

Abstract
Sepsis is a major cause of morbidity and mortality. NO, an endogenous vasodilator, has been associated with the hypotension, catecholamine hyporesponsiveness, and myocardial depression of septic shock. Although iNOS is thought to be responsible for the hypotension and loss of vascular tone occurring several hours after endotoxin administration, little is known on the effects of constitutive eNOS on LPS-induced organ dysfunction. This study assessed the distribution of eNOS and iNOS in various vascular beds in conscious pigs challenged with LPS. Cardiac and regional hemodynamic parameters were recorded over 8 h in the presence and absence of aminoguanidine, a rather selective inhibitor of iNOS activity, and N-methyl-L-arginine, a nonspecific NOS inhibitor. Our data show that LPS-induced cardiac depression was associated with coronary, renal, and mesenteric vasoconstrictions and a hepatic vasodilatation. LPS also induced increases in eNOS in the heart and lungs, whereas iNOS was mostly detected in the liver. Nitrotyrosine formation was mainly detected in the lungs, with traces in the kidney, liver, and gut. Accordingly, our results suggest that the early decrease in blood pressure and cardiac depression are likely due to activated eNOS, whereas both isoforms are involved in the hepatic vasodilation. In contrast, carotid, coronary, mesenteric, and renal vasoconstrictions were significant at 5 and/or 6 h after LPS infusion, suggesting that NO is not the primary mediator, facilitating and/or unmasking the release of vasoconstrictor mediators. Consequently, developing newer tissue- or isoform-specific NOS inhibitors can lead to novel therapeutic agents in septic shock.
Keywords: Septic shock, NOS isoforms, cardiac function, regional blood flow, porcine endotoxin model
INTRODUCTION
Sepsis is a major cause of morbidity and mortality. NO, an endogenous vasodilator, has been associated with the hypotension (1), catecholamine hyporesponsiveness (2), and myocardial depression (3) of septic shock. Furthermore, NO is cytotoxic and may cause direct tissue injury, and it contributes to sepsis-induced multiple organ failure. These potentially harmful effects have led to the development of therapeutic strategies that scavenge NO (4) or block its production (5). However, the ability of NO to dilate blood vessels, block platelet aggregation, inhibit leukocyte adhesion, and scavenge superoxide suggests the possibility that NO also plays a role in endothelium protection and maintenance of microvascular blood flow during sepsis (6). Thus, like many other endogenous mediators of sepsis, NO may have both harmful (7) and beneficial effects (8).
NO is a highly reactive radical synthesized from the amino acid L-arginine by the action of NOS. Several isoforms of NOS have been identified and divided into two categories with different regulation and activities. The constitutive NOS (eNOS and nNOS) isoforms exist in endothelial, neuronal, and various cells and comprise the low output path on demand in homeostatic processes such as neurotransmission or blood pressure regulation (9). In addition, there are inducible isoforms (iNOS) that may be expressed after exposure to endotoxin and certain cytokines (IL-1, TNF, interferon γ) in macrophages, neutrophils, mast cells, endothelial cells, and vascular smooth muscle cells (6). Overproduction of NO after cytokine- or endotoxin-mediated expression of iNOS can result in shock (1). Specifically, iNOS is believed to be responsible for the hypotension and loss of vascular tone requiring gene transcription and posttranscriptional alterations occurring several hours after the administration of endotoxin. To confirm these findings, investigators have reported that mice having a genetically altered nonfunctional iNOS gene (KO model) survive longer than wild-type mice when given LPS or bacteria (10). However, little is known on the effects of eNOS on LPS-induced organ dysfunction. Inasmuch as NO is involved in the physiological regulation of basal vascular tone, it is possible that NO formation might vary depending on the vascular bed. Currently, a large portion of sepsis literature, particularly cellular and molecular data, is based on studies involving rodents and mice.
The specific role of NOS isoforms in the septic responses of species larger than mice and rodents is thus unclear. To overcome these discrepancies, we developed an animal model of sepsis comparable physiologically to human sepsis to assess the cardiac and regional distribution of both eNOS and iNOS isoforms. Because the studies elucidate the cardiac and regional blood flows of a large animal model—the pig—challenged with endotoxin, experiments were performed in conscious animals that do not experience any pain or other stress. Acute surgical instrumentation and anesthesia were avoided, along with possible mediator release and drug interactions, all of which can influence the results of the study. It is now well established that administration of halothane alters the endothelium-mediated vasodilatation in vitro and in vivo likely by affecting the NO pathway (11). In addition, the induction of NOS requires de novo protein synthesis and a delay of several hours, justifying the need for a chronic versus acute animal model. The goal of this study was to reduce tissue NO production during acute endotoxemia using aminoguanidine (AG), a rather selective inhibitor of iNOS activity, without greatly affecting eNOS activity and N-methyl-L-arginine (L-NMA), a nonspecific NOS inhibitor. We investigated the correlation between tissue NO production and changes in cardiovascular function during acute endotoxemia.
MATERIALS AND METHODS
Surgical instrumentation
Twenty-three pigs (25–30 kg) were instrumented as follows: animals were anesthetized with Telazol (2–4 mg kg−1q, i.m.) and isoflurane anesthesia (2%) then intubated and prepared for sterile surgery. The experiments were performed in adherence to the National Institutes of Health Guidelines on the Use of Laboratory Animals. The animals were ventilated with a Harvard ventilator (Harvard Apparatus, So. Natick, Mass). They received an infusion of bretylium (5 mg kg−1) to prevent arrhythmias. Under aseptic conditions, a Tygon catheter was introduced into the descending aorta via the iliac artery. A left thoracotomy between the fourth and fifth intercostal space was performed, the pericardium incised, the circumflex coronary dissected free, and a 20-MHz pulsed Doppler flow probe (2.5–3.0 mm; Baylor College of Medicine, Houston, Tex) applied. A miniature pressure transducer (6.5–7.0 mm; Konigsberg Instruments, Inc., Pasadena, Calif) was inserted into the left ventricle through an apical stab wound. A precalibrated flow probe (14–16 mm; Transonic Systems, Inc., Ithaca, NY) was positioned around the pulmonary artery. Tygon catheters were inserted into the left atrium and the pulmonary artery. Finally, an epicardial wall thickness probe (10 MHz; Baylor College of Medicine) was sutured on the myocardial wall (12). After a median laparotomy, 20-MHz pulsed Doppler flow probes were positioned around the hepatic artery (3.0 mm), portal vein (8.0 mm), and mesenteric artery (4.0–4.5 mm). After appropriate incisions, 20-MHz pulsed Doppler flow probes were positioned around the left common carotid (3.5–4.0 mm) and the left renal arteries (3.5–4.0 mm). In addition, Tygon catheters were introduced into the jugular and femoral veins for drug administration. Probe functions were carefully checked in vitro and in vivo before closure of the incisions. All transducer leads and catheters were tunneled subcutaneously to the dorsum of the neck and secured in place after closure of the thoracotomy.
After surgery, animals were carefully nursed through the first 24 h with intravenous fluids and systemic analgesic as necessary. Antibiotic prophylaxis was initiated before surgery and maintained for 7 days after surgery. Animals were allowed to recover from surgery for at least 10 days. During this period, animals were trained in a sling for acclimatization. All experiments were performed in fasted, awake animals. A blood sample from the underlying catheters was cultured before the experiments. White blood cell count, body temperature, arterial blood gases, hematocrit, and animals’ weight were routinely monitored before each experiment to ascertain the health of the animals.
Hemodynamic measurements
This experimental preparation allowed continuous measurements of left ventricular and aortic pressures; pulmonary arterial pressure; the first derivative of left ventricular pressure (dP/dt); wall thickening; cardiac output (CO); heart rate; and carotid, coronary, hepatic, portal, mesenteric, and renal blood flows. Systemic and regional vascular resistances (VRs) were calculated as the ratio of MAP to CO and/or regional blood flow. Pressures were measured by means of the aortic and left atrial catheters using a Statham P23 DB pressure transducer (Gould Inc., Cleveland, Ohio). Cardiac output was recorded using a Transonic flow meter (T202-S). Ultrasonic flow dimension system allowed simultaneous measurements of pressures and flow. The miniature Konigsberg pressure transducer was calibrated both after implantation and before each study using the aortic and mean left atrial pressures as references. The pulsed Doppler flow signals were calibrated in Doppler frequency shift (12). Baseline zero and the linear relation between volume flow and frequency shift have been previously established in vivo (13). All hemodynamic variables were recorded in phasic and mean modes on a 16-channel Gould brush polygraph.
Experimental protocol
The protocol was approved by The University of Texas Animal Welfare Committee. The experiments were performed in adherence to the National Institutes of Health Guidelines on the Use of Laboratory Animals. Pigs were studied no less than 10 days after surgery, when hematocrit was greater than 30%, and body temperature, appetite, and general appearance were normal. Animals were divided into three groups. Group 1 (n = 8) received LPS from Escherichia coli, titrated to a maximum dose of 200 μg kg−1 (i.v.) over 60 min in 20 mL of phosphate-buffered saline, pH 7.4, according to blood pressure response. In group 2 (n = 8), animals were given AG in a dose of 1 mg kg−1 min−1 (i.v.) in 10 mL of phosphate-buffered saline over 60 min in the presence of LPS. In group 3 (n = 7), animals were given L-NMA in a dose of 300 μg kg−1 min−1 (i.v.) in 10 mL of phosphate-buffered saline over 60 min in the presence of LPS. All animals received 250 mL of 0.9% NaCl containing 5% dextrose in control conditions during LPS infusion and for 6 hours after AG and/or L-NMA infusions. Aminoguanidine and L-NMA dosages were chosen based on previous studies performed in our laboratory (14). In all groups, hemodynamic parameters were continuously recorded in control conditions, during LPS infusion, during 8 hours after LPS infusion (group 1), and for 6 h after AG and/or L-NMA administrations (groups 2 and 3).
Drugs
LPS from E. coli serotype 0128:B12 (Sigma Chemical Co., St. Louis, Mo) and AG hemisulfate (Sigma) was dissolved in isotonic saline. N-Methyl-L-arginine (acetate salt) was synthesized according to the method of Corbin and Reporter (15). LPS, AG, and L-NMA were dissolved in 10 mL of phosphate-buffered saline (pH 7.4).
Nitrite and nitrate production determination
Plasma nitrite/nitrate levels were quantified by a modified chemiluminescence method of Radomski et al. (16) using a Sievers NO analyzer (model 270B; GE Analytical Instruments, Boulder, Colo). Blood samples were immediately centrifuged at 1,200g for 10 min. Serum samples were then removed and stored at −70°C until used for assay. Serum (200 μL) was deproteinized with 30% ZnSO4 and 1 M NaOH at a 1:5 dilution. Samples (20 μL) were injected into a purged vessel of the NO analyzer containing 1% potassium iodide in glacial acetic acid in an atmosphere of nitrogen; under these conditions, all nitrites are reduced to NO. Gaseous NO was purged from the vessel into a reaction chamber containing ozone where NO was detected by the resultant chemiluminescent signal. Nitrite concentrations were quantified by comparison of the unknown signals to that of authentic sodium nitrate (20–400 pmol). Similarly, nitrates were converted to nitrites in plasma samples (200 μL) using the same concentrations of nitrate reductase and nicotinamide adenine dinucleotide phosphate (reduced form) indicated above. Reduced plasma samples (20 μL) were injected into the NO analyzer, and plasma nitrite was quantified in comparison to a standard curve of authentic sodium nitrite.
Homogenization
Frozen tissues were homogenized at a ratio of 1 g per 3–6 mL of ice-cold homogenization buffer. Homogenization was first performed with pestle and mortar and then with a homogenizer in ice- and salt-cold environment. Homogenization buffer (pH 7.4) had the final concentration of 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 100 μM dithiothreitol, 55 μM leupeptin, 100 μg mL−1 phenylmethylsulfonyl fluoride, 2 μg mL−1 aprotinin, 1.4 μM pepstatin A, and 2.5% glycerol in double-distilled water. The homogenate was centrifuged at 10,000 to 11,000 g for 15 to 20 min at 4°C. Supernatants were aliquoted and frozen at −70°C.
NOS activity
Supernatant (50 μL) from the homogenate was added to 10 mL prewarmed (37°C) tubes that contained 100 μL of reaction buffer with the following composition: 50 mM KH2PO4, 60 mM valine, 1.5 mM nicotinamide adenine dinucleotide phosphate (reduced form), 10 mM flavin adenine dinucleotide, 1.2 mM MgCL2, 2 mM CaCL2, 1 mg mL −1 bovine serum albumin, 1 μg mL−1 calmodulin, 10 μM BH4, and 25 μM of 120 μM stock L-[2,3 3H] arginine (150–200 cpm pM−1). The samples were incubated for 30 min at 37°C, and the reaction was terminated by the addition of cold (4°C) stop buffer (pH 5.5), 100 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, and 12 mM EDTA. Dowex 50W resin (8% cross-linked, Na+ form) was added to eliminate excess L-[2,3 3H] arginine. Enzyme activity was expressed in picomoles of L-citrulline produced per total protein (in milligrams) per minute as described in detail by Salter et al. (17). Protein was measured by the Bradford technique with bovine serum albumin as a standard (BioRad, Inc., Richmond, Calif) (17). The supernatant was assayed for L-[3H] citrulline by a liquid scintillation counting. NOS activity was also measured in the presence of 1.5 mM of each EGTA and EDTA that replaced CaCl2 calmodulin in the reaction buffer and in the presence of 1 mM of NG-nitro-L-arginine methyl ester, a NOS inhibitor. Ca2+/calmodulin-independent NOS activity was calculated as the difference between samples assayed in the presence of EGTA/EDTA and those measured in the presence of NG-nitro-L-arginine methyl ester.
Western blot
The potential contribution of iNOS and nitrotyrosine in controlling vascular tone in the presence and in the absence of LPS was performed in collected tissue samples using specific antibodies. Frozen tissue homogenates were thawed on ice and mixed with equal volumes of sample buffer (200 mM Tris-HCl (pH 6.8), 8% sodium dodecyl sulfate, 30% glycerol + 1% β-mecapto ethanol, 0.5% bromophenol blue), followed by heating at 95°C for 5 to 10 min. Tissue homogenates were centrifuged and loaded on 8% Tris/glycine/sodium dodecyl sulfate–polyacrylamide gel for fractionation. Predetermined molecular weight standards (Novex Inc., Encinitas, Calif) were used as markers. Protein on the gel was blotted onto nitrocellulose membranes at 4°C at 25 to 30 V and 370 to 380 mA for 150 min. After transfer, the membranes were incubated with 5% skim milk in Tris-buffered saline containing 0.05% Tween 20 for 2 h at room temperature or overnight at a cold temperature (4°C).
iNOS
iNOS protein was detected by incubating with primary antibody at a concentration of 1:500 to 1:1,000. Two monoclonal and three polyclonal antibodies were used.
Nitrotyrosine
Samples for immunoblot analysis were boiled for 3 min and electrophoresed on 12.5% acrylamide gels. The proteins were electrophoretically transferred to nitrocellulose sheets using a semidry apparatus. The sheets were reacted with a monoclonal mouse antinitrotyrosine antibody (Zymed Laboratories, South San Francisco, Calif). Immune complexes were detected using peroxidase-labeled goat antimouse immunoglobulin G (KPL, Inc., Gaithersburg, Md).
Fluorescence immunostaining of eNOS and iNOS
After completion of all protocols, organs and tissues (e.g., heart, lungs, kidney, and liver) were removed and immediately freeze-clamped and stored at −70°C until studied. Immunohistochemical staining for eNOS and iNOS were performed on postfixed sections in 3.7% formaldehyde for 5 min, and nonspecific staining was blocked by incubating sections with 10% goat serum for 30 min at room temperature. Stock antibody was diluted 1:1,000 in 10% goat serum, and specimens were incubated for 30 to 45 min at 37°C. Cover slips were rinsed two to three times with 0.05% Tween 20. We then proceeded in the dilution of the tagged fluorescent secondary antibody 1:500 in 0.05% Tween 20 in 10% goat serum, followed by 30 min incubation at 37°C. Cover slips were examined on an inverted Nikon Optiphot microscope (Nikon, New York, NY) and scanned with a fluorescence microscope (Issaquah, Wash) fitted with an Olympus IX70 microscope and with deconvolution capabilities. Images were captured at the maximum fluorescence (18).
Statistical analysis
To compare the hemodynamic changes between LPS and control measurements, data were analyzed using a paired t test. To compare the hemodynamic changes induced by AG or L-NMA (treatment) versus LPS (septic shock), data were analyzed by a one-way ANOVA to assess overall significance. When significant, an appropriate multiple comparison method (Fischer) was applied. Data are presented as mean ± SEM. P < 0.05 was considered significant.
RESULTS
Hemodynamic effects of LPS alone (group 1)
LPS infused in a dose of 200 μg kg−1 (i.v.) over 60 min resulted in fever, rigors, vomiting, and diarrhea. LPS induced a significant decrease in MAP at 1, 2, and 3 h by 26%, 36%, and 31%, respectively, followed by a significant increase at 7 and 8 h by 25% and 22%, respectively. Heart rate (HR) increased significantly as early as 1 h by 17% and remained significantly increased throughout the study by 28%. Pulmonary arterial pressure (PAP) increased by 25% within 1 h after LPS administration and remained sustained thereafter. Cardiac output and dP/dt decreased significantly at the end of LPS infusion by 53% and 33%, respectively. dP/dt further decreased significantly at 6, 7, and 8 h by 31%, 39%, and 40%, respectively. Systemic VR (SVR) increased significantly at the end of LPS infusion by 88% and at 6, 7, and 8 h by 117%, 135%, and 133%, respectively (Fig. 1). The LPS-induced cardiac depression was associated with coronary, renal and mesenteric vasoconstrictions and a hepatic vasodilatation. Carotid blood flow decreased significantly at the end of LPS infusion by 45%, whereas renal and mesenteric blood flow decreased significantly at 4 and 5 h (49% and 26%, respectively) and remained decreased for the entire study. In contrast, hepatic blood flow increased significantly 1 h after LPS infusion (192%) and remained elevated throughout the study. Mesenteric and renal VRs significantly increased at 5 h (59% and 258%, respectively), whereas carotid and coronary VRs increased at 6 h after LPS infusion (86% and 67%, respectively). In contrast, hepatic VR significantly decreased at the end of LPS infusion (71%) and remained decreased thereafter (Fig. 2).
Fig. 1.

Hemodynamic changes recorded for 8 h on MAP, CO, SVR, PAP, HR, and dP/dt in conscious pigs subjected to LPS infused at 200 μg kg−1 over 60 min
*P < 0.05 vs. B. B indicates baseline.
Fig. 2.

Hemodynamic changes recorded for 8 h on Car BF, Cor BF, Mes BF, Ren BF, Hep BF, Car VR, Cor VR, Mes VR, Ren VR, and Hep VR in conscious pigs subjected to LPS infused at 200 μg kg−1 over 60 min
*P < 0.05 vs. B. B indicates baseline; Car BF, carotid blood flow; Car VR, carotid resistance; Cor BF, coronary blood flow; Cor VR, coronary vascular resistance; Hep BF, hepatic blood flow; Hep VR, hepatic vascular resistance; Mes BF, mesenteric blood flow; Mes VR; mesenteric vascular resistance; Ren BF, renal blood flow; Ren VR, renal vascular resistance.
Western analysis
There was no evidence of iNOS production by Western analysis. However, we found marked nitrotyrosine formation in the lungs (Fig. 3).
Fig. 3.
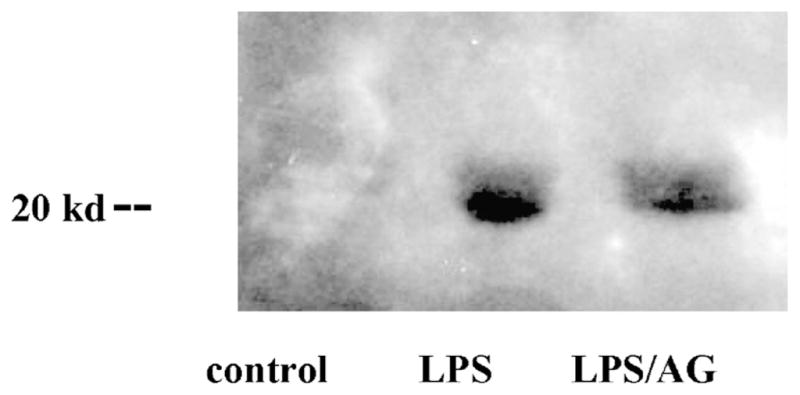
Immunoblot analysis of lung tissue treated with LPS with and without AG, an inducible NOS inhibitor, using a monoclonal mouse antinitrotyrosine.
Nitrates
No significant increase in plasma nitrates was recorded. In this study, systemic blood samples were taken from the aortic catheter. Thus, we think that circulating NO2 levels are an insensitive indicator of NO biosynthesis. “However, nitrate levels may vary when blood samples are taken directly at the level of a specific vascular bed.
NOS activity
iNOS activity was present in all organs mainly in the liver, kidney, and intestine (Fig. 4). Indeed, the determination of NOS activity can be influenced, like NO production, by posttranscriptional alterations and substrate availability.
Fig. 4.

iNOS activity recorded at 8 h in various tissues, for example, liver, kidney, lung, heart, spleen, and large and small intestine in pigs challenged with LPS at 200 μg kg−1 over 60 min.
Immunofluorescence detection of eNOS and iNOS
Although eNOS was detected in all organs in control pigs (in the absence of LPS), eNOS detection was predominant in the heart and lungs in pigs challenged with LPS (Fig. 5). As compared with the heart, lungs, and kidney, iNOS detection was markedly pronounced in the liver in pigs challenged with LPS (Fig. 6).
Fig. 5.

eNOS recorded by immunofluorescence staining in the heart and lung in control pigs (without LPS) and treated with LPS at 200 μg kg−1 over 60 min
Green indicates smooth muscle actin (vasculature); magenta, eNOS; white, eNOS and smooth muscle actin (vasculature).
Fig. 6.
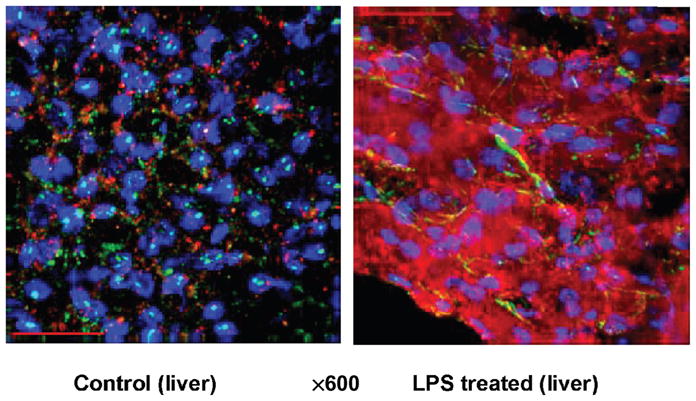
iNOS recorded by immunofluorescence staining in the liver in control pigs (without LPS) and treated with LPS at 200 μg kg−1 over 60 min
Blue indicates 4′,6-diamidino-2-phenylindole (nuclei); green, smooth muscle actin (vasculature); red, iNOS.
Hemodynamic effects of AG in the presence of LPS (group 2)
The goal of this study was to reduce tissue NO production during acute endotoxemia by a rather selective inhibition of iNOS using AG without greatly affecting eNOS activity. We have previously shown that AG was a weak inhibitor of the cNOS isoform in vivo studies (19). Thus, we have investigated the correlation between NO production and changes in cardiovascular function during 6 h after AG infusion in pigs challenged with LPS. Aminoguanidine induced significant increases in MAP and SVR at 2, 4, and 6 h (47%, 67%, and 71% and 82%, 94%, and 78%, respectively). Furthermore, heart rate, CO, PAP, and dP/dt as well as carotid, coronary, hepatic, mesenteric, and renal blood flow remained unchanged. However, AG induced a carotid vasoconstriction at 4 and 6 h (97% and 82%, respectively), and an isolated mesenteric vasoconstriction at 2 h (Figs. 7 and 8). As compared with LPS alone (group 1), AG reversed some of the deleterious effects of LPS. For instance, AG abolished the LPS-induced tachycardia, increase in PAP, and decrease in dP/dt. Aminoguanidine also abolished the LPS-induced hepatic vasodilatation and LPS-induced coronary and renal vasoconstrictions.
Fig. 7.

Effects of AG infused at 1 mg kg−1 min−1 on MAP, CO, SVR, PAP, HR, and dP/dt in eight conscious pigs treated with LPS at 200 μg kg−1 over 60 min
*P < 0.05 vs. LPS; †P < 0.05 LPS vs. B. B indicates baseline.
Fig. 8.

Effects of AG infused at 1 mg kg−1 min−1 on Car BF, Cor BF, Mes BF, Ren BF, Hep BF, Car VR, Cor VR, Mes VR, Ren VR, and Hep VR in eight conscious pigs treated with LPS at 200 μg kg−1 over 60 min
*P < 0.05 vs. LPS;†P < 0.05 LPS vs. B. B indicates baseline; Car BF, carotid blood flow; Car VR, carotid resistance; Cor BF, coronary blood flow; Cor VR, coronary vascular resistance; Hep BF, hepatic blood flow; Hep VR, hepatic vascular resistance; Mes BF, mesenteric blood flow; Mes VR; mesenteric vascular resistance; Ren BF, renal blood flow; Ren VR, renal vascular resistance.
No significant difference was recorded in nitrate levels in the presence of AG as compared with control pigs treated with LPS. However, our data show that AG decreased iNOS activity in all organs (Fig. 9). Furthermore, LPS-induced nitrotyrosine production in lung tissue was also decreased with AG (Fig. 3).
Fig. 9.

iNOS activity recorded in various tissues, for example, liver, kidney, lung, heart, spleen, and large and small intestine in pigs challenged with LPS at 200 μg kg−1 over 60 min in the presence and in the absence of AG.
Hemodynamic effects of L-NMA in the presence of LPS (group 3)
The hemodynamic effects of LPS in the presence of L-NMA are presented in Figures 10 and 11. MAP, SVR, and PAP increased significantly after L-NMA infusion. MAP increased by 50%, reaching a maximum effect at 2 h. SVR and PAP increased at 30 min by 120% and 43%, respectively, and at 1 h by 118% and 40%, respectively, and returned to baseline thereafter. In contrast, in the presence of L-NMA, carotid, coronary, renal, mesenteric, and hepatic blood flow remained unchanged. However, L-NMA reversed the LPS-induced hepatic vasodilatation at 6 h by 68%. Although insignificant, L-NMA decreased the LPS-induced renal vasoconstriction within 1 h and remained further decreased thereafter. As compared with LPS alone, L-NMA abolished the LPS-induced tachycardia and decrease in dP/dt.
Fig. 10.

Effects of L-NMA infused at 300 μg kg−1 min−1 on MAP, CO, SVR, PAP, HR, and dP/dt in seven conscious pigs treated with LPS at 200 μg kg−1 over 60 min
*P < 0.05 vs. LPS; †P < 0.05 LPS vs. B. B indicates baseline.
Fig. 11.

Effects of L-NMA infused at 300 μg kg−1 min−1 on Car BF, Cor BF, Mes BF, Ren BF, Hep BF, Car VR, Cor VR, Mes VR, Ren VR, and Hep VR in seven conscious pigs treated with LPS at 200 μg kg−1 over 60 min
*P < 0.05 vs. LPS; †P < 0.05 LPS vs. B. B indicates baseline; Car BF, carotid blood flow; Car VR, carotid resistance; Cor BF, coronary blood flow; Cor VR, coronary vascular resistance; Hep BF, hepatic blood flow; Hep VR, hepatic vascular resistance; Mes BF, mesenteric blood flow; Mes VR; mesenteric vascular resistance; Ren BF, renal blood flow; Ren VR, renal vascular resistance.
DISCUSSION
Our data demonstrate that LPS-induced hemodynamic changes in conscious pigs are expressed as heterogeneous patterns of vasodilatation and vasoconstriction in different organs. LPS-induced cardiac depression was associated with an increase in pulmonary pressure, carotid, coronary, renal, and mesenteric vasoconstrictions and an isolated hepatic vasodilatation.
Despite a decrease in blood pressure, NO plasma levels remained unchanged after LPS infusion in contrast to previous findings in rats (20). However, the dissociation between hypotension and plasma NO was also noted by van den Berg et al. (21). Furthermore, rodents are more resistant to the effects of endotoxin than larger animals. The marked increase in NO that is seen in rodents as compared with septic patients and/or large animal species might reflect this pronounced resistance. In addition, Mehta et al. (22) also concluded from their porcine endotoxemic shock study that caution should be used in extrapolating from rodents to higher-order animals.
Concomitantly to a decrease in blood pressure, CO and dP/dt dropped significantly 30 min after LPS infusion. We also found markedly increased myocardial eNOS activity. Salvemini et al. (23) showed NO release within 1 min of exposure of endothelial cells to endotoxin, suggesting eNOS activation responsible for early LPS-induced decrease in blood pressure. Furthermore, although not quantified in our study, cytokines such as TNF-α and IL-1 are also released during sepsis and are suggested to exert cardiodepressant effects. Another possible mechanism is that activated leukocytes might exercise cardiodepressant effects as leukocytes transit time increases in the coronary microcirculation during endotoxemia. Concluding from our results, we propose an additional role for eNOS activation in the pathophysiology of both the early blood pressure decrease and cardiac depression.
LPS induced a sustained increase in pulmonary arterial pressure. Although involvement of the cyclooxygenase pathway has been documented in the early phase of pulmonary hypertension, a pathophysiological role for the endothelin system has been shown in the late phase of endotoxin-induced pulmonary hypertension. Furthermore, Wedgwood et al. (24) proposed that endothelin may contribute to rebound hypertension by inducing superoxide synthesis in the pulmonary vasculature.
In contrast to the cardiac and pulmonary vasculature, our data suggest that NO produced locally by the activated iNOS may contribute to individual organ dysfunction induced by endotoxin. Except for the heart, iNOS activity was detected in all organs but predominantly in the liver, which correlates with the profound hepatic vasodilatation. Hepatic vasodilatation appeared as soon as 1 h after LPS infusion and lasted for several hours. As shown in previous studies (25, 26), although early vasodilatation is triggered through eNOS activity, iNOS can contribute to NO production in as early as 2 h after stimulation.
Although eNOS was present in all studied organs from control animals, eNOS activity was more pronounced in heart and lung tissues from pigs challenged with LPS. The generated NO reacts with superoxide to form peroxinitrite, which in turn leads to tissue injuries such as protein nitration (nitrotyrosine formation). The role of iNOS in the formation of nitrotyrosine is also supported by the work of El-Dwairi et al. (27). We found nitrotyrosine formation in multiple tissues both by immunoblotting and immunohistochemistry. More specifically, our data show that nitrotyrosine formation was detected in the lungs, whereas only traces were recorded in the heart, kidneys, liver, and gut in pigs challenged with LPS. It is now well known that peroxynitrite has a relatively long half-life and, therefore, can diffuse and attack distant targets (28). Interestingly, our data show that nitrotyrosine formation was also detected in the kidney and liver in control pigs in the absence of LPS as previously reported by Bian et al. (29). The authors suggested that protein nitration is a normal biological process, and that any perturbation of this balance between protein nitration and denitration may lead to pathological changes in endothelial cells and tissues.
In addition to the hepatic vasodilatation, we also found that LPS induced carotid, coronary, mesenteric, and renal vasoconstriction. Vascular endothelial cells have been shown to synthesize a potent pressor substance called endothelin 1 (ET-1). Endothelin 1 is a potent modulator of regional perfusion and may well contribute to the maldistribution of blood flow seen in sepsis (30). Previous studies on ET-1 have also reported vasoconstriction in splanchnic and renal vascular beds and a reduction of CO (31). Because ET-1 is the most potent coronary vasoconstrictor, we postulate that NO production was deficient, unmasking the release of ET-1, leading to a coronary vasoconstriction. In contrast to the hepatic vasodilatation, the carotid, coronary, mesenteric, and renal vasoconstrictions were significant at 5 and/or 6 h after LPS infusion, suggesting that NO is not the primary mediator and, therefore, facilitate and/or unmask the release of vasoconstrictor mediators.
Our data show that LPS-induced increase in hepatic blood flow was inhibited after AG infusion, and there was no apparent correlation between CO and hepatic blood flow. However, it is well established that multiple parameters such as portal blood flow and autoregulatory mechanisms influence hepatic blood flow. Aminoguanidine abolished coronary and renal vasoconstrictions and partially inhibited the mesenteric vasoconstriction induced by LPS. Aminoguanidine significantly decreased iNOS in all vascular beds and nitrotyrosine formation in the lungs. However, because AG has numerous pharmacological properties, it is difficult to specifically conclude that the beneficial effect of AG on LPS-induced hemodynamic changes is solely due to inhibition of iNOS activity (32).
Despite improving systemic blood pressure, L-NMA infusion exacerbated pulmonary hypertension, which can result from increased smooth muscle sensitivity to ET-1 during decreased NO production. This finding suggests that a certain amount of NO is physiologically important for the regulation of systemic hemodynamics and microvascular perfusion. Our data also demonstrate that regional hemodynamic responses induced by LPS remained unchanged in the presence of L-NMA, suggesting that eNOS is not the primary isoform accountable for the hemodynamic variations induced by LPS. Thus, we propose that the lack of NOS inhibition can result from nonspecificity of L-NMA toward NOS isoforms. We did not find the beneficial effects of L-NMA on mesenteric perfusion observed by Siegemund et al. (33). However, this can be explained by several different variables such as L-NMA and endotoxin dosage used.
In summary, we found organ-specific hemodynamic changes in response to LPS challenge with eNOS activity involved in the early pathophysiology (minutes) and increased iNOS expression at a later-stage (hours) postendotoxin administration. We further demonstrate significant NO associated lung tissue damage as measured by nitrotyrosine formation. Our results differ from previous studies in rodents but are in accordance with findings in pigs by other groups. Thus, the interspecies difference highlights the importance of differences in NO metabolism and is therefore a crucial aspect in choosing the appropriate animal model for clinically relevant studies on NO-associated pathophysiology in sepsis.
References
- 1.Kilbourn RG, Griffith OW. Overproduction of nitric oxide in cytokine-mediated and septic shock. J Natl Cancer Inst. 1992;84:827–831. doi: 10.1093/jnci/84.11.827. [DOI] [PubMed] [Google Scholar]
- 2.Julou-Schaeffer G, Gray GA, Fleming I, Schott C, Parratt JR, Stoclet JC. Loss of vascular responsiveness induced by endotoxin involves L-arginine pathway. Am J Physiol. 1990;259(4 Pt 2):H1038–H1043. doi: 10.1152/ajpheart.1990.259.4.H1038. [DOI] [PubMed] [Google Scholar]
- 3.Klabunde RE, Ritger RC. NG-Monomethyl-L-arginine (NMA) restores arterial blood pressure but reduces cardiac output in a canine model of endotoxic shock. Biochem Biophys Res Commun. 1991;178:1135–1140. doi: 10.1016/0006-291x(91)91010-a. [DOI] [PubMed] [Google Scholar]
- 4.Sharma VS, Traylor TG, Gardiner R, Mizukami H. Reaction of nitric oxide with heme proteins and model compounds of hemoglobin. Biochemistry. 1987;26:3837–3843. doi: 10.1021/bi00387a015. [DOI] [PubMed] [Google Scholar]
- 5.Kilbourn RG, Jubran A, Gross SS, Griffith OW, Levi R, Adams J, Lodato RF. Reversal of endotoxin-mediated shock by NG-methyl-L-arginine, an inhibitor of nitric oxide synthesis. Biochem Biophys Res Commun. 1990;172:1132–1138. doi: 10.1016/0006-291x(90)91565-a. [DOI] [PubMed] [Google Scholar]
- 6.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 7.Parker JL, Adams HR. Selective inhibition of endothelium-dependent vasodilator capacity by Escherichia coli endotoxemia. Circ Res. 1993;72:539–551. doi: 10.1161/01.res.72.3.539. [DOI] [PubMed] [Google Scholar]
- 8.Thiemermann C, Vane J. Inhibition of nitric oxide synthesis reduces the hypotension induced by bacterial lipopolysaccharides in the rat in vivo. Eur J Pharmacol. 1990;182:591–595. doi: 10.1016/0014-2999(90)90062-b. [DOI] [PubMed] [Google Scholar]
- 9.Busse R, Mulsch A. Induction of nitric oxide synthase by cytokines in vascular smooth muscle cells. FEBS Lett. 1990;275:87–90. doi: 10.1016/0014-5793(90)81445-t. [DOI] [PubMed] [Google Scholar]
- 10.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 11.Chelly JE, Doursout MF, Lechevalier T, Liang YY, Chelly F, Hartley C, Kilbourn RG. Cardiac and regional hemodynamic interactions between halothane and nitric oxide synthase activity in dogs. Anesthesiology. 1996;85:142–149. doi: 10.1097/00000542-199607000-00020. [DOI] [PubMed] [Google Scholar]
- 12.Hartley CJ, Cole JS. An ultrasonic pulsed Doppler system for measuring blood flow in small vessels. J Appl Physiol. 1974;37:626–629. doi: 10.1152/jappl.1974.37.4.626. [DOI] [PubMed] [Google Scholar]
- 13.Ishida T, Lewis RM, Hartley CJ, Entman ML, Field JB. Comparison of hepatic extraction of insulin and glucagon in conscious and anesthetized dogs. Endocrinology. 1983;112:1098–1109. doi: 10.1210/endo-112-3-1098. [DOI] [PubMed] [Google Scholar]
- 14.Doursout MF, Hartley CJ, Chelly JE. Comparison of cardiac and regional hemodynamic responses to N-methyl-L-arginine and aminoguanidine infusions in conscious pigs. J Cardiovasc Pharmacol. 2001;37:349–358. doi: 10.1097/00005344-200104000-00001. [DOI] [PubMed] [Google Scholar]
- 15.Corbin JL, Reporter M. N-G-methylated arginines; a convenient preparation of N-G-methylarginine. Anal Biochem. 1974;57:310–312. doi: 10.1016/0003-2697(74)90080-3. [DOI] [PubMed] [Google Scholar]
- 16.Radomski MW, Palmer RM, Moncada S. The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br J Pharmacol. 1987;92:639–646. doi: 10.1111/j.1476-5381.1987.tb11367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 18.Bick RJ, Wood DE, Poindexter B, McMillin JB, Karoly A, Wang D, Bunting R, McCann T, Law GJ, Buja LM. Cytokines increase neonatal cardiac myocyte calcium concentrations: the involvement of nitric oxide and cyclic nucleotides. J Interferon Cytokine Res. 1999;19:645–653. doi: 10.1089/107999099313794. [DOI] [PubMed] [Google Scholar]
- 19.Doursout MF, Kilbourn RG, Hartley CJ, Chelly JE. Effects of N-methyl-L-arginine on cardiac and regional blood flow in a dog endotoxin shock model. J Crit Care. 2000;15:22–29. doi: 10.1053/jcrc.2000.0150022. [DOI] [PubMed] [Google Scholar]
- 20.Stuehr DJ, Marletta MA. Mammalian nitrate biosynthesis: mouse macrophages produce nitrite and nitrate in response to Escherichia coli lipopolysaccharide. Proc Natl Acad Sci U S A. 1985;82:7738–7742. doi: 10.1073/pnas.82.22.7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van den Berg C, van Amsterdam JG, Bisschop A, Piet JJ, Wemer J, de Wildt DJ. Septic shock: no correlation between plasma levels of nitric oxide metabolites and hypotension or lethality. Eur J Pharmacol. 1994;270:379–382. doi: 10.1016/0926-6917(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 22.Mehta S, Javeshghani D, Datta P, Levy RD, Magder S. Porcine endotoxemic shock is associated with increased expired nitric oxide. Crit Care Med. 1999;27:385–393. doi: 10.1097/00003246-199902000-00047. [DOI] [PubMed] [Google Scholar]
- 23.Salvemini D, Seibert K, Masferrer JL, Settle SL, Currie MG, Needleman P. Nitric oxide activates the cyclooxygenase pathway in inflammation. Am J Ther. 1995;2:616–619. doi: 10.1097/00045391-199509000-00007. [DOI] [PubMed] [Google Scholar]
- 24.Wedgwood S, McMullan DM, Bekker JM, Fineman JR, Black SM. Role for endothelin-1-induced superoxide and peroxynitrite production in rebound pulmonary hypertension associated with inhaled nitric oxide therapy. Circ Res. 2001;89:357–364. doi: 10.1161/hh1601.094983. [DOI] [PubMed] [Google Scholar]
- 25.Rees DD, Cellek S, Palmer RM, Moncada S. Dexamethasone prevents the induction by endotoxin of a nitric oxide synthase and the associated effects on vascular tone: an insight into endotoxin shock. Biochem Biophys Res Commun. 1990;173:541–547. doi: 10.1016/s0006-291x(05)80068-3. [DOI] [PubMed] [Google Scholar]
- 26.Lowenstein CJ, Glatt CS, Bredt DS, Snyder SH. Cloned and expressed macrophage nitric oxide synthase contrasts with the brain enzyme. Proc Natl Acad Sci U S A. 1992;89:6711–6715. doi: 10.1073/pnas.89.15.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.el-Dwairi Q, Comtois A, Gou Y, Hussain SN. Endotoxin-induced skeletal muscle contractile dysfunction: contribution of nitric oxide synthases. Am J Physiol. 1998;274(3 Pt 1):C770–C779. doi: 10.1152/ajpcell.1998.274.3.C770. [DOI] [PubMed] [Google Scholar]
- 28.Xu J, Kim GM, Chen S, Yan P, Ahmed SH, Ku G, Beckman JS, Xu XM, Hsu CY. iNOS and nitrotyrosine expression after spinal cord injury. J Neurotrauma. 2001;18:523–532. doi: 10.1089/089771501300227323. [DOI] [PubMed] [Google Scholar]
- 29.Bian K, Davis K, Kuret J, Binder L, Murad F. Nitrotyrosine formation with endotoxin-induced kidney injury detected by immunohistochemistry. Am J Physiol. 1999;277(1 Pt 2):F33–F40. doi: 10.1152/ajprenal.1999.277.1.F33. [DOI] [PubMed] [Google Scholar]
- 30.Wanecek M, Weitzberg E, Rudehill A, Oldner A. The endothelin system in septic and endotoxin shock. Eur J Pharmacol. 2000;407:1–15. doi: 10.1016/s0014-2999(00)00675-0. [DOI] [PubMed] [Google Scholar]
- 31.Ahlborg G, Weitzberg E, Lundberg JM. Circulating endothelin-1 reduces splanchnic and renal blood flow and splanchnic glucose production in humans. J Appl Physiol. 1995;79:141–145. doi: 10.1152/jappl.1995.79.1.141. [DOI] [PubMed] [Google Scholar]
- 32.Laszlo F, Evans SM, Whittle BJ. Aminoguanidine inhibits both constitutive and inducible nitric oxide synthase isoforms in rat intestinal microvasculature in vivo. Eur J Pharmacol. 1995;272:169–175. doi: 10.1016/0014-2999(94)00637-m. [DOI] [PubMed] [Google Scholar]
- 33.Siegemund M, van Bommel J, Schwarte LA, Studer W, Girard T, Marsch S, Radermacher P, Ince C. Inducible nitric oxide synthase inhibition improves intestinal microcirculatory oxygenation and CO2 balance during endotoxemia in pigs. Intensive Care Med. 2005;31:985–992. doi: 10.1007/s00134-005-2664-7. [DOI] [PubMed] [Google Scholar]