

Abstract
Purpose
To characterize the effects of diabetes on the expression of histidine decarboxylase mRNA and on the morphology of the histaminergic centrifugal axons in the rat retina.
Methods
Rats were made diabetic by streptozotocin. After 3 months, retinal histidine decarboxylase expression was analyzed by in situ hybridization in radial sections. Flatmount retinas from a second group of rats were labeled with an antiserum to histamine or an antibody to phosphorylated neurofilament protein.
Results
Histidine decarboxylase mRNA was expressed in cells in the inner and outer nuclear layers of diabetic retinas, but not in normal retinas. However, immunoreactive (IR) histamine was not localized to perikarya in either the normal or the diabetic retinas. Instead, a population of centrifugal axons was labeled. These axons emerged from the optic disc and had varicose terminal branches in the inner plexiform layer (IPL) of the peripheral retina. Some branches ended on large retinal blood vessels and others in dense clusters in the IPL. In rats with streptozotocin-induced diabetes, the centrifugal axon terminals developed many large swellings that contained neurofilament immunoreactivity; these swellings were rare in normal retinas.
Conclusions
Diabetes perturbs the retinal histaminergic system, causing increases in histidine decarboxylase mRNA expression in neurons or glia and abnormal focal swellings on the centrifugal axons.
In mammalian retinas, centrifugal axons contain immunore-active (IR) histamine and originate from neurons in the hypothalamus.1-3 In macaque monkey retinas, histamine-IR axons emerge from the optic nerve head, run in the optic fiber layer, and terminate in the IPL, sometimes adjacent to retinal blood vessels.4 Histamine released from these centrifugal axons may promote the breakdown of the blood–retinal barrier (BRB) in diabetic retinopathy. In patients with diabetes, microaneurysms commonly form in the central retina, temporal to the fovea,5 and this area has the highest density of histamine-IR centrifugal axons in normal macaque retinas.4 In rats, histamine applied intravitreally increases the permeability of the BRB.6 Histamine also decreases the expression of the tight junction protein ZO-1 in cultured bovine retinal vascular endothelial cells, and this effect would be expected to increase vessel permeability.7 Histamine antagonists reduce the thickening of the retinal capillary basement membranes8 and prevent increases in retinal vascular permeability in rats with streptozotocin-induced diabetes (streptozotocin-diabetic rats).9
In patients with insulin-dependent diabetes who have mild, nonproliferative diabetic retinopathy, a combination of histamine H1 and H2 receptor antagonists administered for 6 months significantly decreases the permeability of the BRB.10 However, patients with diabetic macular edema do not benefit from 1 year of treatment with an H1 antagonist alone.11 The activity of retinal histidine decarboxylase, the enzyme that synthesizes histamine, is markedly increased in experimental diabetic rats.12 Therefore, the purposes of this study were to identify the cells that express histidine decarboxylase in diabetic rat retinas and to determine whether diabetes alters retinal histaminergic axons.
Methods
Animals
Male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) were given a single tail vein injection of streptozotocin (65 mg/kg; Sigma Chemical Co., St. Louis, MO), freshly dissolved in 10 mM citrate buffer (pH 4.5). Diabetes was confirmed 3 days later by blood glucose greater than 250 mg/dl (Lifescan, Milpitas, CA). For 3 months, age-matched control and diabetic rats were monitored regularly by weight and blood glucose tests. Rats were housed in accordance with the Institutional Animal Care and Use Committee guidelines, and the study protocol adhered to the ARVO Statement for the Use Animals in Ophthalmic and Vision Research. All rats were group housed in suspended wire-bottomed cages with ad libitum food and water and under a normal 12-hour light-dark schedule.
In Situ Hybridization
A 30-mer oligonucleotide probe complementary to rat histidine decarboxylase cDNA (nucleotides 583-612) was generated with a DNA synthesizer (PE Applied Biosystems; Foster City, CA). This probe is identical with the first 30 nucleotides of a 50-mer oligonucleotide probe, 583-632, which was used previously to label histaminergic neurons in the tuberomammillary nucleus, but not in any other brain areas. This probe is not homologous to any known decarboxylase sequence.13 A second 30-mer oligonucleotide sense probe matching the cDNA nucleotides for the same region was used as a control. The probes were 3′ end labeled using 35S-dATP (NEN, Boston, MA) and terminal deoxynucleotide transferase (Roche Boehringer Mannheim Biochemicals; Indianapolis, IN), according to protocols supplied by the manufacturers.
Eleven diabetic and six control rats were decapitated and their eyes enucleated and frozen in optimal cutting temperature (OCT) compound (Miles Laboratories, Elkhart, IN) using isopentane cooled with dry ice. Cryostat sections were cut at 8 μm, collected on RNAase-free slides, and fixed immediately for 10 minutes in 4% buffered paraformaldehyde. They were then rinsed in phosphate-buffered saline (PBS) and dehydrated in ethanol. Sections were hybridized in 75 μl hybridization buffer (50% formamide, 1× Denhardt’s solution, 3× SSC, 100 μg/ml salmon sperm DNA, 125 μg/ml tRNA, 100 mM dithiothreitol [DTT], 10% dextran sulfate) containing 106 counts per minute (cpm)/ml oligo probe at 37°C overnight in a humidified box. After the hybridization, sections were washed under conditions of high stringency in decreasing concentrations of SSC and dehydrated in ethanol. Dried sections were dipped into photographic emulsion (NTB2, 1:1 with water; Eastman Kodak, Rochester, NY), exposed for 2 weeks, developed in D-19 and fixed. The sections were counterstained with hematoxylin and eosin. The positive controls for this experiment were sections of fetal liver that contain high levels of histidine decarboxylase mRNA.14 Separate sections were stained with acidic toluidine blue to label mast cells in the sclera and choroid.15 They were reviewed in a masked fashion.
Immunohistochemistry of Flatmount Retinas
The rats were deeply anesthetized with ketamine-xylazine (40 mg-4 mg/kg intramuscularly), and blood was taken from the tail vein to determine the final blood glucose level. Rats were perfused transcardially with freshly prepared, ice-cold 4% 1-ethyl-3(3diethylaminopropyl)-carbodiimide in 0.1 M phosphate buffer (pH 7.4) for approximately 6 minutes, and the eyes were hemisected and postfixed overnight at 4°C in the same fixative. Carbodiimide was used for tissue fixation, because histamine has been conjugated to bovine serum albumin with water-soluble carbodiimides to make it antigenic. Thus, the histamine antibody was more sensitive because of the resemblance of the histamine conjugate to the fixed tissue antigen.16 Other rats were decapitated and the posterior halves of their eyes were fixed by immersion in the same fixative overnight at 4°C. Whole retinas were isolated and the vitreous removed with a fine brush. Optic nerves were fixed in the same way, cryoprotected in 30% sucrose, and frozen in OCT. Longitudinal cryostat sections (30 μm) were cut and dried overnight on microscope slides.
Whole retinas were incubated in rabbit anti-histamine primary antiserum (AB134,1:500; Chemicon, Temecula, CA) with 0.3% Triton X-100, 0.25% bovine serum albumin in PBS with 0.3% azide (PBSa) for 8 to 10 days at 4°C. Retinas were incubated in affinity-purified biotinylated goat anti-rabbit secondary antibody (1:100, Vector Laboratories, Burlingame, CA) for 2 days at 4°C, followed by streptavidin-Cy3 (1:00, Jackson ImmunoResearch, West Grove, PA) overnight at 4°C. Retinas were wholemounted in 3:1 glycerol/PBSa with p-phenylenediamine to prevent fading. Optic nerve sections were processed similarly. Alternate optic nerve sections from both normal and diabetic rats were incubated without the histamine antiserum or with the histamine antiserum preabsorbed overnight at 4°C with histamine (1 mg/ml) of diluted antiserum.
Retinas were also labeled with antibody to phosphorylated neurofilament protein. Hemisected eyes were fixed in 2% paraformaldehyde for 10 minutes at room temperature. Retinas were isolated and blocked for 1 to 2 hours with 10% donkey serum (Sigma) in PBS with 0.3% Triton X-100. Retinas were incubated for 3 days in a monoclonal mouse anti-phosphorylated 200-kDa neurofilament antibody (1:1000, clone NE14; Sigma) and Cy2-conjugated donkey anti-mouse F(ab′)2 secondary antibody (1:1000; Jackson ImmunoReaearch) in blocking solution at 4°C overnight. The retinas were mounted on 3-aminopropyltriethoxy silane–coated slides (Aqua/Polymount; Polysciences, Warrington, PA).
Image Analysis
Images of histamine-IR axons were acquired on a confocal laser-scanning microscope (LSM; Carl Zeiss, Thornwood, NY) with a krypton-argon laser at a 512 × 512-pixel image size. All diameters and areas were calculated using software (Zeiss) from reconstructed stacks of optical sections. Axons were drawn and measured using a 40× water-immersion lens with a motorized stage (Neurolucida software, ver. 3.18; MicroBrightField, Colchester, VT). Drawings were rotated about the z-axis by computer (Neuroexplorer, ver. 3.01; MicroBrightField). Axon swellings were counted using a microscope with a ×40 oil-immersion lens (Axiophot; Carl Zeiss). Neurofilament immunoreactivity and autoradiograms were observed with a BH-2 fluorescence microscope (Olympus, Lake Success, NY). Digital images were captured using image-analysis software (Optimus, Seattle, WA) linked to a charge-coupled device camera (Sony, Tokyo, Japan) with a 640 × 480-pixel image size and a resolution of 213 pixels/in. Brightness and contrast of digital images were adjusted with image-management software (Photoshop; Adobe, San Jose, CA), and image resolution was held constant.
Statistical Analysis
To determine the efficacy of the streptozotocin injections, weight and blood glucose were measured in both normal and diabetic rats and compared using the Mann-Whitney test. A t-test was used to compare the number of swellings in control and diabetic retinas. Linear correlations between the numbers of swellings, rat weight, and blood glucose were evaluated on computer (Excel; Microsoft, Redmond, WA). P < 0.05 was considered to be statistically significant.
Results
Localization of Histidine Decarboxylase mRNA in the Rat Retina
The first set of experiments was designed to test the hypothesis that elevated histidine decarboxylase activity in diabetic retinas12 results from an increase in expression of histidine decarboxylase mRNA in neurons and glial cells. Specific hybridization with histidine decarboxylase antisense probe was found in 7 of 11 diabetic retinas. Labeled cells were most common in the outer nuclear layer (ONL; Fig. 1). In some rats, there were patches of diffuse labeling in the vitreal half of the inner nuclear layer (INL). Occasionally, small clusters of cells were also labeled in the INL and in the ganglion cell layer (GCL). No gene expression was found in sections of normal retinas. This pattern of labeling is consistent with histidine decarboxylase mRNA expression in a subset of retinal neurons including photoreceptors and, possibly, bipolar cells, amacrine cells, or both. A subset of labeled cells may have been glia. However, the specific types of cells labeled could not be identified. No specific hybridization was found by using the sense oligomer on diabetic rats (Fig. 1c). Both mast cells and hepatocytes (not illustrated) were intensely labeled. Mast cells were detected in the choroid and sclera, but no mast cells were found in control or diabetic retinas. These data support previous findings of increased retinal histidine decarboxylase activity in diabetes.12
Figure 1.
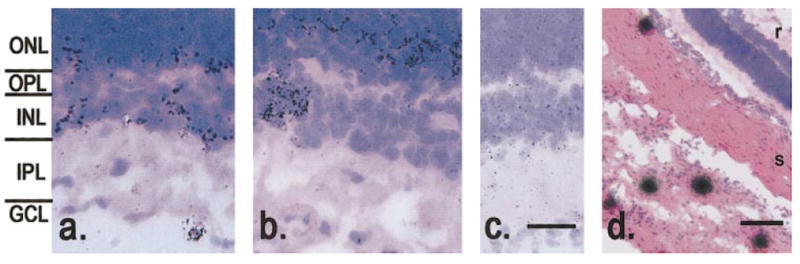
Specific hybridization of histidine decarboxylase mRNA in diabetic retinas. (a) Some retinas had labeled cells in the lower third of the INL; occasionally, a single cell in the GCL was labeled (arrow). (b) In other retinas, clusters of labeled cells were found in the INL (arrow). (c) Section incubated with the sense probe. No specific hybridization was found in any layer of the retina. (d) Labeled mast cells in the choroid and sclera. Scale bar, (c) 20 μm; (d) 100 μm.
Distribution of Histamine Immunoreactivity in Rat Retinas
The second set of experiments was designed to test the hypothesis that centrifugal axons in the normal rat retina contain histamine. Wholemount preparations were labeled with antibodies to histamine by an immunofluorescence technique. No histamine-IR cell bodies were observed in either normal or diabetic retinas. The histamine-IR axons were 0.6 to 1.0 μm in diameter as they emerged from the optic disc in the nerve fiber layer (NFL) and decreased in diameter as they ran to the peripheral retina. Most retinas had only one axon, and it typically supplied terminal branches to the inner plexiform layer (IPL; Fig. 2a, red).
Figure 2.
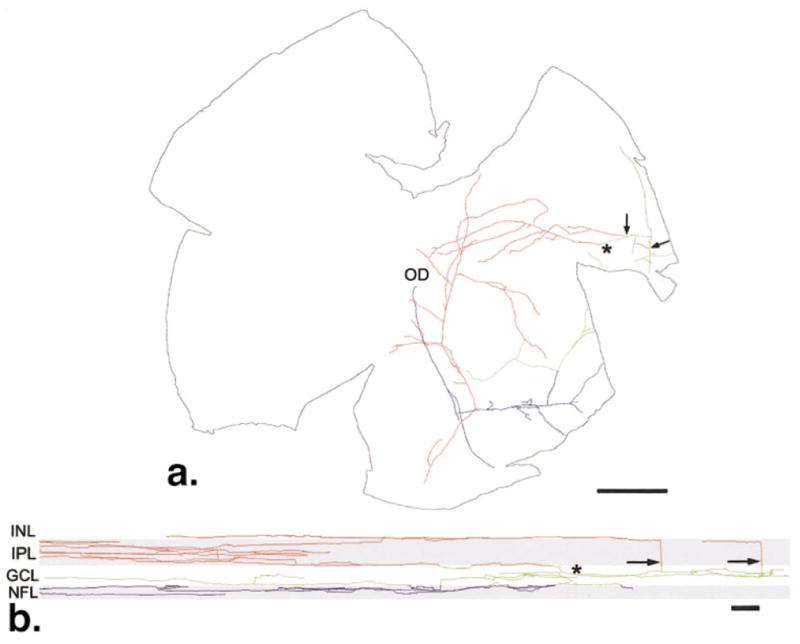
(a) Tracing of a single histamine-IR axon. This axon supplied terminal branches to the IPL (red), GCL (green), and NFL (blue) that covered approximately 12 mm2 of the retina. The primary axon emerged from the optic disc (OD) in the nerve fiber layer and ran to the peripheral retina. The axonal branches (arrows) ran through the GCL into the IPL where they terminated. The majority of terminals in the IPL were supplied by this branch (*). (b) A section of the wholemount rotated 90° as though it were viewed from the top to the bottom. This manipulation of the data set compresses all the branches into two dimensions. Typically, most terminals in the IPL arose from a single branch (*) that gradually ascends from the NFL to the outer half the IPL. Other branches (arrows) ran orthogonally to the border of the INL and IPL but did not contribute many terminal branches there. Scale bar, (a) 1.0 mm; (b) 30 μm.
Some histamine-IR axons terminated in the GCL (green) and NFL (Fig. 2a, blue), but the majority of axons did not have branches there. When the primary axons reached the peripheral retina, they made a perpendicular turn and descended through the GCL to the IPL (Fig. 2b). Most axons branched in a band 20 μm wide in the center of the IPL. Some terminal branches extended into the INL, where they ended in small swellings. Some retinas had one or two additional axons emerging from the optic disc that branched much less extensively and had different branching patterns. Some terminated with a few short branches in the peripheral IPL. Other axons ran to the peripheral retina without branching and returned to the optic disc.
Histamine-IR terminals in the IPL typically had varicosities that ranged from 0.8 to 3.1 μm in diameter (Fig. 3a). Varicosities on branches in the NFL and GCL were smaller and less common. Some branches were completely devoid of varicosities, and the primary axons had very few, if any varicosities. Histamine-IR axons usually terminated in swellings ranging in diameter from 1.0 to 3.3 μm. A few histamine-IR branches were closely associated with large retinal blood vessels in the IPL. Some short branches from the primary axon contacted the surface of blood vessels (Fig. 3b).
Figure 3.
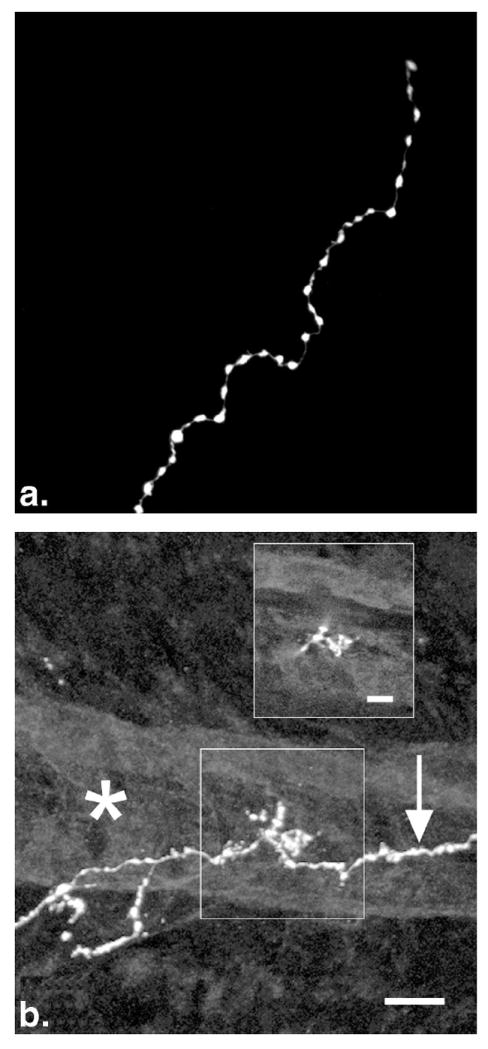
Histamine-IR terminal branches in the IPL of the normal rat retina. (a) Distribution of varicosities at the end of a terminal branch. The varicosities were large and closely spaced. Branches often ended in a small swelling. (b) Contact with a retinal blood vessel. A labeled axon is seen passing below a large blood vessel (*). A single optical section (b, inset) shows that these terminals were in the same focal plane as the surface of the blood vessel. Scale bars, (b) 10 μm; (b, inset) 5 μm.
Some histamine-IR branches made specialized endings in the IPL. One common specialization was a cluster of varicose terminal branches that overlapped extensively within a small region of the retina (Fig. 4a). One histamine-IR axon had as many as 12 clusters, but some axons had none. These clusters were distributed randomly in the peripheral retina. The area covered by each cluster varied, averaging 1200 μm2. Other histamine-IR terminal branches had spines in the IPL (Fig. 4b). These spines extended 1 to 2 μm from the main terminal branch. The larger spines ended in swellings, but the thinner and shorter spines did not. Spines were not found on any branches in the NFL or GCL.
Figure 4.

Specialized centrifugal axon terminals in the IPL. (a) Clusters of histamine-IR terminals were found in the IPL. They are formed by several overlapping varicose branches. No cell bodies or blood vessels were detected within these clusters. (b) Histamine-IR spines in the peripheral retina. These spines ended either with (arrowhead, inset) or without (arrow, inset) a small swelling. Spines were identified only on branches in the IPL. Scale bar, (a) 10 μm; (b, inset) 2 μm.
Histamine-IR axons in the normal and diabetic optic nerve were identical in diameter with the axons labeled in the retina. Small, widely spaced varicosities were also present on these axons. Occasionally, axons bifurcated as they ran toward the retina (Fig. 5). Six or more labeled axons were found in the optic nerve, but the retina usually had only one histamine-IR axon. Apparently, branches in the optic nerve either ended there or returned to the brain. Labeled axons were also observed in the optic chiasm and optic tract (not shown) confirming the results of Auvinen and Panula.17 Control sections with primary antiserum omitted or preadsorbed with histamine showed no evidence of labeled axons.
Figure 5.

Histamine-IR axons in the normal optic nerve. This axon bifurcated and crossed the central retina artery (*). Widely spaced, small varicosities were present on most of these axons. Most branches ran toward the retina, but a few ran at oblique angles (arrow). Labeled axons were not restricted to any region of the optic nerve. Scale bar, 25 μm.
Pathologic Changes in Centrifugal Axons
The third set of experiments was designed to test the hypothesis that centrifugal axons are abnormal in retinas of streptozotocin-diabetic rats. After 3 months of diabetes, wholemount preparations of rat retina were labeled with the same immunofluorescence technique. Numerous swellings 8 to 12 μm in diameter were found on histamine-IR branches in the peripheral retinas of diabetic rats (Fig. 6). Axons in some normal retinas also had a few large swellings on short branches running toward the INL, typically near the ends of terminal branches. Although the majority of swellings were found on axons in the IPL, they occasionally were observed in both the NFL and INL. The swellings were typically spherical (Fig. 6a), but a few were irregular in shape (Fig. 6b). Only the outer portions of the swellings were labeled (Fig. 6, inset).
Figure 6.

Axonal swellings in the IPL of diabetic retinas. (a) Spherical swelling on a histamine-IR branch. Inset: a single optical section through the center of this swelling showing no histamine immunore-activity in the center; only the outer surface was labeled (arrowhead). In some cases, other irregularly shaped structures (arrow) were found on branches within 10 μm of a spherical swelling. (b) Other large swellings on histamine-IR branches that were ovoid in shape also labeled only at the edges (arrowhead, inset). (c) Phosphorylated neurofilament-IR axon (arrow) with two large swellings in proximity to each other. Their centers were intensely immunoreactive. Scale bar, 10 μm.
To determine whether the histamine-IR swellings were more prevalent in diabetic retinas, the swellings were counted in 7 diabetic and 13 control retinas (Table 1). The histamine-IR axons were observed in their entirety. The number of swellings per diabetic retina (7.57 ± 2.25; mean ± SE) was significantly greater than in control retinas (0.5 ± 0.33; P ≤ 0.02). Most normal histamine-IR axons contained no swellings. In diabetic retinas, a histamine-IR axon always had at least one swelling, and some had as many as 15. There was no correlation between the number of swellings and the overall change in blood glucose or weight after a 3-month period in either normal or diabetic rats.
Table 1.
Characteristics of Animals
| Treatment Group | Swellings | Weight (g) | Blood Glucose (mg/dl) |
|---|---|---|---|
| Control (n = 11) | 0.538 ± 0.33 | 506 ± 21 | 82 ± 8 |
| Diabetic (n = 7) | 7.57 ± 2.3* | 322 ± 17† | 396 ± 31† |
All values are means ± SE.
Significant difference (P ≤ 0.02).
Significant difference (P ⪡ 0.001).
Using an antibody to phosphorylated neurofilament in retinas of diabetic and age-matched control rats, many labeled axons and a few ganglion cell bodies were observed. Centrifugal axons could be clearly distinguished from other labeled axons by a more intense immunofluorescence, a larger diameter, and a tendency not to follow the labeled ganglion cell axons in the NFL. Some large swellings, 10 μm in diameter, were found on these phosphorylated neurofilament-IR axons in the IPL of diabetic retinas (Fig. 6c); none were found in the normal retinas. They resembled the large histamine-IR swellings, except that the neurofilament immunoreactivity filled the entire swelling.
Discussion
Diabetes is known to produce glial activation18-20 and degenerative changes in retinal neurons.21 The results of this study provide further evidence for changes in retinal histaminergic system during diabetes.
The results of in situ hybridization for histidine decarboxylase mRNA and histamine immunolabeling were consistent in normal rat retinas. That is, histamine immunoreactivity was found only in centrifugal axons, where no histidine decarboxylase mRNA would be expected. The results of the two techniques in diabetic retinas were inconsistent, however. There were no histamine-IR cell bodies, but histidine decarboxylase mRNA was found in cells in all nuclear layers of the retina. The increase in retinal histidine decarboxylase activity after 3 weeks of streptozotocin-induced diabetes12 and the results of in situ hybridization suggest that there should be additional cells containing immunoreactive histamine in the diabetic retinas, but none were found. Perhaps the enzyme was inactive in vivo, or else the levels of histamine in the retinal cells were below the limit of detection of our immunolabeling technique. The ratio of histidine decarboxylase activity to tissue histamine concentration varies widely between species, and the rat has a particularly low ratio (0.04).22 Another possibility is that the increase in histidine decarboxylase activity was transient, peaking at 3 weeks and declining afterward. There have also been apparently contradictory results using antibodies to histidine decarboxylase and histamine in the guinea pig retina. Histidine decarboxylase-IR horizontal cells were labeled,23 but endogenous histamine was localized only to centrifugal axons.1
The histamine-IR centrifugal axons resembled those described previously in the macaque retina4 in most respects. The major difference was that some of the centrifugal axons in rats had clusters of terminal branches in the IPL. The centrifugal axons were also labeled with an antibody to the phosphorylated 200-kDa subunit of the neurofilament protein in both normal and diabetic rats. Double labeling to confirm that histamine and neurofilaments were colocalized in centrifugal axons was not feasible, because histamine immunolabeling requires carbodiimide fixation16 and neurofilament immunolabeling requires paraformaldehyde fixation. In the mouse retina, two types of centrifugal axons have been labeled with an antibody to the 200-kDa neurofilament protein.24 One type resembled the histamine-IR centrifugal axons in the rat retina.
In streptozotocin-diabetic rat retinas, centrifugal axons developed large swellings filled with phosphorylated 200-kDa neurofilament IR. This suggests that these swellings contain a large number of neurofilaments, such as the dystrophic axons described in human diabetic neuropathy.25-27 Dystrophic axon swellings are also observed in central nervous system disorders that secondarily affect axon integrity, such as diffuse axonal injury28,29 and Parkinson disease.30 Long axonal projections in aged brains are susceptible to axonal disease,27 and this is consistent with the finding in this study of occasional centrifugal axon swellings in normal rat retinas.
There is additional evidence suggesting that centrifugal axon swellings in diabetic rat retinas were pathologic and not sites of histamine release. The centrifugal axon swellings were not filled with histamine immunoreactivity, as would be expected if they were simply large varicosities. The same is true of other types of axon swellings observed in diabetes. In the intraganglionic sympathetic axons of the dorsal root ganglia of humans with diabetes, dystrophic swellings have no clear preor postsynaptic densities and no neurotransmitter granules.27
Pathologic axonal swellings result from progressive accumulation of neurofilaments and other organelles when slow component a (SCa) of anterograde axonal transport is impaired.31 Administration of β, β′-iminodipropionitrile selectively inhibits SCa, resulting in neurofilament-filled swellings in rat sciatic motor axons.32 Impairment of the SCa was also found in experimental diabetic rat sciatic motor axons,33 and this was reversed with insulin treatment.34,35 Axons in the optic nerve showed a decrease of SCa in diabetic rats33 and rabbits36; however, these changes were probably measured from axons of the retinal ganglion cells, not centrifugal axons. No histamine-IR swellings were observed in the optic nerve in this study.
Degenerative changes also occur in other retinal neurons and glia during experimental diabetes. There is a loss of dopaminergic neurons in streptozotocin-diabetic rat retinas,37 with a concomitant loss of tyrosine hydroxylase activity.38 Reduced nicotinamide adenine dinucleotide phospate (NADPH)-diaphorase–positive amacrine cells are also lost,39 but there is a paradoxical increase in nitric oxide synthase activity.40 There is a 50% decrease in the number of neurons in the INL after 4 months of streptozotocin-diabetes in rats.41 Apoptotic cell death is increased after 3 months in streptozotocin-diabetic rats and the thickness of the INL and IPL decreases.21 The levels of the substance P and vasoactive intestinal polypeptide present in the rat retina are significantly reduced in streptozotocin-diabetic rats.42 In addition, Müller glial cells also express more glial fibrillary acidic protein.18-20
In this study of streptozotocin-diabetic rats, histamine-IR centrifugal axons did not contact blood vessels more frequently, and the large swellings did not occur along retinal blood vessels. However, in humans with advanced diabetic retinopathy, silver-stained centrifugal axons proliferate around blood vessels and microaneurysms.43 This discrepancy was not surprising, because streptozotocin-induced diabetes reproduces only the early changes in diabetic retinas, before the development of gross vascular lesions.44 The proliferation of histamine-IR centrifugal axons may be detectable only in later stages of diabetic retinopathy. This hypothesis should be tested in future experiments in retinas from diabetic human donors with advanced retinopathy.
In summary, histidine decarboxylase mRNA expression and histamine immunoreactivity were examined in the retinas of streptozotocin-diabetic and control rats. Histidine decarboxylase mRNA was expressed in several types of cells in diabetic retinas but not in control eyes. These cells were not detected by histamine immunolabeling, however. Instead, histamine was localized to only one or a small number of centrifugal axons in both normal and diabetic rat retinas. In diabetes, these centrifugal axons show development of pathologic axon swellings in the IPL. Their terminal branches did not proliferate in the inner retina, nor were there any additional contacts with retinal blood vessels.
Acknowledgments
Supported by Grants EY12610 and EY12021 from the National Eye Institute, the Robert J. Kleberg, Jr and Helen C. Kleberg Foundation, the American Diabetes Association, the Juvenile Diabetes Research Foundation, Research to Prevent Blindness, and the Pennsylvania Lions Sight Conservation and Eye Research Foundation.
Footnotes
Commercial relationships policy: N.
References
- 1.Airaksinen MS, Panula P. The histaminergic system in the guinea pig central nervous system: an immunocytochemical mapping study using an antiserum against histamine. J Comp Neurol. 1988;273:163–186. doi: 10.1002/cne.902730204. [DOI] [PubMed] [Google Scholar]
- 2.Labandeira-Garcia JL, Guerra-Seijas MJ, Gonzalez F, Perez R, Acuna C. Location of neurons projecting to the retina in mammals. Neurosci Res. 1990;8:291–302. doi: 10.1016/0168-0102(90)90035-d. [DOI] [PubMed] [Google Scholar]
- 3.Manning KA, Wilson JR, Uhlrich DJ. Histamine-immunoreactive neurons and their innervation of visual regions in the cortex, tectum, and thalamus in the primate Macaca mulatta. J Comp Neurol. 1996;373:271–282. doi: 10.1002/(SICI)1096-9861(19960916)373:2<271::AID-CNE9>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 4.Gastinger MJ, O’Brien JJ, Larsen NB, Marshak DW. Histamine immunoreactive axons in the macaque retina. Invest Ophthalmol Vis Sci. 1999;40:487–495. [PMC free article] [PubMed] [Google Scholar]
- 5.Dowler JG, Hamilton P. Clinical features of diabetic eye disease. In: Pickup J, Williams G, editors. Textbook of Diabetes. 2. Oxford, UK: Blackwell Science, Ltd; 1997. pp. 46.1–46.15. [Google Scholar]
- 6.Bamforth SD, Lightman SL, Greenwood J. Interleukin-1 beta-induced disruption of the retinal vascular barrier of the central nervous system is mediated through leukocyte recruitment and histamine. Am J Pathol. 1997;150:329–340. [PMC free article] [PubMed] [Google Scholar]
- 7.Gardner TW, Lesher T, Khin S, et al. Histamine reduces ZO-1 tight-junction protein expression in cultured retinal microvascular endothelial cells. Biochem J. 1996;320:717–721. doi: 10.1042/bj3200717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gardner TW. Histamine, ZO-1 and increased blood-retinal barrier permeability in diabetic retinopathy. Trans Am Ophthalmol Soc. 1995;93:583–621. [PMC free article] [PubMed] [Google Scholar]
- 9.Hollis TM, Gardner TW, Vergis GJ, et al. Antihistamines reverse blood-ocular barrier breakdown in experimental diabetes. J Diabetes Complications. 1988;2:47–49. doi: 10.1016/0891-6632(88)90029-3. [DOI] [PubMed] [Google Scholar]
- 10.Gardner TW, Eller AW, Friberg TR, D’Antonio JA, Hollis TM. Antihistamines reduce blood-retinal barrier permeability in type I (insulin-dependent) diabetic patients with nonproliferative retinopathy: a pilot study. Retina. 1995;15:134–140. [PubMed] [Google Scholar]
- 11.Sander B, Larsen M, Johansen S, et al. The astemizole retinopathy trial: effect of astemizol on diabetic macular edema. Invest Ophthalmol Vis Sci. 2000;41(4):S114. ARVO Abstract, Abstract nr 587. [Google Scholar]
- 12.Carroll WJ, Hollis TM, Gardner TW. Retinal histamine synthesis is increased in experimental diabetes. Invest Ophthalmol Vis Sci. 1988;29:1201–1204. [PubMed] [Google Scholar]
- 13.Castren E, Panula P. The distribution of histidine decarboxylase mRNA in the rat brain: an in situ hybridization study using synthetic oligonucleotide probes. Neurosci Lett. 1990;120:113–116. doi: 10.1016/0304-3940(90)90181-8. [DOI] [PubMed] [Google Scholar]
- 14.Nissinen MJ, Karlstedt K, Castren E, Panula P. Expression of histidine decarboxylase and cellular histamine-like immunoreactivity in rat embryogenesis. J Histochem Cytochem. 1995;43:1241–1252. doi: 10.1177/43.12.8537641. [DOI] [PubMed] [Google Scholar]
- 15.Enerback L, Miller HRP, Mayrhofer G. Methods for the identification and characterization of mast cells by light microscopy. In: Befus AD, Bienenstock J, Denburg JA, editors. Mast Cell Differentiation and Heterogeneity. New York: Raven Press; 1986. pp. 405–417. [Google Scholar]
- 16.Panula P, Happola O, Airaksinen MS, Auvinen S, Virkamaki A. Carbodiimide as a tissue fixative in histamine immunohistochemistry and its application in developmental neurobiology. J Histochem Cytochem. 1988;36:259–269. doi: 10.1177/36.3.3343510. [DOI] [PubMed] [Google Scholar]
- 17.Auvinen S, Panula P. Development of histamine-immunoreactive neurons in the rat brain. J Comp Neurol. 1988;276:289–303. doi: 10.1002/cne.902760211. [DOI] [PubMed] [Google Scholar]
- 18.Lieth E, Barber AJ, Xu B, et al. Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Diabetes. 1998;47:815–820. doi: 10.2337/diabetes.47.5.815. [DOI] [PubMed] [Google Scholar]
- 19.Barber AJ, Antonetti DA, Gardner TW. Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes. Invest Ophthalmol Vis Sci. 2000;41:3561–3568. [PubMed] [Google Scholar]
- 20.Rungger-Brandle E, Dosso AA, Leuenberger PM. Glial reactivity, an early feature of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2000;41:1971–1980. [PubMed] [Google Scholar]
- 21.Barber AJ, Lieth E, Khin SA, et al. Neural apoptosis in the retina during experimental and human diabetes: early onset and effect of insulin. J Clin Invest. 1998;102:783–791. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sawai S, Fukui H, Imamura I, et al. Histamine and its synthesis in mammalian retinas. J Ocul Pharmacol. 1991;7:213–219. [PubMed] [Google Scholar]
- 23.Ando-Yamamoto M, Kiyama H, Hayashi H, et al. Demonstration of histaminergic neurons in horizontal cells of guinea pig retina. Brain Res. 1987;410:269–274. doi: 10.1016/0006-8993(87)90324-6. [DOI] [PubMed] [Google Scholar]
- 24.Drager UC, Edwards DL, Barnstable CJ. Antibodies against filamentous components in discrete cell types of the mouse retina. J Neurosci. 1984;4:2025–2042. doi: 10.1523/JNEUROSCI.04-08-02025.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Byers PD, Page KM. Axonic swellings demonstrated by a supravital methylene blue technique. Acta Neurol Scand. 1969;45:303–308. doi: 10.1111/j.1600-0404.1969.tb01242.x. [DOI] [PubMed] [Google Scholar]
- 26.Schroer JA, Plurad SB, Schmidt RE. Fine structure of presynaptic axonal terminals in sympathetic autonomic ganglia of aging and diabetic human subjects. Synapse. 1992;12:1–13. doi: 10.1002/syn.890120102. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt RE, Dorsey D, Parvin CA, et al. Dystrophic axonal swellings develop as a function of age and diabetes in human dorsal root ganglia. J Neuropathol Exp Neurol. 1997;56:1028–1043. doi: 10.1097/00005072-199709000-00008. [DOI] [PubMed] [Google Scholar]
- 28.Cervos-Navarro J, Lafuente JV. Traumatic brain injuries: structural changes. J Neurol Sci. 1991;103(Suppl):S3–S14. doi: 10.1016/0022-510x(91)90002-o. [DOI] [PubMed] [Google Scholar]
- 29.Grady MS, McLaughlin MR, Christman CW, et al. The use of antibodies targeted against the neurofilament subunits for the detection of diffuse axonal injury in humans. J Neuropathol Exp Neurol. 1993;52:143–152. doi: 10.1097/00005072-199303000-00007. [DOI] [PubMed] [Google Scholar]
- 30.Graham DL, Lantos PL. Greenfields Neuropathology. 6. Vol. 1. New York: Arnold; 1997. p. 1231. [Google Scholar]
- 31.Jakobsen J, Sidenius P, Braendgaard H. A proposal for a classification of neuropathies according to their axonal transport abnormalities. J Neurol Neurosurg Psychol. 1986;49:986–990. doi: 10.1136/jnnp.49.9.986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griffin JW, Hoffman PN, Clark AW, Carroll PT, Price DL. Slow axonal transport of neurofilament proteins: impairment of beta,beta’-iminodipropionitrile administration. Science. 1978;202:633–635. doi: 10.1126/science.81524. [DOI] [PubMed] [Google Scholar]
- 33.Medori R, Autilio-Gambetti L, Monaco S, Gambetti P. Experimental diabetic neuropathy: impairment of slow transport with changes in axon cross-sectional area. Proc Natl Acad Sci USA. 1985;82:7716–7720. doi: 10.1073/pnas.82.22.7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sidenius P, Jakobsen J. Reversibility and preventability of the decrease in slow axonal transport velocity in experimental diabetes. Diabetes. 1982;31:689–693. doi: 10.2337/diab.31.8.689. [DOI] [PubMed] [Google Scholar]
- 35.Larsen JR, Sidenius P. Slow axonal transport of structural polypeptides in rat, early changes in streptozocin diabetes, and effect of insulin treatment. J Neurochem. 1989;52:390–401. doi: 10.1111/j.1471-4159.1989.tb09134.x. [DOI] [PubMed] [Google Scholar]
- 36.Chihara E, Sakugawa M, Entani S. Reduced protein synthesis in diabetic retina and secondary reduction of slow axonal transport. Brain Res. 1982;250:363–366. doi: 10.1016/0006-8993(82)90432-2. [DOI] [PubMed] [Google Scholar]
- 37.Larabi Y, Dahmani Y, Gernigon T, Nguyen-Legros J. Tyrosine hydroxylase immunoreactivity in the retina of the diabetic sand rat Psammomys obesus. J Hirnforsch. 1991;32:525–531. [PubMed] [Google Scholar]
- 38.Gibson CJ. Diurnal alterations in retinal tyrosine level and dopamine turnover in diabetic rats. Brain Res. 1988;454:60–66. doi: 10.1016/0006-8993(88)90803-7. [DOI] [PubMed] [Google Scholar]
- 39.Roufail E, Soulis T, Boel E, Cooper ME, Rees S. Depletion of nitric oxide synthase-containing neurons in the diabetic retina: reversal by aminoguanidine. Diabetologia. 1998;41:1419–1425. doi: 10.1007/s001250051087. [DOI] [PubMed] [Google Scholar]
- 40.do Carmo A, Lopes C, Santos M, et al. Nitric oxide synthase activity and L-arginine metabolism in the retinas from streptozotocin-induced diabetic rats. Gen Pharmacol. 1998;30:319–324. doi: 10.1016/s0306-3623(97)00363-7. [DOI] [PubMed] [Google Scholar]
- 41.Zeng XX, Ng YK, Ling EA. Neuronal and microglial response in the retina of streptozotocin-induced diabetic rats. Vis Neurosci. 2000;17:463–471. doi: 10.1017/s0952523800173122. [DOI] [PubMed] [Google Scholar]
- 42.Troger J, Neyer S, Heufler C, et al. Substance p and vasoactive intestinal polypeptide in the streptozotocin-induced diabetic rat retina. Invest Ophthalmol Vis Sci. 2001;42:1045–1050. [PubMed] [Google Scholar]
- 43.Wolter JR. Diabetic retinopathy. Am J Ophthalmol. 1961;51:1124–1141. doi: 10.1016/0002-9394(61)91802-5. [DOI] [PubMed] [Google Scholar]
- 44.Engerman RL, Kern TS. Retinopathy in animal models of diabetes. Diabetes Metab Rev. 1995;11:109–120. doi: 10.1002/dmr.5610110203. [DOI] [PubMed] [Google Scholar]