

Abstract
The Ras family of GTPases is a collection of molecular switches that link receptors on the plasma membrane to signaling pathways that regulate cell proliferation and differentiation. The accessory GTPase-activating proteins (GAPs) negatively regulate the cell signaling by increasing the slow intrinsic GTP to GDP hydrolysis rate of Ras. Mutants of Ras are found in 25–30% of human tumors. The most dramatic property of these mutants is their insensitivity to the negative regulatory action of GAPs. All known oncogenic mutants of Ras map to a small subset of amino acids. Gln-61 is particularly important because virtually all mutations of this residue eliminate sensitivity to GAPs. Despite its obvious importance for carcinogenesis, the role of Gln-61 in the GAP-stimulated GTPase activity of Ras has remained a mystery. Our molecular dynamics simulations of the p21ras–p120GAP–GTP complex suggest that the local structure around the catalytic region can be different from that revealed by the x-ray crystal structure. We find that the carbonyl oxygen on the backbone of the arginine finger supplied in trans by p120GAP (Arg-789) interacts with a water molecule in the active site that is forming a bridge between the NH2 group of the Gln-61 and the γ-phosphate of GTP. Thus, Arg-789 may play a dual role in generating the nucleophile as well as stabilizing the transition state for P—O bond cleavage.
Low molecular weight GTP-binding proteins, like p21Ras, act as molecular switches in cellular signaling pathways that control cell proliferation and differentiation (for extensive reviews see refs. 1–7). In the GTP-bound on (active) state, Ras interacts with effector molecules and transmits signals to the next downstream component. The hydrolysis of GTP to GDP switches Ras to the off (inactive) state. Guanine nucleotide exchange factors catalyze the dissociation of GDP from Ras and thus promote the loading of GTP to regenerate the active state. The accessory GTPase-activating proteins (GAPs) negatively regulate the GTP-bound state by increasing the slow intrinsic hydrolysis rate of Ras by factors of up to 105 (8–12). Oncogenic mutants of Ras are found in 25–30% of human tumors (3, 13, 14). Mutations of Gln-61 increase the activation barrier for Ras hydrolysis of GTP by 1.5–2 kcal/mol, thereby decreasing the rate by at least one order of magnitude depending on the mutant (15, 16). The most dramatic property of these mutants is their insensitivity to the negative regulatory action of GAPs. Mutations of Gln-61 have been found that reduce the rate of GAP-stimulated hydrolysis by as much as 106-fold (17, 18), which corresponds to an increase in the activation barrier by 8.5 kcal/mol. There is little doubt that the oncogenic properties of Gln-61 mutations are caused by their impact on the intrinsic and GAP-stimulated rates of GTP hydrolysis; however, the biochemical origins of these effects are still not entirely clear.
The proton-transfer step before nucleophilic attack on the
γ-phosphate of GTP is a critical point in understanding the intrinsic
and GAP-stimulated GTPase activity of Ras. It was suggested initially
that Gln-61 facilitates the nucleophilic attack on the terminal
γ-phosphate by acting as the general base in the hydrolysis (19, 20).
Experiments measuring the intrinsic hydrolysis functionality for Ras
mutated at position 61 are not sufficiently conclusive to decide
whether Gln-61 is the base. However, glutamine is a very weak base and
there is no polar group in the vicinity that can significantly alter
its pKa. Therefore, this mechanism was argued to
be highly unlikely (15, 16, 21). It has been suggested that Gln-61 is
not the base but plays a crucial structure-stabilizing role by being
able to act as a hydrogen bond donor and acceptor (18, 21). Because
there is no other likely candidate, it was advocated that the GTP
itself is the general base (18, 22). This hypothesis was supported by
linear free-energy relationships for mutant Ras proteins and by
pKa determinations using a combination of
experimental approaches (18, 22). Later, Sondek et al. (23)
used the x-ray structure of a related protein, transducin-α,
complexed with GTP analog GDP-AlF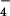 , to propose a
variant of this mechanism in which Gln-61 participates in a proton
shuttle from a water molecule in the active site to the γ-phosphate.
, to propose a
variant of this mechanism in which Gln-61 participates in a proton
shuttle from a water molecule in the active site to the γ-phosphate.
In agreement with experimental results (24), recent computer
simulations using the empirical valence bond method (25) showed that
part of the catalytic effect of RasGAPs derives from direct interaction
between an inserted arginine finger (Arg-789 in the case of p120GAP)
and the charge distribution of the transition state for hydrolysis of
GTP to GDP. The same function is expected for arginine residues of Gα
proteins based on the similarity of their active-site structure (23) to
that of the complex between p120GAP and RasGTP (26). However, the
arginine finger inserted by p120GAP approaches the γ-phosphate of GTP
from the opposite direction of that observed for Arg-178 in the crystal
structure of Gαi1 complexed with GTP analog
GDP-AlF (23). This different orientation allows the
backbone carbonyl of Arg-789 to interact with the
NH2 group of the Gln-61 side chain (26). We
observed this configuration in the early part of our 1-ns molecular
dynamics (MD) simulation of a Ras–GTP–RasGAP complex started from the
resolved part of the crystal structure of p21ras bound by
GDP-AlF3 and complexed with p120GAP (26). After
about 350 ps of simulation, our model underwent a structural
rearrangement to a conformation in which both the backbone carbonyl of
Arg-789 and the side chain NH2 of Gln-61 are
interacting with a crystallographic water molecule believed to be the
precursor of the nucleophile that attacks the γ-phosphate of GTP. We
refer to this water molecule as the “nucleophilic” water.
(23). This different orientation allows the
backbone carbonyl of Arg-789 to interact with the
NH2 group of the Gln-61 side chain (26). We
observed this configuration in the early part of our 1-ns molecular
dynamics (MD) simulation of a Ras–GTP–RasGAP complex started from the
resolved part of the crystal structure of p21ras bound by
GDP-AlF3 and complexed with p120GAP (26). After
about 350 ps of simulation, our model underwent a structural
rearrangement to a conformation in which both the backbone carbonyl of
Arg-789 and the side chain NH2 of Gln-61 are
interacting with a crystallographic water molecule believed to be the
precursor of the nucleophile that attacks the γ-phosphate of GTP. We
refer to this water molecule as the “nucleophilic” water.
Methods
The starting structure used in the MD simulations was taken from the x-ray crystal structure (Protein Data Bank code: 1WQ1) of the p21ras bound by the GTP analog GDP-AlF3 and in complex with the p120GAP (GAP-334; ref. 26). Only the Ras-binding domain of p120GAP (residues 718-1037) was included in our simulations. Because of the lack of resolution, the x-ray structure was reported with residues 981–990 of p120GAP modeled as alanines and tyrosine (Tyr)-952 modeled as a phenylalanine. The missing parts of these residues as well as the polar hydrogens were added to our starting structure by using the internal coordinate definitions of the charmm program package (27). To obtain a model with GTP bound to Ras, AlF3 was converted to be the γ-phosphate PO3 terminal group.
Protonation states of residues were determined by calculating pKa shifts with the algorithm developed by Antosiewitz et al. (28). These pKa shift calculations‡ indicated that all of the aspartic acid, glutamic acid, arginine, and lysine residues were charged as in a standard assignment. Cysteine residues that are not disulfide bonded and tyrosine residues were found to be neutral. All three p21ras histidines were charged (doubly protonated) but only 3 of the 12 histidines (His-812, -986, and -1005) of p120GAP were charged. Ambiguity regarding the pKa of a reference compound makes the protonation state of GTP more uncertain than that of the amino acids. An experimental pKa value of 2.9 ± 0.1 has been measured for the γ-phosphate of GTP in p21ras (18). Because the environment of phosphate oxygens in the p21ras-p120GAP-GTP complex is probably more positively charged than in p21ras alone, a formal charge of −4 for GTP is a reasonable first approximation. Based on this assumption, MD simulations and energy minimization were carried out to relax the starting structure before calculating pKa shifts (from the reference compound) for the two γ-phosphate oxygens not involved in Mg2+ coordination. The resulting pKa shift values of −3.4 and −2.8 were consistent with the assumption that the γ-phosphate in the p21ras–p120GAP–GTP complex is not protonated.
Simulations were performed with the NWChem computational chemistry package (30) by using the amber all-atom force field (31). Parameters for GTP were obtained by using an approach consistent with the amber force-field development.§ Missing hydrogen atoms were placed at standard positions. Ten sodium ions were added to the unit cell to achieve the charge neutrality. The x-ray crystal structure contains 35 detected waters molecules that were kept as part of our starting structure. The system was solvated further by placing the biomolecular complex into a box of solvent molecules and deleting the waters that overlap with the solute molecules. This setup resulted in a system of 7,856 solute atoms and 12,780 solvent molecules for a total of 46,196 atoms. The Particle Mesh Ewald (PME) method was used to deal with the long-range electrostatic interactions. A cutoff radius of 0.9 nm was imposed on nonbonded short-range interactions between atoms. The shake algorithm was used to constrain bond lengths involving hydrogen atoms at most stages of the equilibration and during the data-collection run. After equilibration,¶ data collection was performed for 1 ns using a 2-fs time step.
Results
Structural Rearrangement of the Active Site.
With the exception of the C terminus of the p120GAP,‖ the complex maintained its general shape throughout the simulation. After about 350 ps, the catalytic region underwent a structural rearrangement that significantly changed the hydrogen-bonding pattern in the active site. No further major structural rearrangement was observed during the production run. Detailed investigation of the structure around the catalytic site revealed only one water molecule in the vicinity of the γ-phosphate group of GTP. This water, W230 in x-ray crystal structure (26), did not exchange with the bulk waters during the simulation. The crystal structure (26) of the Ras–GTP–RasGAP complex suggests that hydrogen bonds allow W230 to form a bridge between the side-chain carbonyl of Gln-61 and the γ-phosphate of GTP. By our “not-too-strict” criteria,** this configuration was present during roughly the first 350 ps of our simulation. Fig. 1 shows that this bond formed only occasionally and for short durations after the structural rearrangement of the active site.
Figure 1.

The solid line represents the number of hydrogen bonds between the Gln-61 side-chain carbonyl and the nucleophilic water as a function of simulation time. The dashed lines represent the distance in nm between the Gln-61 side-chain carbonyl oxygen and each of the catalytic-water hydrogen atoms.
The hydrogen-bonding diagram reported by Scheffzek et al. (26) shows the NH2 group of Gln-61 forming a hydrogen bond with the GTP γ-phosphate. Even though multiple equivalent sites (three γ-phosphate oxygens and two hydrogens in the NH2 group of Gln-61) favor this type of interaction, the main tendency that we observed for the NH2 of Gln-61 was an indirect interaction with GTP via a hydrogen-bond chain through the nucleophilic water. The side-chain NH2 group of Gln-61 also contacts the carboxylate of the nearby Glu-63. The crystal structure suggests that a hydrogen bond exists between the backbone carbonyl of Arg-789 and the NH2 group of Gln-61. This bond was observed at the beginning of our simulation, but around 220 ps, Gln-61 moved away from Arg-789 and the hydrogen bond between Gln-61 and Arg-789 was later replaced with a hydrogen bond between the Arg-789 backbone and the nucleophilic water as shown in Fig. 2.
Figure 2.

The solid line represents the number of hydrogen bonds between the Arg-789 backbone carbonyl and the nucleophilic water. The dashed lines represent the distance in nm between the Arg-789 backbone carbonyl oxygen and each of the nucleophilic-water hydrogen atoms.
W230 plays a key role in the local dynamics around the catalytic region by forming an extensive network of hydrogen bonds. At ≈350 ps into the production run of our MD simulation, W230 changed its location relative to the Gln-61 side chain, lost its hydrogen bond with the side-chain carbonyl of Gln-61,** and formed a more persistent hydrogen bond with the NH2 group of Gln-61. Before this transition, W230 interacted mainly with the side-chain carbonyl of Gln-61 and the GTP γ-phosphate oxygens. A weaker and irregular interaction with the backbone of Thr-35 was present also. This pattern of interactions, shown schematically in Fig. 3A, resembles that reported in the analysis of the x-ray crystal structure (26). Simultaneous with switching between the Gln-61 side-chain groups, the nucleophilic water also forms a hydrogen bond with the backbone carbonyl oxygen of Arg-789. The pattern shown in Fig. 3B of water interacting with GTP, Gln-61 NH2, and the Arg-789 backbone carbonyl, in addition to less frequent interactions between Gln-61 NH2 and GTP, dominated the final 500 ps of our simulation.
Figure 3.

Schematic diagrams of interaction patterns during the first 250 ps (A) and last 500 ps (B) of the MD simulation. Thick solid arrows represent the interactions that always exist. Interactions shown by thick broken arrows are strong and almost always exist. Interactions shown by thick dotted arrows are important, and the thin dashed arrows show weaker interactions that are infrequent.
The structural change that occurs in our simulation around 350 ps can be viewed as the Gln-61 side chain swinging away from the active site and allowing the nucleophilic water to approach the arginine residue inserted by RasGAP. This interpretation is consistent with the x-ray crystal structures (19, 21, 26) where residues 61–64 exhibit large B factors, suggesting high mobility and possible multiple side chain positioning. MD simulations of p21Ras (32–35) also have indicated that Gln-61 is very mobile, frequently forming and breaking hydrogen bonds with the nearby groups.
Structural Characterization by Using Cluster Analysis.
To find the dominant hydrogen bond configurations, we have extended the analysis of the MD trajectory by clustering the bonding patterns. Seven key hydrogen bonds were identified in the catalytic region and were used to characterize the state of the system. These seven bonds were between: (i) W230 H and Gln-61 C⩵O (side chain); (ii) Gln-61 NH2 and W230 O; (iii) Gln-61 NH2 and GTP OG (γ-phosphate oxygens); (iv) W230 H and GTP OG; (v) W230 H and Arg-789 C⩵O (backbone); (vi) Gln-61 NH2 and Arg-789 C⩵O (backbone); and (vii) Thr-35 NH (backbone) and W230 O. In all cases, the key hydrogen bonds involve either Gln-61 or the crystallographic water, W230, which remained in the active site throughout our simulation. Because the structural reorganization took place around 350 ps, the MD results between 250 and 500 ps were not used (to avoid statistical corruption). The analysis was performed separately for the first 250-ps and for the last 500-ps parts of the run. For each snapshot of the MD trajectory, a bitmap was generated showing which of the seven key hydrogen bonds existed at that time. All of the equivalent sites (for example, three oxygens of GTP γ-phosphate or two water hydrogens), were included in deciding whether a particular hydrogen bond was present. For seven bonds, there are a total of 27 = 128 possible bonding patterns. Patterns observed in more than 5% of the conformations sampled from the MD trajectory‡‡ are shown in Table 1.
Table 1.
Hydrogen bond (HB) properties during the MD simulation
| Configuration | %* | HB
no. 1
|
HB no. 2
|
HB no. 3
|
HB
no. 4
|
HB no. 5
|
HB no. 6
|
HB
no. 7
|
|---|---|---|---|---|---|---|---|---|
| W230 H–Gln-61 C⩵O | Gln-61 NH2–W230 O | Gln-61 NH2–GTP OG | W230 H–GTP OG | W230 H–Arg-789 C⩵O | Gln-61 NH2–Arg-789 C⩵O | Thr-35 NH–W230 O | ||
| During the first 250 ps of the simulation | ||||||||
| 1 | 32 | Y | N | N | Y | N | Y | N |
| 2 | 28 | Y | N | Y | Y | N | Y | Y |
| 3 | 16 | Y | N | N | Y | N | N | N |
| 4 | 12 | Y | N | N | Y | N | Y | Y |
| 5 | 8 | Y | N | Y | Y | N | Y | N |
| During the last 500 ps of the simulation | ||||||||
| 1 | 56 | N | Y | N | Y | Y | N | N |
| 2 | 10 | Y | Y | N | Y | N | N | N |
| 3 | 7 | N | Y | N | Y | N | N | N |
| 4 | 7 | N | Y | Y | Y | Y | N | N |
| 5 | 7 | N | N | Y | Y | Y | N | N |
| 6 | 6 | N | N | N | Y | Y | N | N |
Y and N respectively denote presence or absence of the hydrogen bond.
Shows the percentage of snapshots in which the configuration was observed.
The five configurations that predominate during the first 250 ps of the simulation (Table 1) are slight variants of each other. Note that hydrogen bonds 1 and 4 are always present whereas 2 and 5 are never seen. Hydrogen bond 6 is always present except in configuration 3. In all cases, crystallographic water W230 is forming a bridge between the γ-phosphate of GTP and the side-chain carbonyl of the Gln-61. Fig. 4 shows the spatial arrangement of the active site corresponding to hydrogen-bond pattern no. 1.
Figure 4.

Conformation of the active site corresponding to the most probable hydrogen-bonding pattern during the first 250 ps of the MD simulation (configuration 1 in Table 1).
Table 1 shows that the local structure of the catalytic site is dominated by a single configuration during the last 500 ps of our MD simulation. The dominant configuration, illustrated by Fig. 5, has the water molecule in the active site interacting with the GTP γ-phosphate group, the NH2 group of Gln-61, and the backbone of Arg-789. The strong interactions of the nucleophilic water with Arg-789 and Gln-61 orient it toward the γ-phosphate so that this water molecule can be said to bridge all three groups. The other configurations in Table 1, which collectively occur with a probability of 37%, are mainly slight variations from the dominant configuration. Configuration 4 has a direct interaction between Gln-61 NH2 and the γ-phosphate, as well as with the water-mediated interaction. Configurations 2 and 3 are missing the W230–Arg-789 interaction whereas configurations 5 and 6 are missing the W230–Gln-61 interaction. The dominance of configurations with direct interaction between Arg-789 and the nucleophilic water (83% total probability) shows that, in addition to charge neutralization and structural transition-state stabilization, Arg-789 also plays a role in the initial proton transfer that generates the nucleophile.
Figure 5.

Conformation of the active site in the dominant hydrogen-bonding pattern during the last 500 ps of the MD simulation (configuration 1 in Table 1).
Discussion
Based on an impressive body of experimental data (23, 24, 26, 36, 37), an “arginine finger” hypothesis has emerged to explain how RasGAPs increase the GTPase activity of p21ras by factors as large as 105. Recent theoretical work (25) has shed considerable light on the mechanisms by which an arginine residue in the active site can influence the energetics of GTP hydrolysis. Charge neutralization of the active site groups is clearly one of the ways that Arg-789 participates in the catalytic process. During our MD simulation, one of the NH2 moieties of the Arg-789 guanidinium group formed strong hydrogen bonds with the γ- and the α-phosphate groups. This bridged configuration, seen in Figs. 4 and 5, was extremely stable and stayed intact throughout the production run. The interaction of the other NH2 group of Arg-789 was less specific; it interacted with GTP α-phosphate group, with the backbone of Thr-785, and with the Glu-31 carbonyl group.
Allosteric effects of complex formation are another important mechanism in the stimulation of GTPase activity by RasGAPs. Glennon et al. (25) showed that the conformation of Ras in the p21ras-p120GAP-GTP complex (26) was more effective in product stabilization than the conformation observed in x-ray structures of p21ras alone (19, 21). Our simulations show a dramatic effect of complex formation on the configuration of Gln-61 and the nucleophilic water in the active site. That RasGAP binding moves Gln-61 closer to the γ-phosphate of GTP is clear from structural data. Sondek et al. (23) proposed that this allosteric effect allowed Gln-61 to act as a proton shuttle from the nucleophilic water to the γ-phosphate. During the second half of our 1-ns MD simulation, crystallographic water W230 resided very close to Arg-789 and interacted strongly with its backbone carbonyl. This configuration, which has not been reported previously, suggests that Arg-789 plays direct role in activating this water molecule for nucleophilic attack on the γ-phosphate.
The possibility for the same residue to participate in formation of the nucleophile as well as to stabilize the transition state for the subsequent bond cleavage may help explain why insertion of an arginine finger enhances GTPase activity of Ras to such a large extent. It is known that the GTP hydrolysis rate of closely related Gα proteins is higher than the intrinsic rate for p21ras but lower than the RasGAP-stimulated rate (8–12). Structural comparison of Arg-178 in Gαi1 and Arg-789 in the p21ras–p120GAP complex shows that the terminal groups on the side chains of these arginine residues are located at almost the same position relative to the γ-phosphate (see for example figure 5C of ref. 26). However, these arginine residues point toward the GTP from different directions in the two systems. The orientation of the backbone of the arginine finger in the p21ras–p120GAP complex allows it to interact with the nucleophilic water, which is not possible for the orientation in Gαi1. Having the same residue participate in both the activation and bond-cleavage steps could increase the efficiency of the overall reaction, thereby making the hydrolysis rate by the Ras–RasGAP complex higher than that of the Gα proteins.
The role of Gln-61 in GAP-stimulated hydrolysis of GTP, suggested by the insensitivity of Gln-61 mutants, remains the greatest area of uncertainty in the catalytic mechanism. The proximity of Gln-61 to the γ-phosphate in the p21ras-p120GAP complex raises the possibility that it could participate in stabilization of the transition state; however, recent work by Glennon et al. (25) appears to rule out stabilization by direct electrostatic interactions. Alternatively, Gln-61 might contribute significantly to stabilizing a conformation of Ras that is favorable for catalysis of the hydrolysis reaction by the inserted arginine finger (25). The configuration that we observe in the last 500 ps of our MD simulation, where the nucleophilic water is linking both Gln-61 and Arg-789 to the γ-phosphate, might be important in this regard.
The prediction by Schweins et al. (22) that the γ-phosphate of GTP in p21ras has a pKa around 3 was confirmed by 31P-NMR experiments (18). Our pKa-shift calculations for the γ-phosphate oxygens of GTP in p21ras alone and in the complex with p120GAP suggest that complex formation decreases the proton affinity of the γ-phosphate oxygen because of the presence of additional positive charge in the active site. The configuration that dominates the later part of our MD simulation might be optimal for proton transfer when Arg-789 is inserted to stabilize the bond-cleavage transition state. Gln-61 could be essential in such a mechanism because its shape and electrostatic properties complement Arg-789. Because Gln-61 is not charged, it would not destabilize the transition state by interacting with Arg-789 and GTP too strongly; however, Gln-61 has enough polarity and the correct size to help orient the nucleophilic water. Clearly, additional work is needed to investigate these speculations about the role of active-site conformations in the catalytic process.
After our study was submitted, Farrar et al. (38) reported that the association of p120GAP induces a conformational change near the metal ion of the active site. By using a combination of electron spin-echo envelope modulation (ESEEM) experiments and short-time MD simulations (50 ps), they observed that the residues, particularly the Gly-13 and -60, which are implicated in oncogenic mutants, in the active-site region can rearrange upon complex formation with the GAP. Conclusions of the study by Farrar et al. and of our study are complementary and support each other in the finding that association of p120GAP can induce a conformation change in the active site that is necessary in the GAP-dependent GTPase reaction.
Acknowledgments
This research was performed at the W. R. Wiley Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the U.S. Department of Energy Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory, which is operated for the Department of Energy by Battelle. The computations were performed by using the supercomputing resources at the National Energy Research Scientific Computer Center (NERSC) and in the Molecular Sciences Computing Facility in the Environmental Molecular Sciences Laboratory.
Abbreviations
- GAP
GTPase-activating protein
- MD
molecular dynamics
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Following the approach detailed in ref. 28, a dielectric constant of 20 was assigned to the region occupied by the proteins, GTP, and the magnesium ion. Calculations were carried out by using the uhbd program (29). The dielectric constant of the surrounding solvent media was assumed to be 80. The united atom charmm force-field charges were used. The probe radius was 1.4 Å, and dielectric boundary smoothing and successive focusing were used. The histidines were modeled as having protonatable groups at their ɛ nitrogen sites. The pH was set to 7.0. The γ-phosphate oxygens of GTP were not protonated. As discussed in Methods, the protonation properties of the oxygens were investigated separately.
Ab initio electronic-structure calculations using the 6–31G* basis set were carried out for GTP at a geometry derived from the x-ray crystal structure of p21Ras bound to the slowly hydrolyzing GTP analogue guanosine 5′-[β,γ-imido]triphosphate (GppNp) (19). The electrostatic potential at the molecular surface was generated from the Hartree–Fock wave function. A restrained electrostatic surface potential (RESP) fit then was used to obtain the atomic charges. The short-range force-field parameters for GTP were assigned using analogy to published results for nucleic acids and proteins (31).
The system was equilibrated in several stages. First, before the system was solvated, the hydrogen atom positions were energy minimized while the heavy atoms were fixed. After solvating the system, the solute atoms were kept fixed and solvent-molecule positions were optimized by energy minimization followed by a short MD simulation. To reduce the bias, the velocities were assigned randomly at regular intervals by using a Boltzmann distribution. Solute-proton and solvent-molecule positions were equilibrated further by running a 5-ps MD simulation and gradually increasing the temperature to 298 K by randomly assigning the velocities at regular intervals. This equilibration step was followed by energy minimization of the whole system and equilibration MD simulations to relax the structure. Once the structure was reasonably relaxed, the Particle Mesh Ewald option was turned on to deal with long-range electrostatics. Constant temperature and pressure equilibration MD simulations were run for 22 ps during which velocities were reassigned at regular intervals. The equilibration run was continued for another 190 ps without reassigning the velocities.
The 32 C-terminal residues lost their helical secondary structure during the production phase of the simulation. Omitting the unresolved parts of p120GAP probably caused an artificial unwinding of the C-terminal helix, which is a small domain on the side of the larger Ras-binding domain and far from the catalytic site. To confirm that unwinding of the C terminus did not have any important effect on the properties of the remainder of the system, we ran a short simulation with the C-terminal helix truncated. This second simulation with a shortened GAP component produced results similar to the first simulation, which assured us that the properties of the catalytic region were unaffected by the unwinding of the C terminus during our production run.
In our analysis of hydrogen bonds, the criterion used to determine whether a bond existed was that the distance between the heavy atoms be less than 3.2 Å and that the donor H-acceptor angle be at least 90°. Use of stricter hydrogen-bond criteria, R < 3.2 Å and a minimum angle of 145°, showed that the hydrogen bond between the Gln-61 side chain C⩵O group and the nucleophilic water formed only occasionally, even during the early part of our MD simulation.
Conformations for H-bond analysis were saved every 10 ps during the first 250 ps of the simulation and every 1 ps during the final 500 ps.
References
- 1.Bourne H R, Sanders D A, McCormick F. Nature (London) 1990;348:125–132. doi: 10.1038/348125a0. [DOI] [PubMed] [Google Scholar]
- 2.Bourne H R, Sanders D A, McCormick F. Nature (London) 1991;349:117–127. doi: 10.1038/349117a0. [DOI] [PubMed] [Google Scholar]
- 3.Lowy D R, Willumsen B M. Annu Rev Biochem. 1993;62:851–891. doi: 10.1146/annurev.bi.62.070193.004223. [DOI] [PubMed] [Google Scholar]
- 4.Wiesmuller L, Wittinghofer A. Cell Signalling. 1994;6:247–267. doi: 10.1016/0898-6568(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 5.Hilgenfeld R. Nat Struct Biol. 1995;2:3–6. doi: 10.1038/nsb0195-3. [DOI] [PubMed] [Google Scholar]
- 6.Maegley K A, Admiraal S J, Herschlag D. Proc Natl Acad Sci USA. 1996;93:8160–8166. doi: 10.1073/pnas.93.16.8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kjeldgaard M, Nyborg J, Clark B C. FASEB J. 1996;10:1347–1368. [PubMed] [Google Scholar]
- 8.Trahey M, Wong G, Halenbeck R, Rubinfeld B, Martin G A, Ladner M, Long C M, Crosier W J, Watt K, Koths K, McCormick F. Science. 1988;242:1697–1700. doi: 10.1126/science.3201259. [DOI] [PubMed] [Google Scholar]
- 9.Gibbs J B, Schaber M D, Alllard W J, Sigal I S, Scolnick E M. Proc Natl Acad Sci USA. 1988;85:5026–5030. doi: 10.1073/pnas.85.14.5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bollag G, McCormick F. Nature (London) 1991;351:576–579. doi: 10.1038/351576a0. [DOI] [PubMed] [Google Scholar]
- 11.Gideon P, John J, Frech M, Lautwein A, Clark R, Scheffler J E, Wittinghofer A. Mol Cell Biol. 1992;12:2050–2056. doi: 10.1128/mcb.12.5.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eccleston J F, Moore K J M, Morgan L, Skinner R H, Lowe P N. J Biol Chem. 1993;268:27012–27019. [PubMed] [Google Scholar]
- 13.Barbacid M. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 14.Bos J. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 15.Chung H-H, Benson D R, Schultz P G. Science. 1993;259:806–808. doi: 10.1126/science.8430333. [DOI] [PubMed] [Google Scholar]
- 16.Langen R, Schweins T, Warshel A. Biochemistry. 1992;31:8691–8696. doi: 10.1021/bi00152a002. [DOI] [PubMed] [Google Scholar]
- 17.Trahey M, McCormick F. Science. 1987;238:542–545. doi: 10.1126/science.2821624. [DOI] [PubMed] [Google Scholar]
- 18.Schweins T, Geyer M, Scheffzek K, Warshel A, Kalbitzer H R, Wittinghofer A. Nat Struct Biol. 1995;2:36–44. doi: 10.1038/nsb0195-36. [DOI] [PubMed] [Google Scholar]
- 19.Pai E F, Krenkel U, Petsko G A, Goody R S, Kabsch W, Wittinghofer A. EMBO J. 1990;9:2351–2359. doi: 10.1002/j.1460-2075.1990.tb07409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krenkel U, Schlichting I, Scherer A, Schumann R, Frech M, John J, Kabsch W, Pai E F, Wittinghofer A. Cell. 1990;62:539–548. doi: 10.1016/0092-8674(90)90018-a. [DOI] [PubMed] [Google Scholar]
- 21.Prive G G, Milburn M V, Tong L, DeVos A M, Yamaizumi Z, Nishimura S, Kim S-H. Proc Natl Acad Sci USA. 1992;89:3649–3653. doi: 10.1073/pnas.89.8.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schweins T, Langen R, Warshel A. Nat Struct Biol. 1994;1:476–484. doi: 10.1038/nsb0794-476. [DOI] [PubMed] [Google Scholar]
- 23.Sondek J, Lambright D G, Noel J P, Hamm H E, Sigler P B. Nature (London) 1994;372:276–279. doi: 10.1038/372276a0. [DOI] [PubMed] [Google Scholar]
- 24.Ahmadian M R, Stege P, Scheffzek K, Wittinghofer A. Nat Struct Biol. 1997;4:686–689. doi: 10.1038/nsb0997-686. [DOI] [PubMed] [Google Scholar]
- 25.Glennon T M, Villa J, Warshel A. Biochemistry. 2000;39:9641–9651. doi: 10.1021/bi000640e. [DOI] [PubMed] [Google Scholar]
- 26.Scheffzek K, Ahmadian M R, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, Wittinghofer A. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 27.Brooks B R, Bruccoleri R E, Olafson B D, States D J, Swaminathan S, Karplus M. Comp Chem. 1983;4:187–217. [Google Scholar]
- 28.Antosiewitz J, McCammon J A, Gilson M. J Mol Biol. 1994;238:415–436. doi: 10.1006/jmbi.1994.1301. [DOI] [PubMed] [Google Scholar]
- 29.Madura J D, Briggs J M, Wade R C, Davis M E, Luty B A, Ilin A, Antosiewicz J, Gilson M K, Bagheri B, Scott L R, McCammon J A. Comput Phys Commun. 1995;91:57–95. [Google Scholar]
- 30.Harrison R J, Nichols J A, Straatsma T P, Dupuis M, Bylaska E J, Fann G I, Windus T L, Apra E, Anchell J, Bernholdt D, et al. nwchem, A Computational Chemistry Package for Parallel Computers. Richland, WA: Pacific Northwest National Laboratory; 2000. , Version 4.0. [Google Scholar]
- 31.Cornell W D, Cieplak P, Bayly C I, Gould I R, Merz K M, Ferguson D M, Spellmeyer D C, Fox T, Caldwell J W, Kollman P A. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 32.Foley C K, Pedersen L G, Charifson P S, Darden T A, Wittinghofer A, Pai E F, Anderson M W. Biochemistry. 1992;31:4951–4959. doi: 10.1021/bi00136a005. [DOI] [PubMed] [Google Scholar]
- 33.Frech M, Darden T A, Pedersen L G, Foley C K, Charifson P S, Anderson M W, Wittinghofer A. Biochemistry. 1994;33:3237–3244. doi: 10.1021/bi00177a014. [DOI] [PubMed] [Google Scholar]
- 34.Diaz J F, Wroblowski B, Engelborghs Y. Biochemistry. 1995;34:12038–12047. doi: 10.1021/bi00037a047. [DOI] [PubMed] [Google Scholar]
- 35.Ma J, Karplus M. Proc Natl Acad Sci USA. 1997;94:11905–11910. doi: 10.1073/pnas.94.22.11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coleman D E, Berghuis A M, Lee E, Linder M E, Gilman A G, Sprang S R. Science. 1994;265:1405–1412. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- 37.Mittal R, Ahmadian M R, Goody R S, Wittinghofer A. Science. 1996;273:115–117. doi: 10.1126/science.273.5271.115. [DOI] [PubMed] [Google Scholar]
- 38.Farrar C T, Ma J, Singel D J, Halkides C J. Structure (London) 2000;8:1279–1287. doi: 10.1016/s0969-2126(00)00532-3. [DOI] [PubMed] [Google Scholar]