], [122], [123], [124]. Miller et al. did not observe any neurological deficits after embolisation of five vagal PGs [17]. The same was true for a group of six HNP patients reported by Frühwirt and coworkers [37]. Persky and coworkers report on 47 patients with 53 HNPs. All tumors in that study underwent pre-operative embolisation. Complications were seen in six patients (12.8%). Those complications included two vagal verve palsies, one jugular foramen syndrome, one facial nerve palsy, one temporary unilateral palsy and one asymptomatic dissection of the vertebral artery [3].
Pre-operative embolisation of cervical paragangliomas
The embolisation of a CBT was first described by Schick in 1980 [125]. The arterial supply of CBTs and vagal PGs typically derives from the external carotid artery. Branches from the ascending pharyngeal artery, the posterior auricular artery as well as the occipital artery usually form the main arterial feeding vessels [37]. It is of great importance to be aware that there may be a great variety of blood supply in cervical HNPs though [3]. The question, whether cervical HNPs should be routinely embolised is a matter of controversial discussion [1], [4], [36], [37], [48], [87], [126], [127]. Many authors are in favor of embolisation of CBTs and vagal PGs [1], [4], [36], [37], [59], [126], [127]. Miller et al. report on a group of 19 patients with vagal PGs. 16 of those tumors were resected and five tumors underwent embolisation before resection. In those five patients intra-operative blood loss was much smaller and time in the operating theatre was shorter. There was also a smaller incidence of post-operative cranial nerve deficits in this group of patients [17]. Wang et al compared a group of 17 CBTs that underwent embolisation with a group of 12 CBTs that were not embolised. The intra-operative blood loss was significantly smaller in the group of CBTs that had undergone embolisation [36]. Based on own experiences with pre-operative embolisation of 28 CBTs, Persky and coworkers also strongly recommend this approach [3].
Pre-operative embolisation of jugulotympanic paragangliomas
The first embolisation of a jugular PG was described by Hekster in 1973 [128]. The arterial tumor supply in jugulotympanic PGs depends on the tumor stage. Class A tympanic PGs are usually supplied via the inferior tympanic artery. Class B tympanic PGs usually get their blood supply via branches of the ascending pharyngeal artery. In class C jugular PGs the main blood supply is via the ascending pharyngeal artery as well as the maxillary, occipital and temporal arteries. Class D jugular PGs usually have a very individual blood supply [59]. It is of great importance to be aware that there may be a great variety of blood supply in patients with jugulotympanic HNPs though.
The pre-operative angiographic embolisation represents the international gold standard in treatment of class C and D jugular PGs [3], [46], [49], [59], [129], [121]. Without embolisation, it would not be possible to completely resect those tumors in the vast majority of cases [46].
Recommendations for pre-operative embolisation of paragangliomas
Our recommendations regarding embolisation of HNPs are summarized in Table 4 (Tab. 4).
Table 4. Recommendations regarding preoperative embolisation.

In class I and II CBTs a pre-operative embolisation is not mandatory. Those patients should be clearly informed about potential benefits and risks of embolisation. We strongly do recommend embolisation in Shamblin class III tumors though.
For vagal paragangliomas the question concerning the need for embolisation may only be answered on an individual basis.
Class A tympanic PGs must not be embolised whereas embolisation may be useful in selected cases of class B tympanic paragangliomas.
Class C and D jugular paragangliomas on the other hand must undergo pre-operative embolisation whenever surgical resection is performed.
2.6 Malignant paragangliomas
Interestingly, there are no histopathological or immunohistochemical criteria for the diagnosis of a malignant paraganglial tumor. Therefore, a malignant HNP or pheochromocytoma may only be diagnosed when there is metastasis to non neuroendocrine tissue [1], [3], [4], [6], [18], [48]. In HNPs, metastases are most frequently found in cervical lymph nodes. Distant metastases are most common in bone, liver and lung [44], [74], [75], [76], [77], [78], [79].
Overall, less than 10% of all HNPs have been cited to be malignant [1], [3], [6], [48]. Malignancy seems to be most common in vagal PGs (16-19%) when compared to CBTs (6%) and jugulotympanic PGs (2-4%) [6].
Malignant HNPs are often reported as single case reports in the literature [44], [52], [74], [75], [77], [78], [130], [131]. Concerning the incidence of malignancy in HNPs, the publication of large groups of PG patients is more interesting [3], [17], [19], [48], [95]. Miller et al. detected three malignant tumors in a group of 19 patients with vagal PGs (15.8%). Two patients presented with regional lymph node metastases, one patient had systemic disease to the sacrum [17]. Patetsios et al. reported on three malignant HNPs in a group of 29 CBT patients (10.3%) [48]. Two patients had cervical lymph node metastases, one presented with intrahepatic metastatic disease. Grotemeyer et al. diagnosed four malignant tumors in a group of 39 HNP patients (10.3%) [95]. In the large series (n=79 HNP patients) reported by Kollert et al. there were no signs of metastatic disease in any patient [19].
The largest study to date dealing with malignant HNPs was published by Lee in 2002 [6]. For this study, the data of the “National Cancer Data Base” from 1985 until 1996 was studied. There were 59 malignant paragangliomas in a group of 355019 patients (0.017%). There were 30 female (50.9%) and 29 male patients (49.1%). Median age at the time of diagnosis was 44 years. 68.7% of patients presented with cervical lymph node metastases, 31.3% of patients had systemic metastases. Five year survival was 59.5%. There was a significant difference between patients with local metastases and patients with systemic disease though. 76.8% of patients with regional lymph node metastases were alive after five years compared to only 11.8% in the group with systemic metastases. Therapy of choice in patients with malignant HNPs is complete tumor resection with resection of all metastases whenever possible [6]. Radiation therapy is a sufficient tool to significantly prolong survival whenever complete resection is not achieved. Median survival time in patients who underwent radiation was 45 months versus 12 months in the group of patients without radiation [6].
Benefits of chemotherapy in the treatment of malignant HNPs are a matter of controversial discussion in the literature. Many authors do not see any benefit for this kind of treatment [6], [44], [74], [76]. Argiris and coworkers reported on their experience with a 33 year old female patient suffering from a CBT with pulmonary and osseous metastases. The patient underwent chemotherapy with cyclophosphamide, adriamycin and dacarbazin leading to partial remission of disease [8]. One has to keep in mind though, that the course of disease is indolent and hard to predict in patients with metastatic HNPs. There are reports of patients with malignant HNPs in the literature who survived for decades without any kind of treatment [132].
There have been promising initial results in the therapy of patients with widespread metastatic disease from paraganglial tumors using radiolabeled somatostatin analogs such as 111In-octreotid, 131I-metaiodobenzylguanidin (131I-MIBG) and 177LU-DOTATATE [133], [134], [135]. Nervertheless, prognosis remains poor for HNP patients with systemic metastases.
Recommendations regarding lymphadenectomy in HNP patients
Cervical lymph node metastases are not an uncommon feature in patients with cervical HNPs. Therefore we perform an ipsilateral selective neck dissection in levels II and III in patients undergoing resection of a cervical HNP. When we resect a jugular paraganglioma we do a lymphadenectomy in the upper region of the ipsilateral jugular vein. In patients with tympanic PGs we usually do not perform any kind of lymph node resection. The risk for lymph node metastases is considered very low for those tumors of the middle ear [4].
2.7 Hereditary paragangliomas and pheochromocytomas
HNPs as well as pheochromocytomas may occur as sporadic or familial entities [136], [137], [138], [139], [140], [141], [142].
It has been well known for decades that familial pheochromocytomas are part of different tumor syndromes [132], [139], [143], [144], [145], [146]. Pheochromocytomas do occur in 30 to 50% of patients with multiple endocrine neoplasia type 2 (MEN2) [132], [146], [147], [148], [149]. 10-20% of patients with von Hippel-Lindau syndrome present with pheochromocytomas [132], [146], [149], [150] and the risk to develop pheochromocytomas is estimated to be 1-5% for patients with neurofibromatosis type 1 (NF 1) and around 1% for subjects with multiple endocrine neoplasia type 1 (MEN1) [146], [149].
The familial clustering of HNPs has also been described in international medical literature for decades. In 1933, Chase first described two sisters who both presented with CBTs [151]. Goekopp mentioned the occurance of jugulotympanic paragangliomas in three sistern the same year [152]. The first description of familial vagal paragangliomas dates back to 1956 [153].
Although so called „paraganglioma syndromes“ (PGL) have been described for many years, they have not been classified based on molecular genetics until the beginning of this century. In 2000, Baysal et al. first identified germline mutations of the succinate dehydrogenase subunit D gene (SDHD gene) as the underlying cause of PGL 1 [139]. PGL 3 is associated with SDHC and PGL 4 with SDHB gene mutations [154], [155]. The genes SDHB, SDHC and SDHD are all tumor suppressor genes [155], [156], [157], [158], [159]. PGL 2 is caused by mutations of the succinate dehydrogenase complex assembly factor 2 gene (SDHAF2 gene) which is also known as SDH5 [160], [161]. PGL 2 has only been characterized on a molecular genetic basis in 2009 [160].
Succinatedehydrogenase, consisting of the subunits SDHA, SDHB, SDHC and SDHD, forms the mitochondrial complex II. It plays an important role in the mitochondrial respiratory chain as well as the tricarboxylic acid cycle [41], [144], [156], [162], [163], [164], [165].
The molecular genetic screening for mutations of the genes SDHB, SDHC and SDHD require 10ml of EDTA anticoagulated blood as well as a written informed consent [9]. The screening for all three genes, SDHB, SDHC and SDHD, costs approximately 2700 US$ per patient. The Dutch group around van Nederveen developed an elegant and cheap immunohistochemical examination for the detection of SDHx mutations. Sensitivity and specifity of the SDHB immunohistochemistry is 100% and 84% respectively [166]. Obviously, this method can only be applied after surgery. It is only possible to predict whether there is an SDHx mutation or not. The method can not tell which kind of SDHx mutation there is.
Table 5 (Tab. 5) shows all tumor syndromes characterized on a molecular genetic basis until October 2010 that are associated with HNPs and/or pheochromocytomas.
Table 5. Tumor syndromes associated with HNPs and pheochromocytomas.

2.7.1 MEN1
Multiple endokrine neoplasia type 1 (MEN1) is an autosomal dominant disease caused by mutations of the MEN1 gene. MEN1 consists of 10 exons and it encodes the 610 amino acid protein menin [174]. Disease penetrance is nearly complete and patients with MEN1 typically develop tumors of the parathyroid glands, the pancreatic islet cells and the anterior pituitary gland. Less prevalent neoplasms associated with MEN1 are carcinoid tumors, facial angiofibromas, lipomas and collagenomas. Pheochromocytomas are only seen in about 1% of patients with MEN1 [146], [149]. HNPs have not been described in association with MEN1 yet.
2.7.2 MEN2
MEN2 is an autosomal dominant syndrome associated with mutations of the RET gene. The RET gene consists of 21 exons [167]. All RET mutations described in the literature to date may be found on http://arup.utah.edu/database/MEN2/MEN2_display.php?sort=1. There are a total of three subtypes. The most common subtype, MEN2A, is characterized by medullary thyroid carcinomas (MTC), pheochromocytomas and parathyroid neoplasia. Patients with MEN2B present with early onset aggressive MTC, pheochromocytomas and characteristic physical features such as a marfanoid habitus as well as neurinomas of the tongue, conjunctivas and the colon. FMTC (familial medullary thyroid cancer) is the third subtype in which MTC is the sole manifestation [167], [168].
About 30 to 50% of MEN2 patients develop pheochromocytomas [132], [146], [147], [148], [149]. HNPs have only been described in the three subjects with MEN2 so far [20], [169], [170].
2.7.3 NF1
Neurofibromatosis type 1 (NF1) is an autosomal dominant tumor syndrome caused by mutations of the NF1 gene [171]. The NF1 gene, consisting of 57 exons, is one of the largest human genes. The disease phenotype is dominated by multiple neurofibromas of the skin, café-au-lait spots, axillary freckling and so called Lisch nodules of the iris. NF1 patients are also at risk for the development of multiple tumors deriving from nerve tissue and endocrine tissues. 1-5% of NF1 mutations carriers present with pheochromocytomas [132], [146], [149], [172], [171]. A HNP in association with NF1 has only been described in one caes yet [173].
2.7.4 VHL
Von Hippel-Lindau syndrome (VHL) is a multisystemic autosomal dominant disease due to mutations of the VHL gene [169], [174], [175], [176], [177]. The VHL gene consists of three exons. The encoded protein has got 213 amino acids. All VHL mutations described up to date may be found at http://www.umd.be/VHL/. VHL displays full penetrance by age 65 although the individual disease phenotype is highly variable. The tumors most commonly seen in VHL mutation carriers are hemangioblastomas, retinal angiomas, renal cell cysts and renal cell carcinomas, pancreatic cysts as well as pheochromocytomas [174]. Occasionally endolymphatic sac tumors (ELST), among others, may also be observed [178].
Between 10 and 20% of all VHL patients present with pheochromocytomas [132], [146], [149], [150]. The fact, that VHL mutation carriers are also at risk for the development of HNPs has found little attention in international medical literature for many years. Until the year 2009 there were only five single case reports of patients with a HNP and VHL syndrome [179], [180], [181], [182], [183]. In 2009, our group conducted a large international multicenter study including a total of 2084 registered VHL patients. Eleven of those patients (0.53%) had a HNP [169]. In a later work, Gaal et al. could demontrate that HNPs are part of VHL and are due to VHL gene inactivation in those patients [175].
2.7.5 Paraganglioma syndromes (PGL)
Since the description of SDHD gene mutations as the cause of PGL 1 by Baysal et al. in the year 2000 [139], the paraganglioma syndromes (PGL) have attracted a lot of scientific attention.
In the meantime, several large international studies could demonstrate that approximately 30% of all seemingly sporadic HNPs are due to genetic mutations [9], [10], [143], [184], [185], [186], [187]. The large majority of those hereditary HNPs is due to the paraganglioma syndromes 1, 3 and 4. HNPs in association with PGL 2 and VHL are less common [160], [161], [169] and in patients with MEN2 and NF1 head and neck paragangliomas are only very rarely seen [169], [173].
The PGL are characterized by distinct clinical features [9], [10], [143], [184], [185], [186], [187], which will be briefly shown.
PGL 1
Paraganglioma syndrome 1 (PGL 1) is caused by SDHD gene mutations [139]. In the SDHD gene, there are 4 exons and the encoded protein has 160 amino acids. All different SDHD gene mutations described in the literature so far may be found at http://chromium.liacs.nl/lovd_sdh/variants.php?action=search_unique&select_db=SDHD.
PGL 1 is the most common paraganglioma syndrome. SDHD mutation carriers are at risk for the development of both HNPs and pheochromocytomas, although HNPs are much more commonly seen. Pasini and Stratakis reviewed a total of 95 international manuscripts dealing with SDHx mutations [142]. There were a total of 395 SDHD patients in this review. 91% of those patients displayed at least one HNP and 79% even presented with multiple HNPs. Malignant paraganglial tumors are seen on a regular basis in SDHD mutation carriers although malignancy is significantly more common in SDHB patients [142]. Neumann et al. presented the results of a large multicenter study in 2009 [10]. A total of 598 patients with apparently sporadic HNPs were screened for mutations of the genes SDHB, SDHC and SDHD. Mutations were detected in a total of 183 subjects (30.6%). There were 94 SDHD mutations carriers as well as 63 SDHB and 26 SDHC patients, respectively. SDHD patients presented with multiple HNPs in 56 out of 94 cases (59.6%). HNPs and pheochromocytomas were seen in 17 cases (18.1%) whereas malignant paraganglial tumors had to be diagnosed in 9 SDHD mutation carriers (9.6%). The family history was positive in 45 patients which is 47.9% of cases.
Tumor penetrance in SDHD mutation carriers is high. Neumann et al. first published data on age related penetrance in SDHD mutation carriers in 2004 [9]. SDHD mutations conferred 50% penetrance by age 31 rising to 86% by the age of 50 in that study. In a 2010 study by Hensen and coworkers, age related penetrance in SDHD patients was 54% by age 40 and 87% by age 70 [186].
The trait of inheritance in SDHD mutation carriers is autosomal dominant with a „parent-of-origin-dependent-inheritance“ [9], [132], [139], [164], [188]. This means that patients with an SDHD mutation are only at risk for the development of paraganglial tumors if they inherited the mutation from their father. Though not at risk for tumor development themselves, SDHD mutation carriers who inherited the mutation through the maternal line will still pass the mutation to their children in 50% of cases.
PGL 2
PGL 2 is associated with mutations of the SDHAF2 gene [160], [161]. SDHAF2, also known as SDH5, consists of 4 exons. It plays an important role in flavination of succinate dehydrogenase subunit A [161].
In 1982, van Baars et al. described a large Dutch family (295 living family members) with familial HNPs [189]. The PGL in this family has later been referred to as PGL 2 [7], [161]. It was not until the year 2009 though, that Hao et al. identified a mutation of the SDHAF2 gene as the underlying cause of PGL 2 [189].
Bayley and coworkers detected another family with PGL 2, this time in Spain [161]. The authors also looked for SDHAF2 mutations in 443 additional HNPs and pheochromocytoma patients without finding another case of PGL 2.
In both families with PGL 2 only HNPs but no pheochromocytomas have been described [161], [189]. As in PGL 1 there is also a “parent-of-origin-dependent-inheritance” in SDHAF2 mutation carriers [161]. Tumor penetrance has been described to be very high in both the Dutch as well as the Spanish family [161], [189].
PGL 3
PGL 3 is caused by SDHC gene mutations [154]. The SDHC gene consists of 6 exons and encodes a 169 amino acid protein [142]. The SDHC mutations described so far are listed at http://chromium.liacs.nl/lovd_sdh/variants.php?action=search_unique&select_db=SDHC.
Nieman et Müller first identified an SDHC mutation as the underlying cause of PGL 3 [154]. Until October 2005, only four patients with SDHC mutations had been described [154], [159], [190], [191]. Many authors concluded that SDHC mutations must be exceedingly rare and therefore, HNP patients were not routinely tested for mutations of the SDHC gene. This only changed when Schiavi et al. reported on a total of 22 SDHC mutation carriers in October 2005 [184]. Due to the following increased rate of SDHC testing, many SDHC mutations could be detected in the years following 2005. Nevertheless, SDHC mutations are still less common than SDHD and SDHB mutations.
In sharp contrast to patients with PGL 1 and PGL 4, SDHC mutation carriers almost exclusively develop benign, single HNPs [10], [184]. Malignant paragangliomas [159] and pheochromocytomas [192], [193], are only very rarely seen in SDHC patients.
In a 2009 study by Neumann and coworkers, there were 26 subjects with SDHC mutations in a total of 598 HNP patients [10]. There were no pheochromocytomas and no malignant HNPs in that SDHC group of patients. 21 out of 26 patients (80.8%) presented with a single HNP, five patients had two HNPs.
SDHC mutations follow an autosomal dominant trait of inheritance [5], [156], [159], [184]. Penetrance is probably rather low. In the study by Neumann et al., family history has only been positive in three out of 26 SDHC patients (11.5%) [10].
PGL 4
PGL 4 is caused by mutations of the SDHB gene [155]. The SDHB gene consists of 8 exons and the protein encoded has 280 amino acids [142]. The site http://chromium.liacs.nl/lovd_sdh/variants.php?action=search_unique&select_db=SDHB lists all SDHB mutations that have been published so far.
The first description of the SDHB mutation as the cause of PGL 4 dates back to 2001 [155].
Patients with PGL 4 frequently develop HNPs and pheochromocytomas. In contrast to SDHD mutation carriers, patients with SDHB mutations develop pheochromocytomas significantly more often than HNPs [142], [185], [193], [194]. Multiple HNPs are significantly less frequent when compared to SDHD patients [10], [142].
The most striking clinical feature of PGL 4 is the very high percentage of malignant pheochromocytomas and malignant HNPs [9], [10], [137], [142], [185]. In the study by Neumann et al. there were malignant paraganglial tumors in 13 out of 63 SDHB mutation carriers (20.6%) [10]. Ricketts et al. reported on 40 SDHB patients with malignant paragangliomas and pheochromocytomas in a group of 163 SDHB mutation carriers (25.2%) [185]. In a literature review by Pasini and Stratakis, malignant paraganglial tumors were diagnosed in 105 out of 256 SDHB mutation carriers (41%) [142]. In addition, PGL 4 patients are at an incresed risk for the development of kidney cancers. In 2004, Vanharanta described two SDHB patients who developed malignant kidney tumors at age 24 and 26 [195]. Ricketts et al. estimate the risk for subjects with PGL 4 to develop a malignant renal tumor by age 70 to be as high as 14%.
SDHB mutations follow an autosomal dominant trait of inheritance [5], [9], [10], [155]. Tumor penetrance is somewhat lower than that seen in SDHD mutation carriers. Recent studies estimated the age related tumor penetrance in SDHB mutation carriers to be at 29% at age 30 rising to 45% at age 40 [196].
Differences between PGL and sporadic HNPs
Patients with paraganglioma syndromes develop paraganglial tumors at a significantly younger age than patients with sporadic HNPs and sporadic pheochromocytomas [5], [142], [197]. Burnichon et al. compared 242 tumor patients with SDHB, SDHC and SDHD mutations with a group of 203 subjects with sporadic paraganglial tumors [197]. The age at first manifestation of a paraganglial tumor was 36.2 years in the PGL group compared to 50.2 years in the group with sporadic tumors (p<0.0001). Multiple paraganglial tumors were also significantly more frequent in SDHx mutation carriers (n=112 versus n=10; p<0.0001). PGL patients also had a significantly higher risk for the development of malignant paraganglial tumors (n=40 versus n=9; p<0.0001), although this difference was greatly due to the very large percentage of malignant tumors in the SDHB subgroup [197].
Tumor development in PGL
Development of sporadic CBTs has been linked to chronic hypoxic stimulation of the glomus caroticum as it can be seen in patients living at high altitudes and in subjects with chronic obstructive lung disease [36], [42], [43].
The role of SDHB, SDHC and SDHD as tumor suppressor genes has been clearly defined. Mutations in one of the three genes lead to complete loss of function of SDH [142], [158]. Thus inactivation of SDHB, SDHC or SDHD with subsequent loss of SDH function results in a pseudohypoxic state. The resulting activation of hypoxia-inducible factors (HIF) pathways has been linked to accumulation of succinate and resulting inhibition of prolyl hydroxyase enzymes that are needed for proteosomal degradation of HIF-alpha subunits. This is believed to lead towords cell proliferation, angiogenesis and tumorgenesis in paraganglial tissue [185].
2.7.6 Additional genes of interest
The majority of familial HNPs is associated with PGL 1, 3 and 4. Pasini and Stratakis reviewed the studies concerning SDHx mutations and paraganglial tumors that were available in 2009 [142]. 87.8% of familial HNPs described in the literature up to then had been due to mutations in the genes SDHB, SDHC and SDHD. The fact, that a certain percantage of clearly familial HNPs does not seem to be associated with PGL 1, 3 and 4, VHL or PGL 2 makes it very likely that other genes probably play a role in the development of HNPs and pheochromocytomas [142], [157].
In this context, two promising genes of interest have been described in 2010.
SDHA
The SDHA gene consists of 15 exons and encodes a 664 amino acid protein that forms the SDHA subunit of SDH [142], [163]. SDHA mutations follow an autosomal recessive trait of inheritance [163]. Interestingly, as the only subunit of SDH, SDHA mutations had not been linked to paraganglial tumors but to a familial neurologic disorder called the Leigh syndrome [161], [198]. Burnichon and coworkers first described a 32 year old SDHA mutation carrier who developed a pheochromocytoma in 2010 [157]. Right now, it is too early to predict what kind of role SDHA mutations with play in the future of hereditary paraganglial tumors though.
TMEM127
Qin et al. described mutations of the TMEM127 gene in patients with hereditary pheochromocytomas in 2010 [199]. Again, it is not possible yet to predict how many patients with paraganglial tumors really do suffer from a TMEM127 mutation.
2.7.7 Carney triad
In 1977, Carney et al. reported on two patients with sarcomas of the stomach (today referred to as gastrointestinal stromal tumor or GIST), extraadrenal pheochromocytomas and pulmonary chondromas [200]. Since the year 1981, this tumor complex is known as the Carney triad [201]. Patients with the triad are also at risk for the development of paragangliomas and esophageal leiomyomas [200].
In 1999, Carney reviewed the literature and found a total of 79 patients who were diagnosed with the Carney triad. Two of those 79 subjects were later outsourced from this group of patients by Carney and Stratakis and were diagnosed with the Carney-Stratakis syndrome [202][. Interestingly, none of the remaining 77 patients had a positive family history for the Carney triad or even for one of its associated tumors. Moreover, none of the 355 first degree relatives had the triad or even one of its tumors [203], [202]. Therefore the potentially hereditary status of the Carney triad remains at best unclear.
2.7.8 “Carney-Stratakis syndrome”
Carney and Stratakis reported on five families in 2002 presenting with GIST, paragangliomas as well as pheochromocytomas [202]. This association of tumors is also referred to as the „Carney-Stratakis syndrome“ in the literature [142], [204]. In the meantime, it could be demonstrated that the majority of patients who had been diagnosed with „Carney-Stratakis syndrome“ really suffer from mutations in one of the genes SDHB, SDHC or SDHD [142]. Therefore, we do not consider the „Carney-Stratakis syndrome“ to be a real syndrome. It is more likely that patients with PGL 1, 3 and 4 also develop GIST occasionally.
2.7.9 Molecular genetic screening
According to the 2003 policy statement from the American Society of Clinical Oncology for cancer genetic testing all patients who are at a risk of more than 10% to harbour a genetic mutation should undergo molecular genetic testing [205].
After the molecular genetic characterization of the paraganglioma syndromes 1, 3 and 4 in the years 2000 and 2001 there was a lack of international standards regarding the question who should be tested for SDHx mutations. In 2002, Young and coworkers suggested a screening for all HNP and pheochromocytoma patients with a positive family history [132]. Neumann et al. published the molecular genetic screening results of 271 patients with apparently sporadic pheochromocytomas in 2002 [143]. In 24% of cases there was a mutation in one of the genes SDHB, SDHD, VHL and RET. Therefore, Neumann strongly recommended a screening of all pheochromocytoma patients for mutations of the genes SDHB, SDHD, VHL and RET [143]. Based on own results, Gimenez-Roqueplo et al. came to the same conclusion in 2003 [158]. Since then it could be demonstrated that up to 30% of all patients with apparently sporadic HNPs suffer from mutations in the genes SDHB, SDHC and SDHD [9], [10], [142], [184], [194], [196]. The molecular genetic screening of all HNP patients is therefore regarded as international standard by the majority of authors.
On the other hand, screening for intragenetic mutations as well as large deletions in all three genes costs approximately 2700 US$ per patient [10]. In many cases, those expenses are not paid for by health insurance companies. Therefore, an algorithm for a cost effective screening would be very helpful. Neumann et al. screened 598 patients with apparently sporadic HNPs for mutations of the genes SDHB, SDHC and SDHD [10]. All patients were screened for mutations in all three genes. Mutations were detected in 183 subjects (30.6%). There were 94 SDHD, 63 SDHB and 26 SDHC gene mutations, respectively. No patient presented with more than one mutation. In the next step, Neumann et al. looked for clinical predictors that were associated with a high probability of an SDHx mutation. A total of six first step predictors could be identified: positive family history, previous pheochromocytomas, multiple HNPs, manifestation of first tumor before age 40, male gender as well as malignant HNPs. Based on those first step predictors, Neumann and coworkers developed an algorithm (Figure 10 (Fig. 10)). Using the approach of testing only patients with first step predictors would have resulted in a cost reduction of approximately 60% in this group of 598 patients. On the other hand, a total of 15 patients with SDHx mutations would not have been detected [10].
Figure 10. General algorithm for cost-effective mutation screening in patients with head and neck paragangliomas (HNPs) according to Neumann et al. [10].
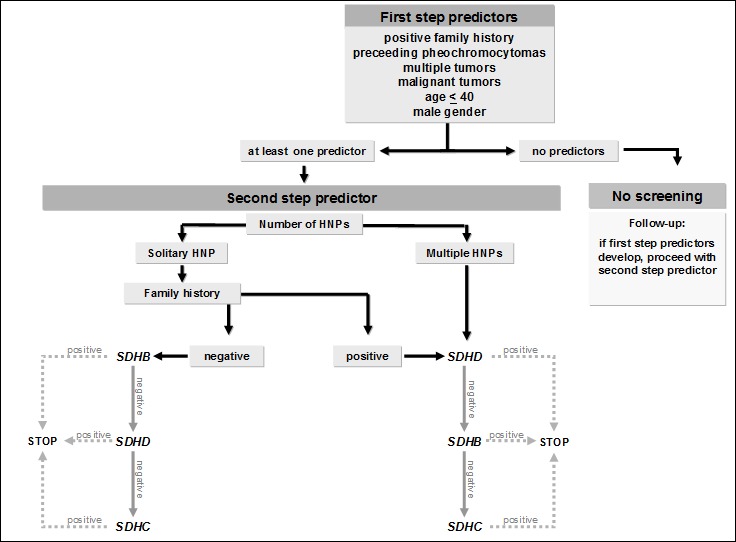
Therefore, we recommend molecular genetic screening of all HNP patients. In order to do this testing in a cost effective way without having the risk of missing a mutation we recommend a step by step approach. Clinical parameters will help to decide which gene should be tested first. Only if this test is negative, the second gene would be testet and so on.
We recommend the following approach for patients with HNPs:
-
multiple HNPs:
1) SDHD 2) SDHB 3) SDHC
-
solitary HNP, positive family history:
1) SDHD 2) SDHB 3) SDHC
-
HNP(s) and pheochromocytoma(s):
1) SDHD 2) SDHB 3) SDHC
-
solitary HNP, negative family history:
1) SDHB 2) SDHD 3) SDHC
-
malignant HNP(s):
1) SDHB 2) SDHD 3) SDHC
This approach should be modified under certain circumstances. If family pedigree confirms a disease transmission from mother to child, showing lack of the parent-of-origin effect seen in SDHD mutations, the SDHB gene should be tested first, followed by the SDHC gene if necessary [10].
A routine screening of HNP patients for mutations of the genes SDHAF2 (PGL 2), VHL (VHL syndrome) und RET (MEN2) is not necessary.
PGL 2 has only been described in two families yet [160], [161]. A screening for SDHAF2 gene mutations would only be useful in an patient with hereditary HNPs in the absence of an SDHB, SDHC and SDHD gene mutation.
HNPs are a part of the VHL tumor syndrome. Nevertheless, they are extremely uncommon compared to hemangioblastomas, retinal angiomas, renal cysts, renal cell cancer, pancreatic cysts and pheochromocytomas [174]. Before developing a HNP, patients with VHL syndrome have usually developed one or multiple of the other neoplasms mentioned [169].
HNPs are exceedingly rare in MEN2 with only three single cases published in the literature [20], [169], [170].
In pheochromocytoma patients without HNPs, the possibility of an RET or VHL mutation have to be kept in mind though. In patients with solitary extraadrenal pheochromocytomas Erlic et al. recommend a screening in the order SDHB, VHL, SDHD and RET [193]. Whenever there is an adrenal pheochromocytoma or in cases with multiple pheochromocytomas the order of choice would be: VHL, RET, SDHB and SDHD [169].
A thorough genetic counselling in combination with molecular genetic screening is essential. Patients and their families have to be familiar of the clinical features of the different hereditary HNP and pheochromocytoma syndromes. Of special interest is the trait of inheritance and here especially the parent-of-origin effect in PGL 1 and PGL 2.
2.7.10 Follow-up for PGL patients
The life-time risk for patients with SDHB and SDHD mutations to develop HNPs and/or pheochromocytomas is very high. Whereas tumor penetrance in SDHB mutation carriers seems to be slightly lower, one has to consider the high risk of PGL 4 patients for the development of malignant paraganglial tumors and malignant renal cell tumors [9], [10], [137], [142], [185], [195].
Those aspects have to be taken into consideration when it comes to follow-up of PGL patients. All mutation carriers should undergo a clinical and radiologic screening after the molecular genetic diagnosis has been established. The only exception is children of female SDHD mutation carriers. In addition to a thorough physical examination the following procedures should be considered a standard program:
MRI with contrast medium of the head and neck, the thorax and the abdomen
Catecholamines or metanephrines in plasma or 24h urine
Additional procedures such as an 18F-DOPA-PET may be of great benefit in selected cases.
In our opinion, patients with SDHC mutations should only undergo MRI of the thorax and abdomen once. Thereafter, MRI follow up of the head and neck region together with a physical examination should be sufficient, since PGL 3 harbours only a very low risk for the development of pheochromocytomas.
SDHD patients who have already developed a paraganglial tumor should undergo the whole standard program once a year. For SDHD patients without any signs of tumor, stretching of intervals to three years seems to be justified.
Due to the high risk for malignant tumors, we suggest the standard examination in SDHB patients once a year. Whenever malignancy is expected, the 18F-DOPA-PET has been an excellent tool in our hands.
SDHC patients with paraganglial tumors should undergo yearly MRI of the head and neck as well as a thorough physical examination. In SDHC patients who have not developed any tumor yet, an MRI of the head and neck together with a physical exam every three to five years should be sufficient.
The percentage of SDHB and SDHD patients who develop a tumor during the first two decades of life is rather high. In the study by Burnichon et al., 21 out of 242 (8.7%) SDHB and SDHD mutation carriers developed a tumor before age 18 [197]. The authors therefore suggest to start examinations in SDHB and SDHD patients at age 6.
Whenever a paraganglial tumor is detected in a PGL patient an individual therapy regimen should be developed. When it comes to HNPs, our general therapy suggestions also apply to PGL patients. Pheochromocytomas of the thorax should be completely resected whenever possible. The same is true for adrenal and extraadrenal abdominal pheochromocytomas. In recent years, a cortical sparing laparoscopic partial adrenalectomy has become the gold standard of surgery [206], [207], [208]. Neumann and coworkers could demonstrate that even in cases of bilateral adrenal tumors enough tissue can be left in place to avoid the need for post-operative hormone therapy. The risk for local recurrences has been shown to be very low for this approach [206], [207].
3. Conclusion
Head and neck paragangliomas (HNPs) are rare, neuroendocrine neoplasms with a strong vascularisation. They are most commonly found as carotid body tumors. Jugulotympanic and especially vagal paragangliomas are less common. Paraganglial tumors in the thorax and the abdomen are referred to as pheochromocytomas.
Post-operative deficits of the lower cranial nerves are the main risk of morbidity after surgical resection. Nevertheless, we recommend surgery as the therapy of choice for the majority of all HNPs since complete tumor resection represents the only curative treatment option. Depending on tumor size, number and localisation, the patients age and general status and pretherapeutic cranial nerve status other treatment options always have to be taken into account. Those alternative options include conventional radiotherapy, stereotactic radiosurgery and a “wait and scan” policy.
Shamblin III CBTs as well as Fisch C and Fisch D jugulotympanic paragangliomas should always undergo pre-operative embolisation whenever a resection is planned. Fisch A jugulotympanic paragangliomas must not be embolised. In all other cases, the need for embolisation should be individually decided.
Both HNPs and pheochromocytomas may occur as sporadic as well as hereditary entities. Up to 30% of apparently sporadic HNPs are associated with different tumor syndroms. Approximately 90% of all hereditary HNPs are related to the paraganglioma syndromes (PGL) 1, 3 and 4. PGL 1 is associated with mutations in the gene encoding succinate dehydrogenase subunit D (SDHD), whereas PGL 3 ist due to SDHC and PGL 4 to SDHB gene mutations. Trait of transmission is generally autosomal dominant for all PGL. In PGL 1 there is a so called “parent-of-origin-dependant-inheritance”. This means that mutation carriers only suffer an increased risk for tumor development if the mutation has been transmitted through the paternal line.
Patients with PGL 1, 3 and 4 develop paraganglial tumors at a significantly younger age when compared to patients with sporadic HNPs and pheochromocytomas. SDHC patients develop benign, single HNPs in the great majority of cases. SDHD mutation carriers frequently suffer from multiple HNPs, whereas SDHB patients do have a significantly increased risk for the development of malignant paraganglial tumors. Malignant renal cell tumors are also detected in SDHB patients on a regular basis.
We recommend a molecular genetic screening of all HNP patients for mutations of the genes SDHB, SDHC and SDHD. The order of testing is performed according to individual clinical features. Whenever a mutation is detected, the affected patient should undergo a thorough clinical examination, an MRI with contrast medium of the head and neck, the thorax and the abdomen as well as catecholamines or metanephrines in plasma or 24h urine.
The identification of patients with PGL will, in addition with follow up examinations, lead to early detection of HNPs and pheochromocytomas in the future. In turn, carriers identified early in the course of their disease can be brought to surgical attention before their disease becomes extensive and potentially life-threatening.
References
- 1.Boedeker CC, Ridder GJ, Neumann HPH, Maier W, Schipper J. Diagnostik, Therapie und Behandlungsergebnisse zervikaler Paragangliome. [Diagnosis and management of cervical paragangliomas: the Freiburg experience]. Laryngo-Rhino-Otol. 2004;83:585–592. doi: 10.1055/s-2004-814466. (Ger). Available from: http://dx.doi.org/10.1055/s-2004-814466. [DOI] [PubMed] [Google Scholar]
- 2.Schipper J, Boedeker CC, Maier W, Neumann HPH. Paragangliome im Kopf-/Halsbereich Teil 1: Systematik und Diagnostik. [Paragangliomas in the head-/neck region. I: Classification and diagnosis]. HNO. 2004;52:569–575. doi: 10.1007/s00106-003-1007-7. (Ger). [DOI] [PubMed] [Google Scholar]
- 3.Persky MS, Setton A, Niimi Y, Hartman J, Frank D, Berenstein A. Combined endovascular and surgical treatment of head and neck paragangliomas - a team approach. Head Neck. 2002;24:423–431. doi: 10.1002/hed.10068. Available from: http://dx.doi.org/10.1002/hed.10068. [DOI] [PubMed] [Google Scholar]
- 4.Boedeker CC, Ridder GJ, Schipper J. Paragangliomas of the head and neck: diagnosis and treatment. Fam Cancer. 2005;4:55–59. doi: 10.1007/s10689-004-2154-z. Available from: http://dx.doi.org/10.1007/s10689-004-2154-z. [DOI] [PubMed] [Google Scholar]
- 5.Boedeker CC, Neumann HPH, Offergeld C, Maier W, Falcioni M, Berlis A, Schipper J. Clinical features of paraganglioma syndromes. Skull Base. 2009;19:17–25. doi: 10.1055/s-0028-1103123. Available from: http://dx.doi.org/10.1055/s-0028-1103123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JH, Barich F, Karnell LH, Robinson RA, Zhen WK, Gantz BJ, Hoffman HAT. National cancer data base report on malignant paragangliomas of the head and neck. Cancer. 2002;94:730–737. doi: 10.1002/cncr.10252. Available from: http://dx.doi.org/10.1002/cncr.10252. [DOI] [PubMed] [Google Scholar]
- 7.Baysal BE. Hereditary paraganglioma targets diverse paraganglia. J Med Genet. 2002;39:617–622. doi: 10.1136/jmg.39.9.617. Available from: http://dx.doi.org/10.1136/jmg.39.9.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Argiris A, Mellott A, Spies S. PET scan assessment of chemotherapy response in metastatic paraganglioma. Am J Clin Oncol. 2003;26:563–566. doi: 10.1097/01.coc.0000037765.75950.3A. Available from: http://dx.doi.org/10.1097/01.coc.0000037765.75950.3A. [DOI] [PubMed] [Google Scholar]
- 9.Neumann HPH, Pawlu C, Peçzkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley T, Hoegerle S, Boedeker CC, Opocher G, Schipper J, Januszewicz A, Eng C. Distinct clinical features characterize paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292:943–951. doi: 10.1001/jama.292.8.943. Available from: http://dx.doi.org/10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- 10.Neumann HPH, Erlic Z, Boedeker CC, Rybicki LA, Robledo M, Hermsen M, Schiavi F, Falcioni M, Kwok P, Bauters C, Lampe K, Fischer M, Edelman E, Benn DE, Robinson BG, Wiegand S, Rasp G, Stuck BA, Hoffmann MM, Sullivan M, Sevilla MA, Weiss MM, Peczkowska M, Kubaszek A, Pigny P, Ward RL, Learoyd D, Croxson M, Zabolotny D, Yaremchuk S, Draf W, Muresan M, Lorenz RR, Knipping S, Strohm M, Dyckhoff G, Matthias C, Reisch N, Preuss SF, Eßer D, Walter MA, Kaftan H, Stöver T, Fottner C, Gorgulla H, Malekpour M, Zarandy MM, Schipper J, Brase C, Glien A, Kühnemund M, Koscielny S, Schwerdtfeger P, Välimäki P, Szyfter W, Finckh U, Zerres K, Cascon A, Opocher G, Ridder GJ, Januszewicz A, Suarez C, Eng C. Clinical Predictors for Germline Mutations in Head and Neck Paraganglioma Patients: Cost Reduction Strategy in Genetic Diagnostic Process as Fall-Out. Can Research. 2009;69:3650–3656. doi: 10.1158/0008-5472.CAN-08-4057. Available from: http://dx.doi.org/10.1158/0008-5472.CAN-08-4057. [DOI] [PubMed] [Google Scholar]
- 11.Maher ER, Eng C. The pressure rises: update on the genetics of pheochromocytoma. Hum Mol Genet. 2002;11:2347–2354. doi: 10.1093/hmg/11.20.2347. Available from: http://dx.doi.org/10.1093/hmg/11.20.2347. [DOI] [PubMed] [Google Scholar]
- 12.Gosepath J, Welkoborsky HJ, Mann W. Untersuchungen zur Biologie und Wachstumsgeschwindigkeit bei Tumoren des Glomus jugolotympanicum und des Glomus caroticum. [Biology and growth velocity of tumors of the globus jugulotympanicum and glomus]. Laryngo-Rhino-Otol. 1998;77:429–433. doi: 10.1055/s-2007-997003. (Ger). Available from: http://dx.doi.org/10.1055/s-2007-997003. [DOI] [PubMed] [Google Scholar]
- 13.Hoegerle S, Nitzsche E, Altehoefer C, Ghanem N, Manz T, Brink I, Reincke M, Moser E, Neumann HPH. Pheochromocytomas: Detection with 18F Dopa whole-body PET - initial results. Radiology. 2002;222:507–512. doi: 10.1148/radiol.2222010622. Available from: http://dx.doi.org/10.1148/radiol.2222010622. [DOI] [PubMed] [Google Scholar]
- 14.D'Acri AM, Ramos-e-Silva M, Basilio-de-Oliveira C, Cerqueira A, Monteiro D, Pretti G, Longo C, Monteiro E. Multiple glomus tumors: recognition and diagnosis. Skinmed. 2002;1:94–98. doi: 10.1111/j.1540-9740.2002.01628.x. Available from: http://dx.doi.org/10.1111/j.1540-9740.2002.01628.x. [DOI] [PubMed] [Google Scholar]
- 15.Masson P. Le glomus myo-neuro-arteriel des regions tactiles et ses tumeurs. Lyon chir. 1924;21. [Google Scholar]
- 16.Mulligan RM. Chemodectoma in the dog. Am J Pathol. 1950;26:680–681. [Google Scholar]
- 17.Miller RB, Boon MS, Atkins JP, Lowry LD. Vagal paraganglioma: The Jefferson experience. Otolaryngol Head Neck Surg. 2000;122:482–487. doi: 10.1016/S0194-5998(00)70088-X. Available from: http://dx.doi.org/10.1016/S0194-5998(00)70088-X. [DOI] [PubMed] [Google Scholar]
- 18.Sobol SM, Dailey JC. Familial multiple cervical paragangliomas: report of a kindred and review of the literature. Otolaryngol Head Neck Surg. 1990;102:382–390. doi: 10.1177/019459989010200413. [DOI] [PubMed] [Google Scholar]
- 19.Kollert M, Minovi A, Mangold R, Hendus J, Draf W, Bockmühl U. Paragangliome des Kopf-Hals-Bereiches - Tumorkontrolle, funktionelle Ergebnisse und Lebensqualität nach Operation. [Paraganglioma of the head and neck--tumor control, functional results and quality of life]. Laryngo-Rhino-Otol. 2006;85:649–656. doi: 10.1055/s-2006-925234. (Ger). Available from: http://dx.doi.org/10.1055/s-2006-925234. [DOI] [PubMed] [Google Scholar]
- 20.Maier W, Marangos N, Laszig R. Paraganglioma as a systemic syndrome: pitfalls and strategies. J Laryngol Otol. 1999;113:978–982. doi: 10.1017/S0022215100145761. Available from: http://dx.doi.org/10.1017/S0022215100145761. [DOI] [PubMed] [Google Scholar]
- 21.Pellitteri PK, Rinaldo A, Myssiorek D, Gary Jackson C, Bradley PJ, Devaney KO, Shaha AR, Netterville JL, Manni JJ, Ferlito A. Paragangliomas of the head and neck. Oral Oncol. 2004;40:563–575. doi: 10.1016/j.oraloncology.2003.09.004. Available from: http://dx.doi.org/10.1016/j.oraloncology.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 22.Biller HF, Lawson W, Som P, Rosenfeld R. Glomus vagale tumors. Ann Otol Rhinol Laryngol. 1989;98:21–26. doi: 10.1177/000348948909800105. [DOI] [PubMed] [Google Scholar]
- 23.Murphy TE, Huvos AG, Frazell E. Chemodectomas of the glomus intravagale and vagal body tumors, non-chromaffin paragangliomas of the nodose ganglion of the vagus nerve. Ann Surg. 1970;172:246–255. doi: 10.1097/00000658-197008000-00011. Available from: http://dx.doi.org/10.1097/00000658-197008000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ketabchi S, Massi D, Santoro R, Franchi A. Paraganglioma of the nasal cavity: a case report. Eur Arch Otolaryngol. 2003;260:336–340. doi: 10.1007/s00405-002-0569-4. Available from: http://dx.doi.org/10.1007/s00405-002-0569-4. [DOI] [PubMed] [Google Scholar]
- 25.Mouadeb DA, Chandra RK, Kennedy DW, Feldman M. Sinonasal paraganglioma: endoscopic resection with a 4-year follow-up. Head Neck. 2003;25:1077–1081. doi: 10.1002/hed.10322. Available from: http://dx.doi.org/10.1002/hed.10322. [DOI] [PubMed] [Google Scholar]
- 26.Connor SE, Gleeson MJ, Odell E. Extracranial glomus faciale tumour. J Laryngol Otol. 2008;122:986–989. doi: 10.1017/S0022215107000126. Available from: http://dx.doi.org/10.1017/S0022215107000126. [DOI] [PubMed] [Google Scholar]
- 27.Birchler MT, Landau K, Went PT, Stoeckli SJ. Paraganglioma of the cervical sympathetic trunk. Ann Otol Rhinol Laryngol. 2002;111:1087–1091. doi: 10.1177/000348940211101205. [DOI] [PubMed] [Google Scholar]
- 28.Moyer JS, Bradford CR. Sympathetic paraganglioma as an unusual cause of Horner's syndrome. Head Neck. 2001;23:338–342. doi: 10.1002/hed.1040. Available from: http://dx.doi.org/10.1002/hed.1040. [DOI] [PubMed] [Google Scholar]
- 29.Del Gaudio JM, Muller S. Diagnosis and treatment of supraglottic laryngeal paraganglioma: report of a case. Head Neck. 2004;26:94–98. doi: 10.1002/hed.10346. Available from: http://dx.doi.org/10.1002/hed.10346. [DOI] [PubMed] [Google Scholar]
- 30.Hinojar AG, Prieto JR, Munoz E, Hinojar AA. Relapsing paraganglioma of the inferior laryngeal paraganglion: case report and review of the literature. Head Neck. 2002;24:95–102. doi: 10.1002/hed.1156. Available from: http://dx.doi.org/10.1002/hed.1156. [DOI] [PubMed] [Google Scholar]
- 31.Maisel R, Schmidt D, Pambuccian S. Subglottic laryngeal paraganglioma. Laryngoscope. 2003;113:401–405. doi: 10.1097/00005537-200303000-00002. Available from: http://dx.doi.org/10.1097/00005537-200303000-00002. [DOI] [PubMed] [Google Scholar]
- 32.Kronz JD, Argani P, Udelsman R, Silverberg L, Westra WH. Paraganglioma of the thyroid: two cases that clarify and expand the clinical spectrum. Head Neck. 2000;22:621–625. doi: 10.1002/1097-0347(200009)22:6<621::AID-HED12>3.0.CO;2-H. Available from: http://dx.doi.org/10.1002/1097-0347(200009)22:6<621::AID-HED12>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 33.Levy MT, Braun JT, Pennant M, Thompson LD. Primary paraganglioma of the parathyroid: a case report and clinicopathologic review. Head Neck Pathol. 2010;4:37–43. doi: 10.1007/s12105-009-0157-7. Available from: http://dx.doi.org/10.1007/s12105-009-0157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harries K, Nunn T, Shah V, Richards D, Manson JM. First reported case of esophageal paraganglioma. A review of the literature of gastrointestinal tract paraganglioma including gangliocytic paraganglioma. Dis Esophagus. 2004;17:191–195. doi: 10.1111/j.1442-2050.2004.00386.x. Available from: http://dx.doi.org/10.1111/j.1442-2050.2004.00386.x. [DOI] [PubMed] [Google Scholar]
- 35.Myssiorek D. Head and neck paragangliomas: an overview. Otolaryngol Clin North Am. 2001;34:829–836. doi: 10.1016/S0030-6665(05)70349-2. Available from: http://dx.doi.org/10.1016/S0030-6665(05)70349-2. [DOI] [PubMed] [Google Scholar]
- 36.Wang SJ, Wang MB, Barauskas TM, Calcaterra TC. Surgical management of carotid body tumors. Otolaryngol Head Neck Surg. 2000;123:202–206. doi: 10.1067/mhn.2000.106709. Available from: http://dx.doi.org/10.1067/mhn.2000.106709. [DOI] [PubMed] [Google Scholar]
- 37.Frühwirth J, Koch G, Klein GE. Die präoperative angiografische Embolisation von Tumoren des Karotisglomus. [Preoperative angiographic embolization of carotid glomus tumors. A method for improving operability]. HNO. 1996;44:510–513. doi: 10.1007/s001060050046. (Ger). Available from: http://dx.doi.org/10.1007/s001060050046. [DOI] [PubMed] [Google Scholar]
- 38.Smit AA, Timmers HJ, Wieling W, Wagenaar M, Marres HA, Lenders JW, van Montfrans GA, Karemaker JM. Long-term effects of carotid sinus denervation on arterial blood pressure in humans. Circulation. 2002;105:1329–1335. doi: 10.1161/hc1102.105744. Available from: http://dx.doi.org/10.1161/hc1102.105744. [DOI] [PubMed] [Google Scholar]
- 39.Timmers HJ, Karemaker JM, Wieling W, Marres HA, Folgering HT, Lenders JW. Baroreflex and chemoreflex function after bilateral carotid body tumor resection. J Hypertens. 2003;21:591–599. doi: 10.1097/00004872-200303000-00026. Available from: http://dx.doi.org/10.1097/00004872-200303000-00026. [DOI] [PubMed] [Google Scholar]
- 40.Strutz J, Mann W. Praxis der HNO-Heilkunde, Kopf- und Halschirurgie. Stuttgart: Thieme; 2000. pp. 1–2008. [Google Scholar]
- 41.Braun S, Riemann K, Pusch CM, Sotlar K, Pfister M, Kupka S. Paragangliome der Kopf-Hals-Region. Ein Überblick über die molekulargenetische Forschung. [Paraganglioma in the area of the head and neck. A review of molecular genetic research]. HNO. 2004;52:11–17. doi: 10.1007/s00106-003-0959-y. (Ger). [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez-Cuevas S, Lopez-Garza J, Labastida-Almendaro S. Carotid body tumors in inhabitants of altitudes higher than 2000 meters above sea level. Head Neck. 1998;20:374–378. doi: 10.1002/(SICI)1097-0347(199808)20:5<374::AID-HED3>3.0.CO;2-V. Available from: http://dx.doi.org/10.1002/(SICI)1097-0347(199808)20:5<374::AID-HED3>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 43.Astrom K, Cohen JE, Willett-Brozick JE, Aston CE, Baysal BE. Altitude is a phenotypic modifier in hereditary paraganglioma type 1: evidence for an oxygen-sensing defect. Hum Genet. 2003;113:228–237. doi: 10.1007/s00439-003-0969-6. Available from: http://dx.doi.org/10.1007/s00439-003-0969-6. [DOI] [PubMed] [Google Scholar]
- 44.Hajnzic TF, Kruslin B, Belicza M. Carotid body paraganglioma in a nine-year-old boy with extensive pulmonary metastases. Med Pediatr Oncol. 1999;32:399–400. doi: 10.1002/(SICI)1096-911X(199905)32:5<399::AID-MPO20>3.0.CO;2-V. Available from: http://dx.doi.org/10.1002/(SICI)1096-911X(199905)32:5<399::AID-MPO20>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 45.Westerband A, Hunter GC, Cintora I, Coulthard SW, Hinni ML, Gentile AT, Devine J, Mills JL. Current trends in the detection and management of carotid body tumors. Vasc Surg. 1998;28:84–93. doi: 10.1016/S0741-5214(98)70203-4. Available from: http://dx.doi.org/10.1016/S0741-5214(98)70203-4. [DOI] [PubMed] [Google Scholar]
- 46.Gstoettner W, Matula C, Hamzavi J, Kornfehl J, Czerny C. Long-term results of different treatment modalities in 37 patients with glomus jugulare tumors. Eur Arch Otorhinolaryngol. 1999;256:351–355. doi: 10.1007/s004050050162. Available from: http://dx.doi.org/10.1007/s004050050162. [DOI] [PubMed] [Google Scholar]
- 47.Hinerman RW, Mendenhall WM, Amdur RJ, Stringer SP, Antonelli PJ, Cassisi NJ. Definitive radiotherapy in the management of chemodectomas arising in the temporal bone, carotid body and glomus vagale. Head Neck. 2001;23:363–371. doi: 10.1002/hed.1045. Available from: http://dx.doi.org/10.1002/hed.1045. [DOI] [PubMed] [Google Scholar]
- 48.Patetsios P, Gable DR, Garrett WV, Lamont JP, Kuhn JA, Shutze WP, Kourlis H, Grimsley B, Pearl GJ, Smith BL, Talkington CM, Thompson JE. Management of carotid body paragangliomas and review of a 30-year experience. Ann Vasc Surg. 2002;16:331–338. doi: 10.1007/s10016-001-0106-8. Available from: http://dx.doi.org/10.1007/s10016-001-0106-8. [DOI] [PubMed] [Google Scholar]
- 49.Schick B, Draf W, Kahle G. Jugulotympanale Paragangliome: Therapiekonzepte in der Entwicklung. [Jugulotympanic paraganglioma: therapy concepts under development]. Laryngo-Rhino-Otol. 1998;77:434–443. doi: 10.1055/s-2007-997004. (Ger). Available from: http://dx.doi.org/10.1055/s-2007-997004. [DOI] [PubMed] [Google Scholar]
- 50.Gehrking E, Gliemroth J, Missler U, Remmert S. Hauptsymptom: „Pulssynchrones Ohrgeräusch”. [Main symptom: "pulse-synchronous tinnitus"]. Laryngo-Rhino-Otol. 2000;79:510–516. doi: 10.1055/s-2000-6944. (Ger). Available from: http://dx.doi.org/10.1055/s-2000-6944. [DOI] [PubMed] [Google Scholar]
- 51.Groblewski JC, Thekdi A, Carrau RL. Secreting vagal paraganglioma. Am J Otolaryngol. 2004;25:295–300. doi: 10.1016/j.amjoto.2004.02.007. Available from: http://dx.doi.org/10.1016/j.amjoto.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 52.Koch CA, Rodbard JS, Brouwers FM, Eisenhofer G, Pacak K. Hypotension in a woman with a metastatic dopamine-secreting carotid body tumor. Endocr Pract. 2003;9:310–314. doi: 10.4158/EP.9.4.310. [DOI] [PubMed] [Google Scholar]
- 53.Kaftan H, Draf W. Erfahrungen mit der farbkodierten Duplexsonographie im HNO-Bereich. [Experiences with color-coded duplex ultrasound in the ENT area]. HNO. 1999;47:287–291. doi: 10.1007/s001060050398. (Ger). Available from: http://dx.doi.org/10.1007/s001060050398. [DOI] [PubMed] [Google Scholar]
- 54.Stoeckli SJ, Schuknecht B, Alkadhi H, Fisch U. Evaluation of paragangliomas presenting as a cervical mass on color-coded doppler sonography. Laryngoscope. 2002;112:143–146. doi: 10.1097/00005537-200201000-00025. Available from: http://dx.doi.org/10.1097/00005537-200201000-00025. [DOI] [PubMed] [Google Scholar]
- 55.Alkadhi H, Schuknecht B, Stoeckli SJ, Valavanis A. Evaluation of topography and vascularization of cervical paragangliomas by magnetic resonance imaging and color duplex sonography. Neuroradiology. 2002;44:83–90. doi: 10.1007/s002340100681. Available from: http://dx.doi.org/10.1007/s002340100681. [DOI] [PubMed] [Google Scholar]
- 56.Thabet MH, Kotob H. Cervical paragangliomas: diagnosis, management and complications. J Laryngol Otol. 2001;115:467–474. doi: 10.1258/0022215011908180. Available from: http://dx.doi.org/10.1258/0022215011908180. [DOI] [PubMed] [Google Scholar]
- 57.Hoegerle S, Ghanem N, Altehoefer C, Schipper J, Brink I, Moser E, Neumann HPH. 18F-Dopa positron emission tomography for the detection of glomus tumors. Eur J Nucl Med Mol Imaging. 2003;30:689–694. doi: 10.1007/s00259-003-1115-3. Available from: http://dx.doi.org/10.1007/s00259-003-1115-3. [DOI] [PubMed] [Google Scholar]
- 58.Shamblin WR, Re Mine WH, Sheps SG, Harrison EG., Jr Carotid body tumour. Clinicopathologic analysis of ninety cases. Am J Surg. 1971;122:732–739. doi: 10.1016/0002-9610(71)90436-3. Available from: http://dx.doi.org/10.1016/0002-9610(71)90436-3. [DOI] [PubMed] [Google Scholar]
- 59.Schipper J, Boedeker CC, Maier W, Neumann HPH. Paragangliome im Kopf-/Halsbereich Teil 2: Therapie und Nachsorge. [Paragangliomas of the head and neck. Part 2: Therapy and follow-up]. HNO. 2004;52:651–662. doi: 10.1007/s00106-003-1006-8. (Ger). [DOI] [PubMed] [Google Scholar]
- 60.Fisch U, Mattox D. Microsurgery of the skull base. Stuttgart, New York: Thieme; 1988. pp. 149–153. [Google Scholar]
- 61.Aschenbach R, Esser D. Magnetresonanztomografie im Kopf-Hals-Bereich. [Magnetic resonance angiography of the head and neck area]. HNO. 2004;52:77–89. doi: 10.1007/s00106-003-0982-z. (Ger). Available from: http://dx.doi.org/10.1007/s00106-003-0982-z. [DOI] [PubMed] [Google Scholar]
- 62.Bustillo A, Telischi F, Weed D, Civantos F, Angeli S, Serafini A, Whiteman M. Octreotide scintigraphy in the head and neck. Laryngoscope. 2004;114:434–440. doi: 10.1097/00005537-200403000-00010. [DOI] [PubMed] [Google Scholar]
- 63.Bustillo A, Telischi F. Octreotide scintigraphy in the detection of recurrent paragangliomas. Otolaryngol Head Neck Surg. 2004;130:479–482. doi: 10.1016/j.otohns.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 64.King KS, Whatley MA, Alexopoulos DK, Reynolds JC, Chen CC, Mattox DE, Jacobs S, Pacak K. The use of functional imaging in a patient with head and neck paragangliomas. J Clin Endocrinol Metab. 2010;95:481–482. doi: 10.1210/jc.2009-2214. Available from: http://dx.doi.org/10.1210/jc.2009-2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luster M, Karges W, Zeich K, Pauls S, Verburg FA, Dralle H, Glatting G, Buck AK, Solbach C, Neumaier B, Reske SN, Mottaghy FM. Clinical value of 18F-fluorodihydroxyphenylalanine positron emission tomography/computed tomography (18F-DOPA PET/CT) for detecting pheochromocytoma. Eur J Nucl Med Mol Imaging. 2010;37:484–493. doi: 10.1007/s00259-009-1294-7. Available from: http://dx.doi.org/10.1007/s00259-009-1294-7. [DOI] [PubMed] [Google Scholar]
- 66.Kayani I, Bomanji JB, Groves A, Conway G, Gacinovic S, Win T, Dickson J, Caplin M, Ell PJ. Functional imaging of neuroendocrine tumors with combined PET/CT using 68Ga-DOTATATE (DOTA-DPhe1,Tyr3-octreotate) and 18F-FDG. Cancer. 2008;112:2447–2455. doi: 10.1002/cncr.23469. Available from: http://dx.doi.org/10.1002/cncr.23469. [DOI] [PubMed] [Google Scholar]
- 67.Srirajaskanthan R, Kayani I, Quigley AM, Soh J, Caplin ME, Bomanji J. The role of 68Ga-DOTATATE PET in patients with neuroendocrine tumors and negative or equivocal findings on 111In-DTPA-octreotide scintigraphy. vJ Nucl Med. 2010;51:875–882. doi: 10.2967/jnumed.109.066134. [DOI] [PubMed] [Google Scholar]
- 68.Chuah KL, Tan PH, Chong YY. Test and teach. Number ninety-three: Part 1. Carotid body paraganglioma. Pathology. 1999;31:215–216, 273. doi: 10.1080/003130299104990. [DOI] [PubMed] [Google Scholar]
- 69.Jaffer S, Harpaz N. Mesenteric paraganglioma: a case report and review of the literature. Arch Pathol Lab Med. 2002;126:362–364. doi: 10.5858/2002-126-0362-MP. [DOI] [PubMed] [Google Scholar]
- 70.Kontogeorgos G, Scheithauer BW, Kovacs K, Horvath E, Melmed S. Growth factors and cytokines in paragangliomas and pheochromocytomas, with special reference to sustentacular cells. Endocr Pathol. 2002;13:197–206. doi: 10.1385/EP:13:3:197. Available from: http://dx.doi.org/10.1385/EP:13:3:197. [DOI] [PubMed] [Google Scholar]
- 71.Shibahara J, Goto A, Niki T, Tanaka M, Nakajima J, Fukayama M. Primary pulmonary paraganglioma: report of a functioning case with immunohistochemical and ultrastructural study. Am J Surg Pathol. 2004;28:825–829. doi: 10.1097/01.pas.0000116832.81882.0b. Available from: http://dx.doi.org/10.1097/01.pas.0000116832.81882.0b. [DOI] [PubMed] [Google Scholar]
- 72.Douwes Dekker PB, Corver WE, Hogendoorn PC, van der Mey AG, Cornelisse CJ. Multiparameter DNA flow-sorting demonstrates diploidy and SDHC wild-type gene retention in the sustentacular cell compartment of head and neck paragangliomas: chief cells are the only neoplastic component. J Pathol. 2004;202:456–462. doi: 10.1002/path.1535. Available from: http://dx.doi.org/10.1002/path.1535. [DOI] [PubMed] [Google Scholar]
- 73.Effat KG, Karam M. Jugulotympanic paraganglioma (glomus tumour) presenting with recurrent epistaxis. J Laryngol Otol. 2004;118:153–155. doi: 10.1258/002221504772784658. Available from: http://dx.doi.org/10.1258/002221504772784658. [DOI] [PubMed] [Google Scholar]
- 74.Au WY, Chan AC, Wong KK, Leung CY, Liang R, Kwong YL. Multiple osseous metastases from occult paraganglioma: a diagnostic pitfall. Histopathology. 1998;33:287–288. doi: 10.1046/j.1365-2559.1998.0482d.x. Available from: http://dx.doi.org/10.1046/j.1365-2559.1998.0482d.x. [DOI] [PubMed] [Google Scholar]
- 75.Kawai A, Healey JH, Wilson SC, Huvos AG, Yeh SD. Carotid body paraganglioma metastatic to bone: report of two cases. Skeletal Radiol. 1998;27:103–107. doi: 10.1007/s002560050346. Available from: http://dx.doi.org/10.1007/s002560050346. [DOI] [PubMed] [Google Scholar]
- 76.Pacheco-Ojeda L. Malignant carotid body tumors: report of three cases. Ann Otol Rhinol Laryngol. 2001;110:36–40. doi: 10.1177/000348940111000107. [DOI] [PubMed] [Google Scholar]
- 77.Righini Ch, Pecher M, Halimi S, Magne JL, Reyt E. Malignant carotid paraganglioma. A case report. Ann Otolaryngol Chir Cervicofac. 2003;120:103–108. [PubMed] [Google Scholar]
- 78.Ferris EJ, Smith PL, Boyd CM, Wetzel WJ. Malignant non-chromaffin paraganglioma with metastases to the kidneys. Br J Urol. 1983;55:332–334. doi: 10.1111/j.1464-410X.1983.tb03310.x. Available from: http://dx.doi.org/10.1111/j.1464-410X.1983.tb03310.x. [DOI] [PubMed] [Google Scholar]
- 79.Wesche WA, Khare VK, Chesney TM, Jenkins JJ. Non-hematopoietic cutaneous metastases in children and adolescents: thirty years experience at St. Jude Children's Research Hospital. J Cutan Pathol. 2000;27:485–492. doi: 10.1034/j.1600-0560.2000.027010485.x. Available from: http://dx.doi.org/10.1034/j.1600-0560.2000.027010485.x. [DOI] [PubMed] [Google Scholar]
- 80.Kumaki N, Kajiwara H, Kameyama K, DeLellis RA, Asa SL, Osamura RY, Takami H. Prediction of malignant behavior of pheochromocytomas and paragangliomas using immunohistochemical techniques. Endocr Pathol. 2002;13:149–156. doi: 10.1385/EP:13:2:149. Available from: http://dx.doi.org/10.1385/EP:13:2:149. [DOI] [PubMed] [Google Scholar]
- 81.Zaharopoulos P. Diagnostic challenges in the fine-needle aspiration diagnosis of carotid body paragangliomas: report of two cases. Diagn Cytopathol. 2000;23:202–207. doi: 10.1002/1097-0339(200009)23:3<202::AID-DC13>3.0.CO;2-S. Available from: http://dx.doi.org/10.1002/1097-0339(200009)23:3<202::AID-DC13>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 82.Monabati A, Hodjati H, Kumar PV. Cytologic findings in carotid body tumors. Acta Cytol. 2002;46:1101–1104. doi: 10.1159/000327114. Available from: http://dx.doi.org/10.1159/000327114. [DOI] [PubMed] [Google Scholar]
- 83.Jahnke V, Klingebiel R. Pulssynchroner Tinnitus. HNO. 2004;52:440–442. doi: 10.1007/s00106-003-0947-2. Available from: http://dx.doi.org/10.1007/s00106-003-0947-2. [DOI] [PubMed] [Google Scholar]
- 84.Schikschneit M, Maier W, Kayser G, Berlis A, Boedeker CC. Amputation neuroma of the middle ear mimicking glomus tympanicum tumor. Otolaryngol Head Neck Surg. 2007;137:843–844. doi: 10.1016/j.otohns.2007.05.007. Available from: http://dx.doi.org/10.1016/j.otohns.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 85.Feigenberg SJ, Mendenhall WM, Hinerman RW, Amdur RJ, Friedman WA, Antonelli PJ. Radiosurgery for paraganglioma of the temporal bone. Head Neck. 2002;24:384–389. doi: 10.1002/hed.10064. Available from: http://dx.doi.org/10.1002/hed.10064. [DOI] [PubMed] [Google Scholar]
- 86.Foote RL, Pollock BE, Gorman DA, Schomberg PJ, Stafford SL, Link MJ, Kline RW, Strome SE, Kasperbauer JL, Olsen KD. Glomus jugulare tumor: tumor control and complications after stereotactic radiosurgery. Head Neck. 2002;24:332–338. doi: 10.1002/hed.10005. Available from: http://dx.doi.org/10.1002/hed.10005. [DOI] [PubMed] [Google Scholar]
- 87.Litle VR, Reilly LM, Ramos TK. Preoperative embolisation of carotid body tumors: when is it appropriate? Ann Vasc Surg. 1996;10:464–468. doi: 10.1007/BF02000594. Available from: http://dx.doi.org/10.1007/BF02000594. [DOI] [PubMed] [Google Scholar]
- 88.Kollert M, Minovi AA, Draf W, Bockmühl U. Cervical paragangliomas - tumor control and long-term functional results after surgery. Skull Base. 2006;16:185–191. doi: 10.1055/s-2006-950386. Available from: http://dx.doi.org/10.1055/s-2006-950386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liapis CD, Evangelidakis EL, Papavassiliou VG, Kakisis JD, Gougoulakis AG, Polyzos AK, Sechas MN, Gogas JG. Role of malignancy and preoperative embolization in the management of carotid body tumors. World J Surg. 2000;24:1526–1530. doi: 10.1007/s002680010272. Available from: http://dx.doi.org/10.1007/s002680010272. [DOI] [PubMed] [Google Scholar]
- 90.Boyle JO, Shimm DS, Coulthard SW. Radiation therapy for paragangliomas of the temporal bone. Laryngoscope. 1990;100:896–901. doi: 10.1288/00005537-199008000-00018. Available from: http://dx.doi.org/10.1288/00005537-199008000-00018. [DOI] [PubMed] [Google Scholar]
- 91.Elshaikh MA, Mahmoud-Ahmed AS, Kinney SE, Wood BG, Lee JH, Barnett GH, Suh JH. Recurrent head-and-neck chemodectomas: A comparison of surgical and radiotherapeutic results. Int J Rad Oncol Biol Phys. 2002;52:953–956. doi: 10.1016/S0360-3016(01)02751-1. Available from: http://dx.doi.org/10.1016/S0360-3016(01)02751-1. [DOI] [PubMed] [Google Scholar]
- 92.Powell S, Peters N, Harmer C. Chemodectoma of the head and neck: Results of treatment in 84 patients. Int J Radiat Oncol Biol Phys. 1992;22:919–924. doi: 10.1016/0360-3016(92)90788-J. Available from: http://dx.doi.org/10.1016/0360-3016(92)90788-J. [DOI] [PubMed] [Google Scholar]
- 93.Hinerman RW, Mendenhall WM, Amdur RJ, Feigenberg SJ. Editorial comment: Stereotactic radiosurgery for glomus jugulare tumor. Head Neck. 2002;24:338–339. [Google Scholar]
- 94.Matthews FS. Surgery of the neck. In: Johnson AB, editor. Operative therapeutics. Vol. 3. East Norwalk, Conn.: Appleton-Century-Croft; 1915. p. 315. [Google Scholar]
- 95.Grotemeyer D, Loghmanieh SM, Pourhassan S, Sagban TA, Iskandar F, Reinecke P, Sandmann W. Dignität von Glomus-caroticum-Tumoren: Literaturübersicht und klinische Erfahrungen. [Dignity of carotid body tumors. Review of the literature and clinical experiences]. Chirurg. 2009;80:854–863. doi: 10.1007/s00104-009-1724-x. (Ger). Available from: http://dx.doi.org/10.1007/s00104-009-1724-x. [DOI] [PubMed] [Google Scholar]
- 96.Erickson D, Kudva YC, Ebersold MJ, Thompson GB, Grant CS, van Heerden JA, Young WF., Jr Benign paragangliomas. Clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab. 2001;86:5210–5216. doi: 10.1210/jc.86.11.5210. Available from: http://dx.doi.org/10.1210/jc.86.11.5210. [DOI] [PubMed] [Google Scholar]
- 97.Tewari M, Dixit A, Mongha R, Shukla HS. Control of intra-operative hemorrhage during excision of carotid body tumor. J Surg Oncol. 2004;85:55–57. doi: 10.1002/jso.20004. Available from: http://dx.doi.org/10.1002/jso.20004. [DOI] [PubMed] [Google Scholar]
- 98.van der Mey AG, Jansen JC, van Baalen JM. Management of carotid body tumors. Otolaryngol Clin North Am. 2001;34:907–924. doi: 10.1016/S0030-6665(05)70354-6. Available from: http://dx.doi.org/10.1016/S0030-6665(05)70354-6. [DOI] [PubMed] [Google Scholar]
- 99.Robbins KT, Clayman G, Levine PA, Medina J, Sessions R, Shaha A, Som P, Wolf GT. American Head and Neck Society. Neck dissection classification update: revisions proposed by the American Head and Neck Society and the American Academy of Otolaryngology - Head and Neck Surgery. Arch Otolaryngol Head Neck Surg. 2002;128:751–758. doi: 10.1001/archotol.128.7.751. [DOI] [PubMed] [Google Scholar]
- 100.Anand VK, Alemar GO, Sanders TS. Management of the internal carotid artery during carotid body tumour surgery. Laryngoscope. 1995;105:231–235. doi: 10.1288/00005537-199503000-00001. Available from: http://dx.doi.org/10.1288/00005537-199503000-00001. [DOI] [PubMed] [Google Scholar]
- 101.Kim JA, Elkon D, Lim ML, Constable WC. Optimum dose of radiotherapy for chemodectomas of the middle ear. Int J Radiat Oncol Biol Phys. 1980;6:815–819. doi: 10.1016/0360-3016(80)90317-X. Available from: http://dx.doi.org/10.1016/0360-3016(80)90317-X. [DOI] [PubMed] [Google Scholar]
- 102.Lalwani AK, Jackler RK, Gutin PH. Lethal fibrosarcoma complicating radiation therapy for benign glomus jugulare tumor. Am J Otol. 1993;14:398–402. [PubMed] [Google Scholar]
- 103.Fisch U. Infratemporal fossa approach for glomus tumors of the temporal bone. Ann Otol Rhinol Laryngol. 1982;91:474–479. doi: 10.1177/000348948209100502. [DOI] [PubMed] [Google Scholar]
- 104.Fisch U. Infratemporal fossa approach for lesions in the temporal bone and base of the skull. Adv Otorhinolaryngol. 1984;34:254–266. doi: 10.1159/000409856. [DOI] [PubMed] [Google Scholar]
- 105.Hawthorne MR, Makek MS, Harris JP, Fisch U. The histopathological and clinical features of irradiated and nonirradiated temporal paragangliomas. Laryngoscope. 1998;98:325–331. doi: 10.1288/00005537-198803000-00018. [DOI] [PubMed] [Google Scholar]
- 106.Lenarz T, Plinkert PK. Glomustumoren des Felsenbeins - operatives Konzept und Ergebnisse. [Glomus tumors of the temporal bone--surgical concept and results]. Laryngo-Rhino-Otol. 1992;71:149–157. doi: 10.1055/s-2007-997267. (Ger). Available from: http://dx.doi.org/10.1055/s-2007-997267. [DOI] [PubMed] [Google Scholar]
- 107.Manolidis S, Shohet JA, Jackson G, Glasscock ME. Malignant glomus tumors. Laryngoscope. 1999;109:30–33. doi: 10.1097/00005537-199901000-00007. Available from: http://dx.doi.org/10.1097/00005537-199901000-00007. [DOI] [PubMed] [Google Scholar]
- 108.Gjuric M, Seidinger L, Wigand ME. Long-term results of surgery for temporal bone paraganglioma. Skull Base Surg. 1996;6:147–152. doi: 10.1055/s-2008-1058638. Available from: http://dx.doi.org/10.1055/s-2008-1058638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jackson CG, Haynes DS, Walker PA, Glasscock ME, III, Storper IS, Josey AF. Hearing conservation in surgery for glomus jugulare tumors. Am J Otolaryngol. 1996;17:425–437. [PubMed] [Google Scholar]
- 110.Murphy TP, Brackmann DE. Effects of preoperative embolization on glomus jugulare tumors. Laryngoscope. 1989;99:1244–1247. doi: 10.1288/00005537-198912000-00007. Available from: http://dx.doi.org/10.1288/00005537-198912000-00007. [DOI] [PubMed] [Google Scholar]
- 111.O'Leary MJ, Shelton C, Giddings NA, Kwartler J, Brackmann DE. Glomus tympanicum tumors: A clinical perspective. Laryngoscope. 1991;101:1038–1043. doi: 10.1288/00005537-199110000-00002. [DOI] [PubMed] [Google Scholar]
- 112.Forest JA, III, Jackson CG, McGrew BM. Long-term control of surgically treated glomus tympanicum tumors. Otol Neurotol. 2001;22:232–236. doi: 10.1097/00129492-200103000-00020. Available from: http://dx.doi.org/10.1097/00129492-200103000-00020. [DOI] [PubMed] [Google Scholar]
- 113.Glasscock ME, Harris PF, Newsome G. Glomus tumors: diagnosis and treatment. Laryngoscope. 1974;84:2006–2032. doi: 10.1002/lary.5540841116. [DOI] [PubMed] [Google Scholar]
- 114.Chino JP, Sampson JH, Tucci DL, Brizel DM, Kirkpatrick JP. Paraganglioma of the head and neck: long-term local control with radiotherapy. Am J Clin Oncol. 2009;32:304–307. doi: 10.1097/COC.0b013e318187dd94. Available from: http://dx.doi.org/10.1097/COC.0b013e318187dd94. [DOI] [PubMed] [Google Scholar]
- 115.Steinberg N, Holz WG. Glomus jugularis tumors. Arch Otolaryngol. 1965;82:387–394. doi: 10.1001/archotol.1965.00760010389009. Available from: http://dx.doi.org/10.1001/archotol.1965.00760010389009. [DOI] [PubMed] [Google Scholar]
- 116.Schulz-Ertner D, Nikoghosyan A, Thilmann C, Haberer T, Jakel O, Karger C, Kraft G, Wannenmacher M, Debus J. Results of carbon ion radiotherapy in 152 patients. Int J Radiat Oncol Biol Phys. 2004;58:631–640. doi: 10.1016/j.ijrobp.2003.09.041. Available from: http://dx.doi.org/10.1016/j.ijrobp.2003.09.041. [DOI] [PubMed] [Google Scholar]
- 117.Karger CP, Jäkel O. Current status and new developments in ion therapy. Strahlenther Onkol. 2007;183:295–300. doi: 10.1007/s00066-007-1645-x. Available from: http://dx.doi.org/10.1007/s00066-007-1645-x. [DOI] [PubMed] [Google Scholar]
- 118.Laramore GE. Role of particle radiotherapy in the management of head and neck cancer. Curr Opin Oncol. 2009;21:224–231. doi: 10.1097/CCO.0b013e328329b716. Available from: http://dx.doi.org/10.1097/CCO.0b013e328329b716. [DOI] [PubMed] [Google Scholar]
- 119.Velegrakis G, Kalikakis G, Karampekios S, Stefanaki K, Helidonis E. Bilaterales Paragangliom des Nervus vagus. [Bilateral paraganglioma of the vagus nerve]. HNO. 2001;49:471–475. doi: 10.1007/s001060170099. (Ger). Available from: http://dx.doi.org/10.1007/s001060170099. [DOI] [PubMed] [Google Scholar]
- 120.Marangos N, Schumacher M. Management eines großen bilateralen Glomus jugulare Tumors. [Management of large bilateral glomus jugulare tumoors]. HNO. 1999;47:816–820. doi: 10.1007/s001060050465. (Ger). Available from: http://dx.doi.org/10.1007/s001060050465. [DOI] [PubMed] [Google Scholar]
- 121.Tasar M, Yetiser S. Glomus tumors: therapeutic role of selective embolization. J Craniofac Surg. 2004;15:497–505. doi: 10.1097/00001665-200405000-00031. Available from: http://dx.doi.org/10.1097/00001665-200405000-00031. [DOI] [PubMed] [Google Scholar]
- 122.Marangos N, Schumacher M. Facial palsy after glomus jugulare tumour embolization. J Laryngol Otol. 1999;113:268–270. doi: 10.1017/S0022215100143762. Available from: http://dx.doi.org/10.1017/S0022215100143762. [DOI] [PubMed] [Google Scholar]
- 123.Horowitz M, Whisnant RE, Jungreis C, Snyderman C, Levy EI, Kassam A. Temporary balloon occlusion and ethanol injection for preoperative embolization of carotid-body tumor. Ear Nose Throat J. 2002;81:536–538, 540, 542. [PubMed] [Google Scholar]
- 124.Herdman RCD, Gillespie JE, Ramsden RT. Facial palsy after glomus tumour embolization. J Laryngol Otol. 1993;107:963–966. doi: 10.1017/S0022215100124934. Available from: http://dx.doi.org/10.1017/S0022215100124934. [DOI] [PubMed] [Google Scholar]
- 125.Schick PM, Hieshima GB, White RA, Fiaschetti FL, Mehringer CM, Grinnell VS, Everhart FR. Arterial catheter embolization followed by surgery for large chemodectoma. Surgery. 1980;87:459–464. [PubMed] [Google Scholar]
- 126.Kafie FE, Freischlag JA. Carotid body tumors: the role of preoperative embolization. Ann Vasc Surg. 2001;15:237–242. doi: 10.1007/s100160010058. Available from: http://dx.doi.org/10.1007/s100160010058. [DOI] [PubMed] [Google Scholar]
- 127.Rödl W, Manzke E. Angiographic differential diagnosis of tumors of the glomus caroticum and jugulo-tympanicum and of arterio-venous shunts in the head and neck. Ann Radiol. 1980;23:732–739. [PubMed] [Google Scholar]
- 128.Hekster RE, Luyendijk W, Matricali B. Transfemoral catheter embolization: a method of treatment of glomus jugulare tumors. Neuroradiology. 1973;5:208–214. doi: 10.1007/BF00394737. Available from: http://dx.doi.org/10.1007/BF00394737. [DOI] [PubMed] [Google Scholar]
- 129.Green JD, Brackmann DE, Nguyen CD, Arriage MA, Telischi FF, De La Cruz A. Surgical management of previously untreated glomus jugulare tumors. Laryngoscope. 1994;104:917–921. doi: 10.1288/00005537-199408000-00001. Available from: http://dx.doi.org/10.1288/00005537-199408000-00001. [DOI] [PubMed] [Google Scholar]
- 130.Ansari MS, Goel A, Goel S, Durairajan LN, Seth A. Malignant paraganglioma of the urinary bladder. A case report. Int Urol Nephrol. 2001;33:343–345. doi: 10.1023/A:1015253427161. Available from: http://dx.doi.org/10.1023/A:1015253427161. [DOI] [PubMed] [Google Scholar]
- 131.Carroll W, Stenson K, Stringer S. Malignant carotid body tumor. Head Neck. 2004;26:301–306. doi: 10.1002/hed.20017. Available from: http://dx.doi.org/10.1002/hed.20017. [DOI] [PubMed] [Google Scholar]
- 132.Young AL, Baysal BE, Deb A, Young WF., Jr Familial malignant catecholamine-secreting paraganglioma with prolonged survival associated with mutation in the succinate dehydrogenase B gene. J Clin Endocrinol Metab. 2002;87:4101–4105. doi: 10.1210/jc.2002-020312. Available from: http://dx.doi.org/10.1210/jc.2002-020312. [DOI] [PubMed] [Google Scholar]
- 133.Tenenbaum F, Schlumberger M, Lumbroso J, Parmentier C. Beneficial effects of octreotide in a patient with a metastatic paraganglioma. Eur J Cancer. 1996;32A:737. doi: 10.1016/0959-8049(95)00617-6. [DOI] [PubMed] [Google Scholar]
- 134.Troncone L, Rufini V. Nuclear medicine therapy of pheochromocytoma and paraganglioma. Q J Nucl Med. 1999;43:344–355. [PubMed] [Google Scholar]
- 135.Garkavij M, Nickel M, Sjögreen-Gleisner K, Ljungberg M, Ohlsson T, Wingårdh K, Strand SE, Tennvall J. 177Lu-[DOTA0,Tyr3] octreotate therapy in patients with disseminated neuroendocrine tumors: Analysis of dosimetry with impact on future therapeutic strategy. Cancer. 2010;116(4 Suppl):1084–1092. doi: 10.1002/cncr.24796. Available from: http://dx.doi.org/10.1002/cncr.24796. [DOI] [PubMed] [Google Scholar]
- 136.Boedeker CC, Neumann HPH, Ridder GJ, Maier W, Schipper J. Paragangliomas in patients with mutations of the SDHD gene. Otolaryngol Head Neck Surg. 2005;132:467–470. doi: 10.1016/j.otohns.2004.09.024. Available from: http://dx.doi.org/10.1016/j.otohns.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 137.Boedeker CC, Neumann HPH, Bausch B, Maier W, Schipper J, Ridder GJ. Malignant paragangliomas in SDHB mutation carriers. Otolaryngol Head Neck Surg. 2007;137:126–129. doi: 10.1016/j.otohns.2007.01.015. Available from: http://dx.doi.org/10.1016/j.otohns.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 138.Baysal BE. Clinical and molecular progress in hereditary paraganglioma. J Med Genet. 2008;45:689–694. doi: 10.1136/jmg.2008.058560. Available from: http://dx.doi.org/10.1136/jmg.2008.058560. [DOI] [PubMed] [Google Scholar]
- 139.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW, 3rd, Cornelisse CJ, Devilee P, Devlin B. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. Available from: http://dx.doi.org/10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 140.Baysal BE, Willett-Brozick JE, Lawrence EC, Drovdlic CM, Savul SA, McLeod DR, Yee HA, Brackmann DE, Slattery WH, 3rd, Myers EN, Ferrell RE, Rubinstein WS. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet. 2002;39:178–183. doi: 10.1136/jmg.39.3.178. Available from: http://dx.doi.org/10.1136/jmg.39.3.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Baysal BE. On the association of succinate dehydrogenase mutations with hereditary paraganglioma. Trends Endocrinol Metab. 2003;14:453–459. doi: 10.1016/j.tem.2003.08.004. Available from: http://dx.doi.org/10.1016/j.tem.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 142.Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med. 2009;266:19–42. doi: 10.1111/j.1365-2796.2009.02111.x. Available from: http://dx.doi.org/10.1111/j.1365-2796.2009.02111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reineke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peczkowska M, Szmigielski C, Eng C Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–1466. doi: 10.1056/NEJMoa020152. Available from: http://dx.doi.org/10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 144.Gimm O, Armanios M, Dziema H, Neumann HPH, Eng C. Somatic and occult germline mutations in SDHD, a mitochondrial complex II gene, in non-familial pheochromocytomas. Cancer Res. 2000;60:6822–6825. [PubMed] [Google Scholar]
- 145.Gimm O, Koch CA, Januszewicz A, Opocher G, Neumann HP. The genetic basis of pheochromocytoma. Front Horm Res. 2004;31:45–60. doi: 10.1159/000074657. Available from: http://dx.doi.org/10.1159/000074657. [DOI] [PubMed] [Google Scholar]
- 146.Yip L, Lee JR, Shapiro SE, Waguespack SG, Sherman SI, Hoff AO, Gagel RF, Arens JF, Evans DB. Surgical management of hereditary pheochromocytoma. J Am Coll Surg. 2004;198:525–535. doi: 10.1016/j.jamcollsurg.2003.12.001. Available from: http://dx.doi.org/10.1016/j.jamcollsurg.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 147.Eng C, Clayton D, Schuffenecker I, Lenoir G, Cote G, Gagel RF, van Amstel HK, Lips CJ, Nishisho I, Takai SI, Marsh DJ, Robinson BG, Frank-Raue K, Raue F, Xue F, Noll WW, Romei C, Pacini F, Fink M, Niederle B, Zedenius J, Nordenskjold M, Komminoth P, Hendy GN, Mulligan LM. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA. 1996;276:1575–1579. doi: 10.1001/jama.1996.03540190047028. Available from: http://dx.doi.org/10.1001/jama.1996.03540190047028. [DOI] [PubMed] [Google Scholar]
- 148.Frank-Raue K, Hoppner W, Frilling A, Kotzerke J, Dralle H, Haase R, Mann K, Seif F, Kirchner R, Rendl J, Deckart HF, Ritter MM, Hampel R, Klempa J, Scholz GH, Raue F. Mutations of the ret protooncogene in German multiple endocrine neoplasia families: relation between genotype and phenotype. German Medullary Thyroid Carcinoma Study Group. J Clin Endocrinol Metab. 1996;81:1780–1783. doi: 10.1210/jc.81.5.1780. Available from: http://dx.doi.org/10.1210/jc.81.5.1780. [DOI] [PubMed] [Google Scholar]
- 149.Shapiro SE, Cote GC, Lee JE, Gagel RF, Evans DB. The role of genetics in the surgical management of familial endocrinopathy syndromes. J Am Coll Surg. 2003;197:818–831. doi: 10.1016/j.jamcollsurg.2003.07.001. Available from: http://dx.doi.org/10.1016/j.jamcollsurg.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 150.Walther MM, Reiter R, Keiser HR, Choyke PL, Venzon D, Hurley K, Gnarra JR, Reynolds JC, Glenn GM, Zbar B, Linehan WM. Clinical and genetic characterization of pheochromocytoma in von Hippel-Lindau families: comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. J Urol. 1999;162:659–664. doi: 10.1097/00005392-199909010-00004. Available from: http://dx.doi.org/10.1097/00005392-199909010-00004. [DOI] [PubMed] [Google Scholar]
- 151.Chase WH. Familial and bilateral tumors of the carotid body. J Pathol Bact. 1933;36:1–12. doi: 10.1002/path.1700360102. Available from: http://dx.doi.org/10.1002/path.1700360102. [DOI] [Google Scholar]
- 152.Goekoop C. Fibrohaemangioma des Felsenbeines und des Mittelohres bei drei Schwestern. Acta Otolaryngol. 1933;18:153–162. doi: 10.3109/00016483309134897. Available from: http://dx.doi.org/10.3109/00016483309134897. [DOI] [Google Scholar]
- 153.Linn H, Proctor B. Tumour of the gangliom nodosum of the vagus nerve. Laryngoscope. 1956;66:1577–1581. doi: 10.1288/00005537-195612000-00003. Available from: http://dx.doi.org/10.1288/00005537-195612000-00003. [DOI] [PubMed] [Google Scholar]
- 154.Niemann S, Müller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nature Genet. 2000;26:268–270. doi: 10.1038/81551. Available from: http://dx.doi.org/10.1038/81551. [DOI] [PubMed] [Google Scholar]
- 155.Astuti D, Latif F, Dallol A, Dahia PLM, Douglas F, George E, Sköldberg F, Husebye ES, Eng C, Maher ER. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49–54. doi: 10.1086/321282. Available from: http://dx.doi.org/10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cascon A, Ruiz-Llorente, Cebrian A, Telleria D, Rivero JC, Diez JJ, Lopez-Ibarra PJ, Jaunsolo MA, Benitez J, Robledo M. Identification of novel SDHD mutations in patients with phaeochromocytoma and/or paraganglioma. Eur J Hum Gen. 2002;10:457–461. doi: 10.1038/sj.ejhg.5200829. Available from: http://dx.doi.org/10.1038/sj.ejhg.5200829. [DOI] [PubMed] [Google Scholar]
- 157.Burnichon N, Brière JJ, Libé R, Vescovo L, Rivière J, Tissier F, Jouanno E, Jeunemaitre X, Bénit P, Tzagoloff A, Rustin P, Bertherat J, Favier J, Gimenez-Roqueplo AP. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–3020. doi: 10.1093/hmg/ddq206. Available from: http://dx.doi.org/10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, Khau Van Kien P, Corvol P, Plouin PF, Jeunemaitre X COMETE Network. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003;63:5615–5621. [PubMed] [Google Scholar]
- 159.Niemann S, Müller U, Engelhardt D, Lohse P. Autosomal dominant malignant and catecholamine-producing paraganglioma caused by a splice donor site mutation in SDHC. Hum Genet. 2003;113:92–94. doi: 10.1007/s00439-003-0938-0. [DOI] [PubMed] [Google Scholar]
- 160.Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG, Gygi SP, Winge DR, Kremer H, Rutter J. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–1142. doi: 10.1126/science.1175689. Available from: http://dx.doi.org/10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bayley JP, Kunst HP, Cascon A, Sampietro ML, Gaal J, Korpershoek E, Hinojar-Gutierrez A, Timmers HJ, Hoefsloot LH, Hermsen MA, Suárez C, Hussain AK, Vriends AH, Hes FJ, Jansen JC, Tops CM, Corssmit EP, de Knijff P, Lenders JW, Cremers CW, Devilee P, Dinjens WN, de Krijger RR, Robledo M. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010;11:366–372. doi: 10.1016/S1470-2045(10)70007-3. Available from: http://dx.doi.org/10.1016/S1470-2045(10)70007-3. [DOI] [PubMed] [Google Scholar]
- 162.Eng C, Kiuru M, Fernandez MJ, Aaltonen LA. A role for mitochondrial enzymes in inherited neoplasia and beyond. Nature Rev Cancer. 2003;3:193–202. doi: 10.1038/nrc1013. Available from: http://dx.doi.org/10.1038/nrc1013. [DOI] [PubMed] [Google Scholar]
- 163.Kantorovich V, King KS, Pacak K. SDH-related pheochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab. 2010;24:415–442. doi: 10.1016/j.beem.2010.04.001. Available from: http://dx.doi.org/10.1016/j.beem.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Renard L, Godfraind C, Boon LM, Vikkula M. A novel mutation in the SDHD gene in a family with inherited paragangliomas - implications of genetic diagnosis for follow-up and treatment. Head Neck. 2003;25:146–151. doi: 10.1002/hed.10220. Available from: http://dx.doi.org/10.1002/hed.10220. [DOI] [PubMed] [Google Scholar]
- 165.Scheffler IE. Molecular genetics of succinate : quinolone oxidoreductase in eukaryotes. Prog Nucleic Acid Res Mol Biol. 1998;60:267–315. doi: 10.1016/S0079-6603(08)60895-8. Available from: http://dx.doi.org/10.1016/S0079-6603(08)60895-8. [DOI] [PubMed] [Google Scholar]
- 166.van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EM, Sleddens HF, Derkx P, Rivière J, Dannenberg H, Petri BJ, Komminoth P, Pacak K, Hop WC, Pollard PJ, Mannelli M, Bayley JP, Perren A, Niemann S, Verhofstad AA, de Bruïne AP, Maher ER, Tissier F, Méatchi T, Badoual C, Bertherat J, Amar L, Alataki D, Van Marck E, Ferrau F, François J, de Herder WW, Peeters MP, van Linge A, Lenders JW, Gimenez-Roqueplo AP, de Krijger RR, Dinjens WN. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 2009;10:764–771. doi: 10.1016/S1470-2045(09)70164-0. Available from: http://dx.doi.org/10.1016/S1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wohllk N, Schweizer H, Erlic Z, Schmid KW, Walz MK, Raue F, Neumann HP. Multiple endocrine neoplasia type 2. Best Pract Res Clin Endocrinol Metab. 2010;24:371–387. doi: 10.1016/j.beem.2010.02.001. Available from: http://dx.doi.org/10.1016/j.beem.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 168.Raue F, Frank-Raue K. Update multiple endocrine neoplasia type 2. Fam Cancer. 2010;9:449–457. doi: 10.1007/s10689-010-9320-2. Available from: http://dx.doi.org/10.1007/s10689-010-9320-2. [DOI] [PubMed] [Google Scholar]
- 169.Boedeker CC, Erlic Z, Richard S, Kontny U, Gimenez-Roqueplo AP, Cascon A, Robledo M, de Campos JM, van Nederveen FH, de Krijger RR, Burnichon N, Gaal J, Walter MA, Reschke K, Wiech T, Weber J, Rückauer K, Plouin PF, Darrouzet V, Giraud S, Eng C, Neumann HP. Head and Neck Paragangliomas in Von Hippel-Lindau Disease and Multiple Endocrine Neoplasia Type 2. J Clin Endocrinol Metab. 2009;94:1938–1944. doi: 10.1210/jc.2009-0354. Available from: http://dx.doi.org/10.1210/jc.2009-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kennedy DW, Nager GT. Glomus tumor and multiple endocrine neoplasia. Otolaryngol Head Neck Surg. 1986;94:644–648. doi: 10.1177/019459988609400621. [DOI] [PubMed] [Google Scholar]
- 171.Bausch B, Borozdin W, Neumann HP. European-American Pheochromocytoma Study Group. Clinical and genetic characteristics of patients with neurofibromatosis typeand pheochromocytoma. N Engl J Med. 2006;354:2729–2731. doi: 10.1056/NEJMc066006. Available from: http://dx.doi.org/10.1056/NEJMc066006. [DOI] [PubMed] [Google Scholar]
- 172.Walther MM, Herring J, Enquist E, Keiser HR, Linehan WM. von Recklinghausen's disease and pheochromocytomas. J Urol. 1999;162:1582–1586. doi: 10.1016/S0022-5347(05)68171-2. Available from: http://dx.doi.org/10.1016/S0022-5347(05)68171-2. [DOI] [PubMed] [Google Scholar]
- 173.DeAngelis LM, Kelleher MB, Post KD, Fetell MR. Multiple paragangliomas in neurofibromatosis: a new neuroendocrine neoplasia. Neurology. 1987;37:129–133. doi: 10.1212/wnl.37.1.129. [DOI] [PubMed] [Google Scholar]
- 174.Neumann HPH, Cybulla M, Glaesker S, Coulin C, van Velthoven V, Berlis A, Hader C, Schaefer O, Treier M, Brink I, Schultze-Seemann W, Leiber C, Rückauer K, Junker B, Agostini HT, Hetzel A, Boedeker CC. Von-Hippel-Lindau-Erkrankung: Interdisziplinäre Patientenversorgung. [Von-Hippel-Lindau disease. Interdisciplinary patient care]. Der Ophthalmologe. 2007;104:119–126. doi: 10.1007/s00347-006-1470-0. (Ger). Available from: http://dx.doi.org/10.1007/s00347-006-1470-0. [DOI] [PubMed] [Google Scholar]
- 175.Gaal J, van Nederveen FH, Erlic Z, Korpershoek E, Oldenburg R, Boedeker CC, Kontny U, Neumann HP, Dinjens WNM, de Krijger RR. Parasympathetic paragangliomas are part of the von Hippel-Lindau syndrome. J Clin Endocrinol Metab. 2009;94:4367–4371. doi: 10.1210/jc.2009-1479. Available from: http://dx.doi.org/10.1210/jc.2009-1479. [DOI] [PubMed] [Google Scholar]
- 176.Bausch B, Boedeker CC, Berlis A, Brink I, Cybulla M, Walz MK, Januszewicz A, Letizia C, Opocher G, Eng C, Neumann HPH. Genetic and clinical investigation of pheochromocytoma: A 22-year experience, from Freiburg, Germany to international effort. Ann N Y Acad Sci. 2006;1073:122–137. doi: 10.1196/annals.1353.013. Available from: http://dx.doi.org/10.1196/annals.1353.013. [DOI] [PubMed] [Google Scholar]
- 177.Eisenhofer G, Lenders JW, Linehan WM, Walther MM, Goldstein DS, Keiser HR. Plasma normetanephrine and metanephrine for detecting pheochromocytoma in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. N Engl J Med. 1999;340:1872–1879. doi: 10.1056/NEJM199906173402404. Available from: http://dx.doi.org/10.1056/NEJM199906173402404. [DOI] [PubMed] [Google Scholar]
- 178.Schipper J, Maier W, Rosahl S, van Velthoven V, Berlis A, Boedeker CC, Laszig R, Teszler CB, Ridder GJ. Endolymphatic sac tumors: surgical management. J Otolaryngol. 2006;35:387–394. doi: 10.2310/7070.2006.0082. Available from: http://dx.doi.org/10.2310/7070.2006.0082. [DOI] [PubMed] [Google Scholar]
- 179.Ercolino T, Becherini L, Valeri A, Maiello M, Gagliano MS, Parenti G, Ramazzotti M, Piscitelli E, Simi L, Pinzani P, Nesi G, Degl'Innocenti D, Console N, Bergamini C, Mannelli M. Uncommon clinical presentations of pheochromocytoma and paraganglioma in two different patients affected by two distinct novel VHL germline mutations. Clin Endocrinol (Oxf) 2008;68:762–768. doi: 10.1111/j.1365-2265.2007.03131.x. Available from: http://dx.doi.org/10.1111/j.1365-2265.2007.03131.x. [DOI] [PubMed] [Google Scholar]
- 180.Hull MT, Roth LM, Glover JL, Walker PD. Metastatic carotid body paraganglioma in von Hippel-Lindau disease. An electron microscopic study. Arch Pathol Lab Med. 1982;106:235–239. [PubMed] [Google Scholar]
- 181.Gross DJ, Avishai N, Meiner V, Filon D, Zbar B, Abeliovich D. Familial pheochromocytoma associated with a novel mutation in the von Hippel-Lindau gene. J Clin Endocrinol Metab. 1996;81:147–149. doi: 10.1210/jc.81.1.147. Available from: http://dx.doi.org/10.1210/jc.81.1.147. [DOI] [PubMed] [Google Scholar]
- 182.Schimke RN, Collins DL, Rothberg PG. Functioning carotid paraganglioma in the von Hippel-Lindau syndrome. Am J Med Genet. 1998;80:533–534. doi: 10.1002/(SICI)1096-8628(19981228)80:5<533::AID-AJMG21>3.0.CO;2-C. Available from: http://dx.doi.org/10.1002/(SICI)1096-8628(19981228)80:5<533::AID-AJMG21>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 183.Zanelli M, van der Walt JD. Carotid body paraganglioma in von Hippel-Lindau disease: a rare association. Histopathology. 1996;29:178–181. doi: 10.1046/j.1365-2559.1996.d01-505.x. Available from: http://dx.doi.org/10.1046/j.1365-2559.1996.d01-505.x. [DOI] [PubMed] [Google Scholar]
- 184.Schiavi F, Boedeker CC, Bausch B, Peçzkowska M, Fuentes Gomez C, Strassburg T, Pawlu C, Buchta M, Salzmann M, Hoffmann MM, Berlis A, Brink I, Cybulla M, Muresan M, Walter MA, Forrer F, Välimäki M, Kawecki A, Szutkowski Z, Schipper J, Walz MK, Pigny P, Bauters C, Willett-Brozick JE, Baysal BE, Januszewicz A, Eng C, Opocher G, Neumann HPH. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA. 2005;294:2057–2063. doi: 10.1001/jama.294.16.2057. Available from: http://dx.doi.org/10.1001/jama.294.16.2057. [DOI] [PubMed] [Google Scholar]
- 185.Ricketts CJ, Forman JR, Rattenberry E, Bradshaw N, Lalloo F, Izatt L, Cole TR, Armstrong R, Kumar VK, Morrison PJ, Atkinson AB, Douglas F, Ball SG, Cook J, Srirangalingam U, Killick P, Kirby G, Aylwin S, Woodward ER, Evans DG, Hodgson SV, Murday V, Chew SL, Connell JM, Blundell TL, Macdonald F, Maher ER. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat. 2010;31:41–51. doi: 10.1002/humu.21136. Available from: http://dx.doi.org/10.1002/humu.21136. [DOI] [PubMed] [Google Scholar]
- 186.Hensen EF, Jansen JC, Siemers MD, Oosterwijk JC, Vriends AH, Corssmit EP, Bayley JP, van der Mey AG, Cornelisse CJ, Devilee P. The Dutch founder mutation SDHD.D92Y shows a reduced penetrance for the development of paragangliomas in a large multigenerational family. Eur J Hum Genet. 2010;18:62–66. doi: 10.1038/ejhg.2009.112. Available from: http://dx.doi.org/10.1038/ejhg.2009.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hes FJ, Weiss MM, Woortman SA, de Miranda NF, van Bunderen PA, Bonsing BA, Stokkel MP, Morreau H, Romijn JA, Jansen JC, Vriends AH, Bayley JP, Corssmit EP. Low penetrance of a SDHB mutation in a large Dutch paraganglioma family. BMC Med Genet. 2010;11:92. doi: 10.1186/1471-2350-11-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hensen EF, Jordanova ES, van Minderhout IJ, Hogendoorn PC, Taschner PE, van der Mey AG, Devilee P, Cornelisse CJ. Somatic loss of maternal chromosomecauses parent-of-origin-dependent inheritance in SDHD-linked paraganglioma and phaeochromocytoma families. Oncogene. 2004;23:4076–4083. doi: 10.1038/sj.onc.1207591. [DOI] [PubMed] [Google Scholar]
- 189.van Baars F, Cremers C, van den Brock P, Geerts S, Veldman J. Genetic aspects of nonchromaffin paraganglioma. Hum Genet. 1982;60:305–309. doi: 10.1007/BF00569208. Available from: http://dx.doi.org/10.1007/BF00569208. [DOI] [PubMed] [Google Scholar]
- 190.Bauters C, Vantyghem MC, Leteurtre E, Odou MF, Mouton C, Porchet N, Wemeau JL, Proye C, Pigny P. Hereditary phaeochromocytomas and paragangliomas: a study of five susceptibility genes. J Med Genet. 2003;40:e75. doi: 10.1136/jmg.40.6.e75. Available from: http://dx.doi.org/10.1136/jmg.40.6.e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Baysal BE, Willett-Brozick JE, Filho PA, Lawrence EC, Myers EN, Ferrell RE. An Alu-mediated partial SDHC deletion causes familial and sporadic paraganglioma. J Med Genet. 2004;41:703–709. doi: 10.1136/jmg.2004.019224. Available from: http://dx.doi.org/10.1136/jmg.2004.019224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Mannelli M, Ercolino T, Giachè V, Simi L, Cirami C, Parenti G. Genetic screening for pheochromocytoma: should SDHC gene analysis be included? J Med Genet. 2007;44:586–587. doi: 10.1136/jmg.2007.051045. Available from: http://dx.doi.org/10.1136/jmg.2007.051045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Erlic Z, Rybicki L, Peczkowska M, Golcher H, Kann PH, Brauckhoff M, Müssig K, Muresan M, Schäffler A, Reisch N, Schott M, Fassnacht M, Opocher G, Klose S, Fottner C, Forrer F, Plöckinger U, Petersenn S, Zabolotny D, Kollukch O, Yaremchuk S, Januszewicz A, Walz MK, Eng C, Neumann HP European-American Pheochromocytoma Study Group. Clinical predictors and algorithm for the genetic diagnosis of pheochromocytoma patients. Clin Cancer Res. 2009;15:6378–6385. doi: 10.1158/1078-0432.CCR-09-1237. Available from: http://dx.doi.org/10.1158/1078-0432.CCR-09-1237. [DOI] [PubMed] [Google Scholar]
- 194.Bayley JP, van Minderhout I, Weiss MM, Jansen JC, Oomen PH, Menko FH, Pasini B, Ferrando B, Wong N, Alpert LC, Williams R, Blair E, Devilee P, Taschner PE. Mutation analysis of SDHB and SDHC: novel germline mutations in sporadic head and neck paraganglioma and familial paraganglioma and/or pheochromocytoma. BMC Med Genet. 2006;7: Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peczkowska M, Morrison CD, Lehtonen R, Januszewicz A, Jarvinen H, Juhola M, Mecklin JP, Pukkala E, Herva R, Kiuru M, Nupponen NN, Aaltonen LA, Neumann HP, Eng C. Early onset renal cell carcinoma as novel extra-paraganglial component of SDHB-associated hereditable paraganglioma. Am J Hum Genet. 2004;74:153–159. [Google Scholar]
- 195.Vanharanta S, Buchta M, McWhinney SR, Virta SK, Peczkowska M, Morrison CD, Lehtonen R, Januszewicz A, Jarvinen H, Juhola M, Mecklin JP, Pukkala E, Herva R, Kiuru M, Nupponen NN, Aaltonen LA, Neumann HP, Eng C. Early onset renal cell carcinoma as novel extra-paraganglial component of SDHB-associated hereditable paraganglioma. Am J Hum Genet. 2004;74:153–159. doi: 10.1086/381054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Benn DE, Gimenez-Roqueplo AP, Reilly JR, Bertherat J, Burgess J, Byth K, Croxson M, Dahia PL, Elston M, Gimm O, Henley D, Herman P, Murday V, Niccoli-Sire P, Pasieka JL, Rohmer V, Tucker K, Jeunemaitre X, Marsh DJ, Plouin PF, Robinson BG. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91:827–836. doi: 10.1210/jc.2005-1862. Available from: http://dx.doi.org/10.1210/jc.2005-1862. [DOI] [PubMed] [Google Scholar]
- 197.Burnichon N, Rohmer V, Amar L, Herman P, Leboulleux S, Darrouzet V, Niccoli P, Gaillard D, Chabrier G, Chabolle F, Coupier I, Thieblot P, Lecomte P, Bertherat J, Wion-Barbot N, Murat A, Venisse A, Plouin PF, Jeunemaitre X, Gimenez-Roqueplo AP PGL.NET network. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab. 2009;94:2817–2827. doi: 10.1210/jc.2008-2504. Available from: http://dx.doi.org/10.1210/jc.2008-2504. [DOI] [PubMed] [Google Scholar]
- 198.Birch-Machin MA, Taylor RW, Cochran B, Ackrell BA, Turnbull DM. Late-onset optic atrophy, ataxia and myopathy associated with a mutation of a complex II gene. Ann Neurol. 2000;48:330–335. doi: 10.1002/1531-8249(200009)48:3<330::AID-ANA7>3.0.CO;2-A. Available from: http://dx.doi.org/10.1002/1531-8249(200009)48:3<330::AID-ANA7>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 199.Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, Lechleiter JD, Sass M, Aronin N, Schiavi F, Boaretto F, Opocher G, Toledo RA, Toledo SP, Stiles C, Aguiar RC, Dahia PL. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42:229–233. doi: 10.1038/ng.533. Available from: http://dx.doi.org/10.1038/ng.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Carney JA, Sheps SG, Go VL, Gordon H. The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma. N Engl J Med. 1977;296:1517–1518. doi: 10.1056/NEJM197706302962609. Available from: http://dx.doi.org/10.1056/NEJM197706302962609. [DOI] [PubMed] [Google Scholar]
- 201.Grace MP, Batist G, Grace WR, Gillooley JF. Aorticopulmonary paraganglioma and gastric leiomyoblastoma in a young woman. Am J Med. 1981;70:1288–1292. doi: 10.1016/0002-9343(81)90840-8. Available from: http://dx.doi.org/10.1016/0002-9343(81)90840-8. [DOI] [PubMed] [Google Scholar]
- 202.Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132–139. doi: 10.1002/ajmg.10235. Available from: http://dx.doi.org/10.1002/ajmg.10235. [DOI] [PubMed] [Google Scholar]
- 203.Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney Triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543–552. doi: 10.4065/74.6.543. Available from: http://dx.doi.org/10.4065/74.6.543. [DOI] [PubMed] [Google Scholar]
- 204.McWhinney SR, Pasini B, Stratakis CA International Carney Triad and Carney-Stratakis Syndrome Consortium. Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med. 2007;357:1054–1056. doi: 10.1056/NEJMc071191. Available from: http://dx.doi.org/10.1056/NEJMc071191. [DOI] [PubMed] [Google Scholar]
- 205.Statement of the American Society of Clinical Oncology: genetic testing for cancer susceptibility, adopted on February 20, 1996. J Clin Oncol. 1996;14:1730–1736. doi: 10.1200/JCO.1996.14.5.1730. [DOI] [PubMed] [Google Scholar]
- 206.Neumann HPH, Bender BU, Reincke M, Eggstein S, Laubenberger J, Kirste G. Adrenal sparing surgery for pheochromocytoma. Brit J Surg. 1999;84:94–97. doi: 10.1046/j.1365-2168.1999.00974.x. Available from: http://dx.doi.org/10.1046/j.1365-2168.1999.00974.x. [DOI] [PubMed] [Google Scholar]
- 207.Neumann HPH, Reincke M, Bender BU, Elsner R, Janetschek G. Preserved adrenocortical function after laparoscopic bilateral adrenal sparing surgery for hereditary Pheochromocytoma. J Clin Endocrinol Metab. 1999;84:2608–2610. doi: 10.1210/jc.84.8.2608. Available from: http://dx.doi.org/10.1210/jc.84.8.2608. [DOI] [PubMed] [Google Scholar]
- 208.Walz MK, Peitgen K, Neumann HPH, Janssen OE, Philipp T, Mann K. Endoscopic treatment of solitary, bilateral multiple, and recurrent pheochromocytomas and paragangliomas. World J Surg. 2002;26:1005–1012. doi: 10.1007/s00268-002-6632-x. Available from: http://dx.doi.org/10.1007/s00268-002-6632-x. [DOI] [PubMed] [Google Scholar]
- 209.Rinaldo A, Myssiorek D, Devaney KO, Ferlito A. Which paragangliomas of the head and neck have a higher rate of malignancy? Oral Oncol. 2004;40:458–460. doi: 10.1016/j.oraloncology.2003.08.018. Available from: http://dx.doi.org/10.1016/j.oraloncology.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 210.Schipper J, Arapakis I, Ridder GJ, Maier W, Spetzger U. Resektion von Foramen-jugulare-Tumoren mit vollständigem Hörerhalt - Mikrochirurgie ohne Transposition des N. facialis. [Microsurgical resection of jugular foramen tumors with hearing preservation and without facial nerve palsy]. HNO. 2003;51:721–727. doi: 10.1007/s00106-003-0805-2. (Ger). Available from: http://dx.doi.org/10.1007/s00106-003-0805-2. [DOI] [PubMed] [Google Scholar]
- 211.Plukker JT, Brongers EP, Vermey A, Krikke A, van den Dungen JJ. Outcome of surgical treatment for carotid body paraganglioma. Br J Surg. 2001;88:1382–1386. doi: 10.1046/j.0007-1323.2001.01878.x. Available from: http://dx.doi.org/10.1046/j.0007-1323.2001.01878.x. [DOI] [PubMed] [Google Scholar]
- 212.Tikkakoski T, Luotonen J, Leinonen S, Siniluoto T, Heikkilä O, Päivänsalo M, Hyrynkangas K. Preoperative embolization in the management of neck paragangliomas. Laryngoscope. 1997;107:821–826. doi: 10.1097/00005537-199706000-00018. Available from: http://dx.doi.org/10.1097/00005537-199706000-00018. [DOI] [PubMed] [Google Scholar]
- 213.Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) Best Pract Res Clin Endocrinol Metab. 2010;24:355–370. doi: 10.1016/j.beem.2010.07.003. Available from: http://dx.doi.org/10.1016/j.beem.2010.07.003. [DOI] [PubMed] [Google Scholar]