

Abstract
Purpose of review
Statins are widely utilized for low-density lipoprotein lowering and for prevention of atherosclerotic cardiovascular disease. Although these drugs have a good safety record, increased risk of developing diabetes during extended use has recently garnered attention. Here we review clinical trial evidence related to statin use and incident diabetes, and the potential mechanisms for this association.
Recent findings
The increased incidence of diabetes with rosuvastatin treatment in Justification for the Use of Statins in Primary Prevention: an intervention Trial Evaluating Rosuvastatin (JUPITER) reignited attention on the link between statin therapy and diabetes. The JUPITER findings are supported by two recent meta-analyses of large-scale placebo-controlled and standard care-controlled trials, which, respectively, observed a 9% [odds ratio 1.09; 95% confidence interval (CI) 1.02–1.17] and 13% (risk ratio 1.13; 95% CI 1.03–1.23) increased risk for incident diabetes associated with statin therapy. However, the underlying mechanisms for this association remain unclear. Experimental evidence supports a paradigm implicating inhibition of β-cell glucose transporters, delayed ATP production, pro-inflammatory and oxidative β-cell effects of plasma-derived cholesterol, inhibition of calcium channel-dependent insulin secretion, and β-cell apoptosis.
Summary
The aggregate of large clinical trials supports the notion that statins modestly increase the risk of incident diabetes. Because diabetes is a risk equivalent condition for coronary and peripheral arterial diseases, these findings create a paradox whereby needed statin therapy may be withheld to avoid excess risk of diabetes while representing the strongest cardiovascular risk reduction tool in diabetics. We simply recommend regular glucose monitoring in patients taking statins.
Keywords: β-cells, cardiovascular disease, diabetes, low-density lipoprotein cholesterol, statins
Introduction
Over the last two decades, multiple clinical trials totaling 170 000 participants have demonstrated the beneficial effects of statins, or 3-hydroxy-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, in the reduction of cardiovascular event rates [1•]. Consequently, statins have become permanent fixtures in our armamentarium for the primary and secondary prevention of atherosclero-tic cardiovascular disease, including stroke [2–5]. However, concerns have arisen regarding the potential risk for developing type 2 diabetes mellitus (T2DM) during extended statin use. It is important to point out that the first report on an association between statin use and incident diabetes was derived from post-hoc analyses of the West of Scotland Coronary Prevention Study (WOSCOP) reported in 2001 [6]. In this study, a 30% [risk ratio 0.7; 95% confidence interval (CI) 0.50–0.99] risk reduction for incident diabetes was reported in association with the use of 40 mg of pravastatin in a randomized placebo-controlled trial of 5974 patients [6]. This protective effect was not corroborated by later trials of simvastatin [7,8], pravastatin [9–12], and atorvastatin [13]. The 2008 report of increased incident diabetes among patients taking rosuvastatin in the Justification for the Use of Statins in Primary Prevention: an intervention Trial Evaluating Rosuvastatin (JUPITER) [14] reignited attention and triggered discussion on the direction and strength of the association between statin therapy and diabetes. WOSCOP and JUPITER constitute book-ends of the spectrum of trials that provide conflicting evidence on the diabetogenicity of statins, and mirror the uncertainties in the basic science arena surrounding the potential mechanisms of statin-induced impairment of glucose metabolism. Conceivably, a loss of the functional integrity of the islet β-cells may be central to this process. Although clinical trial evidence for an effect of statins on insulin sensitivity is conflicting, there is experimental evidence to support a paradigm that implicates inhibition of glucose transporters, delayed ATP production, pro-inflammatory and oxidative intracellular effects of plasma-derived cholesterol, inhibition of calcium channel-dependent insulin secretion, and apoptosis. In this article, we discuss the evidence for the diabetogenic effects of extended statin use based on the aggregate of large-scale placebo-controlled and standard care-controlled trials of statin therapy on cardiovascular outcomes. In addition, we propose a mechanistic paradigm for the diabetogenic effects of statins.
Insights from clinical trials
The account from WOSCOP introduced the notion that extended statin use – 40 mg of pravastatin – may be associated with decreased incidence of diabetes [6]. Of note, this was a post-hoc analysis of 5974 patients of whom 139 developed new-onset diabetes as defined by at least 2 mmol/l (36 mg/dl) rise in blood glucose above baseline values – a criterion for diabetes inconsistent with conventional clinical practice [6]. Even so, the reported risk ratio of 0.7 for incident diabetes was only marginally statistically significant, as reflected by the upper limit of the confidence interval (95% CI 0.5–0.99), the width of which reflects the small number of cases of incident diabetes and limited precision in the risk estimate [6]. The WOSCOP investigators acknowledged that their criterion for diabetes restricted the number of patients classified as newly diabetic. In the aftermath of the WOSCOP report, numerous placebo-controlled and standard care-controlled trials of statin therapy have provided conflicting reports regarding statin use and diabetes. Such conflicting observations could stem from the fact that some individual studies lacked the statistical power and event rate (of incident diabetes) required to demonstrate the association. Given such a potential scenario, a systematic review of multiple studies with quantitative synthesis of data – meta-analysis – can be particularly helpful for determining the statistical robustness of any association. In this context, a meta-analysis of 13 [6–18] randomized placebo-controlled and standard care-controlled trials was recently conducted using published and unpublished data. Sattar et al. [19••] demonstrated a 9% increased risk of incident diabetes (odds ratio 1.09; 95% CI 1.02–1.17) in their analysis of 91 140 participants, among whom 4278 developed diabetes. Furthermore, metaregression analysis demonstrated that age was associated with the risk of incident diabetes, with stronger statin-attributable risk found in studies with elderly patients [19••]. Neither change in low-density lipoprotein (LDL)-cholesterol during treatment nor baseline BMI was found associated with the risk of new-onset diabetes. There was little heterogeneity among the component studies of the meta-analysis.
In another study, Rajpathak et al. [20] reported similar findings in their synthesis of five trials [8,13,14,16,17] involving 51 619 participants, among whom 1943 developed diabetes. There was no heterogeneity among the component studies. The risk of incident diabetes was 13% (risk ratio 1.13; 95% CI 1.03–1.23); however, upon adding the WOSCOP study results, heterogeneity among the component studies became significant, and attenuation of the effect size resulted in loss of statistical significance [20]. Notably, Sattar et al. [19••] included the WOSCOP study in their meta-analysis but eliminated the nonstandard criteria for diabetes employed by Freeman et al. [6] in their original post-hoc analyses of WOSCOP. However, Sattar et al. found that even the inclusion of WOSCOP data, as in the 2001 report, did not change the outcome of their meta-analysis, perhaps due to greater statistical power in their study [6,19••]. Collectively, the consistency in the findings from two independent meta-analyses suggests that statin use is indeed associated with increased risk of incident diabetes, although small and of no clear practical relevance. Furthermore, the fact that there was no significant heterogeneity between the studies synthesized suggests that the increased incidence of diabetes is secondary to a class effect as opposed to any apparent differences between drugs, such as their compartmentalization (lipophilic versus hydrophilic), metabolism [Cytochrome P450 (CYP)3A4, CYP2C9, UDP glucuronosyltransferase (UGT), etc.], potency, and half-life. Contrary to the findings of the above-discussed meta-analyses, another meta-analysis from Coleman et al. [21] reported that statins as a class are not associated with increased risk (risk ratio 1.03; 95% CI 0.89–1.19) of new-onset diabetes. However, in their meta-analysis of five prospective randomized controlled trials involving 39 791 patients, among whom 1407 developed diabetes, they found significant statistical heterogeneity. Notably, as in the study by Rajpathak et al., upon removal of the WOSCOP study from their analysis there was little or no heterogeneity, and a significant increase in the relative risk of developing diabetes was noted (risk ratio 1.14; 95% CI 1.02–1.28) [21]. It is also important to note that this meta-analysis was performed before the publication of JUPITER.
The above three meta-analyses, published sequentially, provide incremental advances in our understanding of the clinical trial evidence for the association between statins and incident diabetes. To put these papers in a chronologic perspective, the reports by Coleman et al. [21] (pre-JUPITER) and that by Rajpathak et al. [20] (post-JUPITER) both demonstrate increased risk of incident diabetes with statin use, an effect that in each report was attenuated upon inclusion of WOSCOP due to heterogeneity despite the increase in sample size. The most recent report by Sattar et al. [19••] appears to have conclusively addressed the controversy of statin use and risk of incident diabetes, as it represents the most comprehensive synthesis of data to date – devoid of heterogeneity – with adequate statistical power. Thus, we believe that extended statin use is associated with a modest increase in the risk of incident diabetes. Despite this conclusion, a cautionary note is important. Although meta-analyses are based on rigorous statistical methodology, they are not controlled experiments, but rather observational studies that rely on previously published data. Consequently, meta-analyses are, at best, hypothesis-generating studies. However, there are mechanistic and experimental studies that support the notion that extended statin use may alter glucose metabolism, thus lending a biologic platform to the conclusion that must be derived from the meta-analyses.
Practical clinical considerations
The development of diabetes during extended use of statin is clinically concerning because diabetes is a risk equivalent condition for cardiovascular disease. Thus, questions arise about the potential clinical predictors of incident diabetes, the effect of statins on blood sugar levels in diabetic and nondiabetic patients, and the clinical efficacy of statins in patients with diabetes or precursor conditions such as insulin resistance and metabolic syndrome. A recent analysis of 345 000 patients in the Veteran Affairs Healthcare System indicates that, after adjustment for age and use of aspirin, β-blockers, and angiotensin-converting enzyme inhibitors, the statin-attributable change in fasting plasma glucose was 2 mg/dl (7 mg/dl increase, versus 5 mg/dl in nonstatin users, P < 0.0001) for nondiabetic users and 7 mg/dl (39 mg/dl increase, versus 32 mg/dl in nonstatin users, P < 0.0001) for diabetic users [22]. The strong statistical significance reported in this study – really a measure of strength in the sample size – should be balanced by the clinically small relevance of such changes. Herein, the insight from Sattar et al. [19••] is very relevant: the absolute risk of developing diabetes was one case per 1000 patient-years of treatment, which is very small relative to the reduction in vascular events. Regarding the latter, the efficacy of statins is now well established even at low LDL-cholesterol concentrations (<77 mg/dl) [1•]. Furthermore, the benefit of statins in diabetics has been established and is independent of the type of diabetes, lipid profile, history of vascular disease, and baseline characteristics [23,24]. Thus, the widespread benefits of statin coverage far outweigh the small risk of incident diabetes. It is important to note that in JUPITER, the trial that more than any other has implicated statin use with development of T2DM, the patients who at baseline had impaired fasting glucose showed a 34% cardiovascular risk reduction attributable to rosuvastatin. Unlike the arena of atypical antipsychotic use, in which it is recommended to assess the likelihood of developing diabetes and severe metabolic disease by paying attention to baseline predictors, such as obesity and hyperglycemia [25,26], no biochemical or clinical indicators for the occurrence of incident diabetes during extended statin use were identified in the meta-analyses, except for age of the patient [19••]. Therefore, it may at best be prudent to encourage caution with elderly patients, particularly those not falling in the high cardiovascular risk category. The use of statins in low cardiovascular risk patients without dyslipidemia should be balanced against the small risk of incident diabetes or potential consequences of prolonged hyperglycemia. In such low-risk patients, traditional lifestyle modification should remain the staple of intervention, as recommended by current guidelines.
Mechanistic insights
Baker et al. [27•] recently evaluated the collective and individual impact of commonly prescribed statins on insulin sensitivity in a systematic review and meta-analysis of 16 randomized controlled trials involving 1146 nondiabetic patients. These trials employed various methods to measure insulin sensitivity. However, for the purposes of the meta-analysis, insulin sensitivity was analyzed as standardized mean differences (SMDs) between statin and placebo/control groups. Overall, statins had no class effect on insulin sensitivity (SMD −0.084; 95% CI −0.210 to 0.042) [27•]. However, pravastatin was noted to improve insulin sensitivity significantly (SMD 0.342; 95% CI 0.032–0.621), whereas simvastatin worsened it (SMD −0.321; 95% CI −0.526 to −0.117). Regardless, a known parabolic relationship exists between insulin secretion and insulin sensitivity, such that an alteration in the latter may not necessarily translate to increased risk of impaired glucose tolerance. It is thus conceivable that the diabetogenic effects of statins may center on altered islet β-cell insulin secretion via the convergence of multiple mechanisms that compromise the integrity and function of the β-cells, resulting in impaired glucose metabolism (Fig. 1).
Figure 1. Hypothetical paradigm for statin-induced impairment of glucose metabolism.
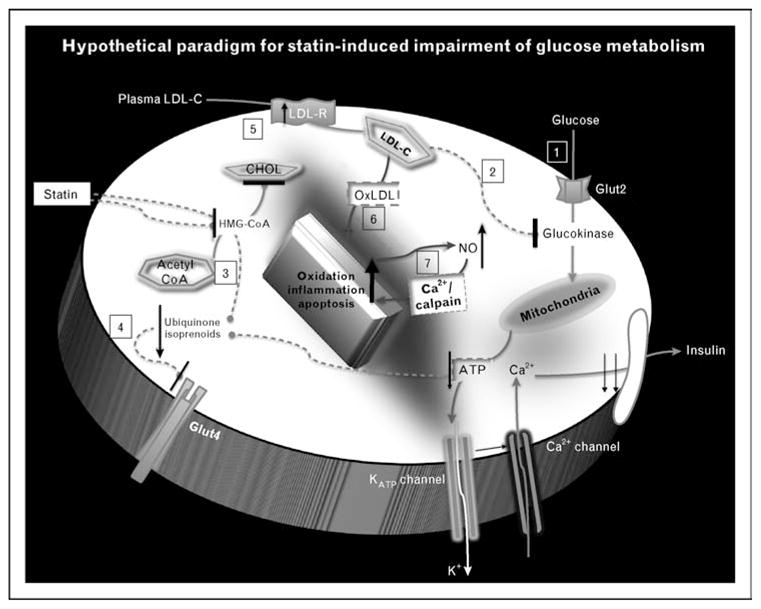
(1) Intracellular arrival of glucose via glucose transporter (Glut2 in β-cells) leads to phosphorylation by glucokinase, and routing to the metabolic pathway. The resulting cascade of closure of ATP-dependent potassium channel, depolarization, and calcium influx leads to insulin secretion; this process may be inhibited by statins [23,24]. (2) Glucokinase is inhibited by abundance of plasma cholesterol [28•], and thus is conceivably affected by statin-induced inhibition of de-novo cholesterol synthesis with increased uptake of plasma LDL. (3) Statin inhibition of HMG-CoA reductase suppresses synthesis of ubiquinone (CoQ10), an essential factor in the mitochondrial electron-transfer system, resulting in inhibition of insulin secretion due to reduced production of ATP [25]. (4) Statin inhibition of HMG-CoA reductase suppresses the synthesis of isoprenoids, thus causing downregulation of Glut4 expression on adipocyte cells, leading to impaired glucose uptake. (5) The inhibition of HMG-CoA reductase causes upregulation of LDL receptors, leading to enhanced uptake of LDL-cholesterol in an effort to replenish intracellular stores. However, the intracellular fate of plasma-derived LDL-cholesterol may be distinct from that of de-novo synthesized cholesterol. (6) The oxidation of LDL-cholesterol may incite an inflammatory cascade that compromises the functional – for example insulin secretion apparatus – and ultimately structural integrity of the islet β-cells. (7) Furthermore, cytokine-induced over-production of nitric oxide (NO) has been shown to induce β-cell apoptosis via the activation of calpain – a calcium-dependent protease [29]. ATP, adenosine triphosphate; CHOL, cholesterol (de-novo synthesized); Glut2, glucose transporter 2; Glut4, glucose transporter 4; HMG-CoA, 3-hydroxy-methylglutaryl coenzyme A; LDL, low-density lipoprotein; LDL-C, low-density lipoprotein cholesterol (plasma-derived); LDL-R, LDL receptor; OxLDL, oxidized LDL; NO, nitric oxide.
Impaired cellular glucose uptake, energy production, and insulin secretion
Glucose is transported by glucose transporter 2 (Glut2) into β-cells, in which it is routed to the metabolic pathway after phosphorylation to glucose-6-phospate by glucokinase. The metabolic cascade that ensues involves closing of the ATP-dependent potassium channel, cell membrane depolarization, and L-type calcium channel-mediated calcium influx, resulting in the secretion of insulin by exocytosis of insulin-containing granules [28•]. Statins have been shown to inhibit this glucose-induced calcium signaling-dependent insulin secretion [30]. In addition, statins suppress the synthesis of ubiquinone (CoQ10), an essential factor in the mitochondrial electron-transfer system, resulting in inhibition of insulin secretion due to reduced production of ATP [31]. Furthermore, Nakata et al. [32] demonstrated that statins decrease the expression of another glucose transporter in adipocytes (Glut4), resulting in impaired glucose tolerance. The report by Chamberlain [33] suggests that the downregulation of Glut4 expression by statins occurs via inhibition of the synthesis of isoprenoids. On a different note, cholesterol loading has been shown to inhibit the activity of glucokinase – the rate-limiting enzyme for intracellular glucose metabolism – thus impairing the glucose-induced calcium signaling required for insulin secretion [34]. This represents a mechanism by which plasma-derived LDL-cholesterol, whose cellular entry is highly increased by statins, can impair the glucose-regulating function of β-cells [28•,35•].
β-Cell inflammation, oxidation, and apoptosis
Although statins have well characterized anti-inflammatory actions, the inhibition of de-novo synthesis of cholesterol could have deleterious immune inflammatory consequences within the islet β-cells. The inhibition of HMG-CoA causes upregulation of LDL receptors for enhanced uptake of LDL-cholesterol in an effort to replenish intracellular stores. However, the intracellular fate of plasma-derived LDL-cholesterol may be distinct from that of de-novo synthesized cholesterol. The oxidation of plasma-derived LDL-cholesterol could incite the innate and adaptive arms of the intracellular immune response system, leading to an inflammatory cascade that compromises the functional and structural integrity of the islet β-cells and leads to reduced insulin secretion. A contribution of plasma-derived LDL-cholesterol to inflammation of the β-cells is thus conceivable [36–40]. Furthermore, cytokine-induced over-production of nitric oxide has been shown to induce β-cell apoptosis via the activation of calpain – a calcium-dependent protease [29]. In this context, because statins exert favorable effects on endothelial function by upregulating the production of nitric oxide [41–43] – an important mediator of endothelial function – it is not certain whether this process negatively affects the β-cell. In addition, HDL protects β-cells against apoptosis, whereas LDL has the ability to induce apoptosis, more so following oxidative modification [44–46]. Overall, the interplay between inflammation, oxidation, and apoptosis – all potentially triggered by increased abundance of plasma-derived LDL-cholesterol due to statin-induced blockade of de-novo cholesterol synthesis – may contribute to the pathogenesis of diabetes during extended statin use. These deleterious effects may become more significant in older patients given the age-dependent loss of β-cells. This could explain the observed increase in new-onset diabetes among older participants in clinical trials [19••].
Benefit versus risk
The war against disease, like any other war, has unintended consequences; all drugs have side effects. In the arena of cardiovascular disease treatment, statins have emerged as the undisputed heavyweight champions for lowering cholesterol and inflammation with excellent reduction in cardiovascular event rates. Therefore, the finding that statins increase the risk of incident diabetes should be weighed against the remarkable cardiovascular benefits that have been exhaustively established by clinical trials. Furthermore, it is important to note that statins are not the only beneficial cardiovascular drugs with unfavorable effects on the risk of diabetes. Notably, atypical antipsychotics, β-blockers, thiazide diuretics, and niacin have all been linked to increased incidence of diabetes but remain in use due to their favorable benefit to risk ratio. In the case of statins, data from the Cholesterol Treatment Trialists [47] indicate that 255 (95% CI 150–852) patients have to be treated for 4 years before a statin-induced case of incident diabetes occurs [19••]. However, a composite of nine vascular events (death, myocardial infarction, stroke, and coronary revascularization) would be avoided by treating 255 patients over the same period of time; this clearly outweighs the risk of incident diabetes (9:1 benefit versus risk ratio). Consequently, the potential risk of incident diabetes should not negate the use of statins for primary or secondary prevention in any patient with moderate to high cardiovascular risk. Furthermore, it is important to note that statins are highly effective in preventing cardiovascular events in individuals with T2DM [23,24]. However, because diabetes in and of itself is a risk equivalent condition for coronary and peripheral arterial disease, the potential mechanisms by which statins impair glucose metabolism require further delineation. This may provide insight for the tailored use and monitoring of statin therapy. Traditional lifestyle modification should be primary in low cardiovascular risk populations when the benefits may not offset the associated risk of new-onset diabetes.
Conclusion
Statin drugs are ubiquitously employed in contemporary approaches for primary and secondary prevention of atherosclerotic cardiovascular disease. The use of statins to modulate levels of cholesterol and inflammation has led to remarkable reduction in cardiovascular endpoints at the relatively small but potential risk of new-onset diabetes. The controversy surrounding the diabetogenic effects of statins pales in comparison with the favorable benefit to risk ratio associated with their use in diabetics and nondiabetic patients with moderate to high cardiovascular risk. It is debatable whether lower-risk individuals, particularly if older than 65, may represent a group in which the benefits of therapy do not outweigh the potential risk of incident diabetes.
Key points.
Sustained statin use appears to moderately increase the risk of developing diabetes.
Patients older than 65 may be particularly susceptible to this unwanted effect of therapy.
The mechanism may be related more to induction of β-cell dysfunction and apoptosis than to reduction of insulin sensitivity.
The favorable ratio between cardiovascular benefits of therapy and risk of diabetes (9:1) supports the current approach of widespread statin use in high-risk populations.
For low-risk patients, particularly if older than 65, risk of diabetes should be added to the list of considerations in deciding to start a statin.
Acknowledgments
Funding support for U.K.S. was provided in part by the Harold Amos Medical Faculty Development Award of the Robert Wood Johnson Foundation and the Vanderbilt Clinical and Translational Scholars Award. Drs S.F. and M.F.L. were partially supported by NIH grants HL65709, HL086988, and HL105375.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (p. 361).
- 1•.Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. The strongest available meta-analysis on the value of LDL lowering in the prevention of ischemic events. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith SC, Jr, Allen J, Blair SN, et al. AHA/ACC guidelines for secondary prevention for patients with coronary and other atherosclerotic vascular disease: 2006 update endorsed by the National Heart, Lung, and Blood Institute. J Am Coll Cardiol. 2006;47:2130–2139. doi: 10.1016/j.jacc.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 3.Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110:227–239. doi: 10.1161/01.CIR.0000133317.49796.0E. [DOI] [PubMed] [Google Scholar]
- 4.Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III) JAMA. 2001;285:2486–2497. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 5.Graham I, Atar D, Borch-Johnsen K, et al. European guidelines on cardiovascular disease prevention in clinical practice: executive summary: Fourth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (Constituted by representatives of nine societies and by invited experts) Eur Heart J. 2007;28:2375–2414. doi: 10.1093/eurheartj/ehm316. [DOI] [PubMed] [Google Scholar]
- 6.Freeman DJ, Norrie J, Sattar N, et al. Pravastatin and the development of diabetes mellitus: evidence for a protective treatment effect in the West of Scotland Coronary Prevention Study. Circulation. 2001;103:357–362. doi: 10.1161/01.cir.103.3.357. [DOI] [PubMed] [Google Scholar]
- 7.Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–1389. [PubMed] [Google Scholar]
- 8.Collins R, Armitage J, Parish S, et al. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes: a randomised placebo-controlled trial. Lancet. 2003;361:2005–2016. doi: 10.1016/s0140-6736(03)13636-7. [DOI] [PubMed] [Google Scholar]
- 9.Results of the low-dose (20 mg) pravastatin GISSI Prevenzione trial in 4271 patients with recent myocardial infarction: do stopped trials contribute to overall knowledge? GISSI Prevenzione Investigators (Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico) Ital Heart J. 2000;1:810–820. [PubMed] [Google Scholar]
- 10.Major outcomes in moderately hypercholesterolemic, hypertensive patients randomized to pravastatin vs usual care: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT-LLT) JAMA. 2002;288:2998–3007. doi: 10.1001/jama.288.23.2998. [DOI] [PubMed] [Google Scholar]
- 11.Nakamura H, Arakawa K, Itakura H, et al. Primary prevention of cardiovascular disease with pravastatin in Japan (MEGA Study): a prospective randomised controlled trial. Lancet. 2006;368:1155–1163. doi: 10.1016/S0140-6736(06)69472-5. [DOI] [PubMed] [Google Scholar]
- 12.Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360:1623–1630. doi: 10.1016/s0140-6736(02)11600-x. [DOI] [PubMed] [Google Scholar]
- 13.Sever PS, Dahlof B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361:1149–1158. doi: 10.1016/S0140-6736(03)12948-0. [DOI] [PubMed] [Google Scholar]
- 14.Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 15.Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279:1615–1622. doi: 10.1001/jama.279.20.1615. [DOI] [PubMed] [Google Scholar]
- 16.Keech A, Colquhoun D, Best J, et al. Secondary prevention of cardiovascular events with long-term pravastatin in patients with diabetes or impaired fasting glucose: results from the LIPID trial. Diabetes Care. 2003;26:2713–2721. doi: 10.2337/diacare.26.10.2713. [DOI] [PubMed] [Google Scholar]
- 17.Kjekshus J, Apetrei E, Barrios V, et al. Rosuvastatin in older patients with systolic heart failure. N Engl J Med. 2007;357:2248–2261. doi: 10.1056/NEJMoa0706201. [DOI] [PubMed] [Google Scholar]
- 18.Tavazzi L, Maggioni AP, Marchioli R, et al. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:1231–1239. doi: 10.1016/S0140-6736(08)61240-4. [DOI] [PubMed] [Google Scholar]
- 19••.Sattar N, Preiss D, Murray HM, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735–742. doi: 10.1016/S0140-6736(09)61965-6. The most recent and comprehensive meta-analysis of statin trials providing evidence of significant, though modest, association between treatment and incident diabetes. [DOI] [PubMed] [Google Scholar]
- 20.Rajpathak SN, Kumbhani DJ, Crandall J, et al. Statin therapy and risk of developing type 2 diabetes: a meta-analysis. Diabetes Care. 2009;32:1924–1929. doi: 10.2337/dc09-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coleman CI, Reinhart K, Kluger J, White CM. The effect of statins on the development of new-onset type 2 diabetes: a meta-analysis of randomized controlled trials. Curr Med Res Opin. 2008;24:1359–1362. doi: 10.1185/030079908x292029. [DOI] [PubMed] [Google Scholar]
- 22.Sukhija R, Prayaga S, Marashdeh M, et al. Effect of statins on fasting plasma glucose in diabetic and nondiabetic patients. J Investig Med. 2009;57:495–499. doi: 10.2310/JIM.0b013e318197ec8b. [DOI] [PubMed] [Google Scholar]
- 23.Kearney PM, Blackwell L, Collins R, et al. Efficacy of cholesterol-lowering therapy in 18,686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet. 2008;371:117–125. doi: 10.1016/S0140-6736(08)60104-X. [DOI] [PubMed] [Google Scholar]
- 24.Ting RZ, Yang X, Yu LW, et al. Lipid control and use of lipid-regulating drugs for prevention of cardiovascular events in Chinese type 2 diabetic patients: a prospective cohort study. Cardiovasc Diabetol. 2010;9:77. doi: 10.1186/1475-2840-9-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marder SR, Essock SM, Miller AL, et al. Physical health monitoring of patients with schizophrenia. Am J Psychiatry. 2004;161:1334–1349. doi: 10.1176/appi.ajp.161.8.1334. [DOI] [PubMed] [Google Scholar]
- 26.Consensus development conference on antipsychotic drugs and obesity and diabetes. J Clin Psychiatry. 2004;65:267–272. doi: 10.4088/jcp.v65n0219. [DOI] [PubMed] [Google Scholar]
- 27•.Baker WL, Talati R, White CM, Coleman CI. Differing effect of statins on insulin sensitivity in nondiabetics: a systematic review and meta-analysis. Diabetes Res Clin Pract. 2010;87:98–107. doi: 10.1016/j.diabres.2009.10.008. An authoritative review on the effects of statin therapy on insulin sensitivity. [DOI] [PubMed] [Google Scholar]
- 28•.Kruit JK, Brunham LR, Verchere CB, Hayden MR. HDL and LDL cholesterol significantly influence beta-cell function in type 2 diabetes mellitus. Curr Opin Lipidol. 2010;21:178–185. doi: 10.1097/MOL.0b013e328339387b. A review of the current mechanistic views on the effects of plasma lipoproteins on β-cell function. [DOI] [PubMed] [Google Scholar]
- 29.Nakata M, Uto N, Maruyama I, Yada T. Nitric oxide induces apoptosis via Ca2+-dependent processes in the pancreatic beta-cell line MIN6. Cell Struct Funct. 1999;24:451–455. doi: 10.1247/csf.24.451. [DOI] [PubMed] [Google Scholar]
- 30.Yada T, Nakata M, Shiraishi T, Kakei M. Inhibition by simvastatin, but not pravastatin, of glucose-induced cytosolic Ca2+ signalling and insulin secretion due to blockade of L-type Ca2+ channels in rat islet beta-cells. Br J Pharmacol. 1999;126:1205–1213. doi: 10.1038/sj.bjp.0702397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mabuchi H, Higashikata T, Kawashiri M, et al. Reduction of serum ubiquinol-10 and ubiquinone-10 levels by atorvastatin in hypercholesterolemic patients. J Atheroscler Thromb. 2005;12:111–119. doi: 10.5551/jat.12.111. [DOI] [PubMed] [Google Scholar]
- 32.Nakata M, Nagasaka S, Kusaka I, et al. Effects of statins on the adipocyte maturation and expression of glucose transporter 4 (SLC2A4): implications in glycaemic control. Diabetologia. 2006;49:1881–1892. doi: 10.1007/s00125-006-0269-5. [DOI] [PubMed] [Google Scholar]
- 33.Chamberlain LH. Inhibition of isoprenoid biosynthesis causes insulin resistance in 3T3-L1 adipocytes. FEBS Lett. 2001;507:357–361. doi: 10.1016/s0014-5793(01)03007-1. [DOI] [PubMed] [Google Scholar]
- 34.Hao M, Head WS, Gunawardana SC, et al. Direct effect of cholesterol on insulin secretion: a novel mechanism for pancreatic beta-cell dysfunction. Diabetes. 2007;56:2328–2338. doi: 10.2337/db07-0056. [DOI] [PubMed] [Google Scholar]
- 35•.Kruit JK, Kremer PH, Dai L, et al. Cholesterol efflux via ATP-binding cassette transporter A1 (ABCA1) and cholesterol uptake via the LDL receptor influences cholesterol-induced impairment of beta cell function in mice. Diabetologia. 2010;53:1110–1119. doi: 10.1007/s00125-010-1691-2. Experimental evidence that cellular cholesterol fluxes regulate insulin export by the β-cell. [DOI] [PubMed] [Google Scholar]
- 36.Donath MY, Boni-Schnetzler M, Ellingsgaard H, Ehses JA. Islet inflammation impairs the pancreatic beta-cell in type 2 diabetes. Physiology (Bethesda) 2009;24:325–331. doi: 10.1152/physiol.00032.2009. [DOI] [PubMed] [Google Scholar]
- 37.Ehses JA, Lacraz G, Giroix MH, et al. IL-1 antagonism reduces hyperglycemia and tissue inflammation in the type 2 diabetic GK rat. Proc Natl Acad Sci U S A. 2009;106:13998–14003. doi: 10.1073/pnas.0810087106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ehses JA, Perren A, Eppler E, et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes. 2007;56:2356–2370. doi: 10.2337/db06-1650. [DOI] [PubMed] [Google Scholar]
- 39.Boni-Schnetzler M, Thorne J, Parnaud G, et al. Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta-cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and auto-stimulation. J Clin Endocrinol Metab. 2008;93:4065–4074. doi: 10.1210/jc.2008-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 41.Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–1135. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- 42.Laufs U, Liao JK. Posttranscriptional regulation of endothelial nitricoxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 43.Kureishi Y, Luo Z, Shiojima I, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abderrahmani A, Niederhauser G, Favre D, et al. Human high-density lipoprotein particles prevent activation of the JNK pathway induced by human oxidised low-density lipoprotein particles in pancreatic beta cells. Diabetologia. 2007;50:1304–1314. doi: 10.1007/s00125-007-0642-z. [DOI] [PubMed] [Google Scholar]
- 45.Cnop M, Hannaert JC, Grupping AY, Pipeleers DG. Low density lipoprotein can cause death of islet beta-cells by its cellular uptake and oxidative modification. Endocrinology. 2002;143:3449–3453. doi: 10.1210/en.2002-220273. [DOI] [PubMed] [Google Scholar]
- 46.Roehrich ME, Mooser V, Lenain V, et al. Insulin-secreting beta-cell dysfunction induced by human lipoproteins. J Biol Chem. 2003;278:18368–18375. doi: 10.1074/jbc.M300102200. [DOI] [PubMed] [Google Scholar]
- 47.Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]