

Abstract
The combined structural and biochemical studies on Lac repressor bound to operator DNA have demonstrated the central role of the hinge helices in operator bending and the induction mechanism. We have constructed a covalently linked dimeric Lac-headpiece that binds DNA with four orders of magnitude higher affinity as compared with the monomeric form. This enabled a detailed biochemical and structural study of Lac binding to its cognate wild-type and selected DNA operators. The results indicate a profound contribution of hinge helices to the stability of the protein–DNA complex and highlight their central role in operator recognition. Furthermore, protein–DNA interactions in the minor groove appear to modulate hinge helix stability, thus accounting for affinity differences and protein-induced DNA bending among the various operator sites. Interestingly, the in vitro DNA-binding affinity of the reported dimeric Lac construct can de readily modulated by simple adjustment of redox conditions, thus rendering it a potential artificial gene regulator.
Central to the biological function of the Lac repressor is a polypeptide linker that connects the DNA-binding domain (residues 1–49) to the core domain (residues 62–357) and is referred to as the hinge region. NMR and x-ray studies of Lac and its related Pur repressor have demonstrated that the hinge region is disordered in free protein and forms an α-helix only when bound to DNA (1–4). Once formed, the hinge helices penetrate into the minor groove of the DNA and force the operator to bend toward the major groove and away from the protein. Additionally, the hinge region holds a central role in the induction mechanism of the Lac repressor. Inducer binding to the core domain is believed to alter the network of interactions between the core and the DNA-binding domain, thereby destabilizing the binding of the hinge helices to the minor groove of the operator (5). So far, however, the formation of hinge helices has been unambiguously demonstrated only in the case of an artificial symmetric operator (Fig. 1), which lacks the central base pair and is a palindrome of the left-operator site (1, 3–5). Therefore, the question whether the hinge helices are present and their relative stability in the complex with its cognate wild-type operator has not yet been addressed in detail (6). Moreover, a complete assessment of the role of hinge helices to operator recognition and its contribution to the overall binding is still missing.
Figure 1.

The Lac operator sequences used for the present studies. For the wild-type operator the asymmetric regions between the two sites relative to the central base pair are indicated in bold. For the two symmetric operators the spacing of the two half-sites relative to wild-type operator is indicated in parentheses.
One of the main obstacles in studying Lac binding to variant operators has been the low affinity of the isolated headpiece (HP) for DNA, which is due to the absence of dimerization interface. In fact, the ability to form dimeric or higher-order aggregates is of crucial importance for DNA-binding proteins because it dramatically increases their binding affinity. Extensive genetic studies on LacI have also emphasized this point. Mutations at positions that disturb the dimerization interface result in monomeric protein that completely abolishes the ability to repress (7, 8). Therefore, the design and production of engineered DNA-binding domains that could restore the binding affinity of the intact repressor is of great importance. In the present work we demonstrate that a designed V52C mutant with a Cys52–Cys52′ disulfide bond in the hinge region of Lac-HP62, which comprises the first 62 residues of the Lac headpiece, provides a large increase in the stability of Lac-HP62–DNA complex. Remarkably, the new covalently linked Lac-HP62–V52C construct binds DNA as tightly as the intact Lac repressor. The high affinity and the relatively small size of this dimeric Lac headpiece render it an ideal system to investigate in detail the binding of repressor and hinge helix formation in various operator sequences. Interestingly, the in vitro DNA-binding affinity of the reported system can be readily regulated through redox adjustment, thus rendering it a potential artificial gene regulator.
Materials and Methods
Protein Engineering.
The HP62-V52C mutant was amplified from the corresponding Lac I genes by the PCR and expressed in Escherichia coli by using a T7 polymerase-based system. The protein was purified analogously to wild-type HP62 (6) in the presence of 10 mM of DTT. To produce the oxidized (dimeric) form of HP62-V52C, the protein sample was dialyzed at room temperature against buffer [60 mM KPi (pH 7.5), 400 mM KCl, 1 mM EDTA, 1 g/l NaN3] in the absence of DTT for 48 h to allow air oxidation and formation of the disulfide bond. Oxidizing or reducing conditions could also be produced by adding 10 mM DTT (or glutathione) in either the oxidized or reduced form. Nonreducing SDS/PAGE was used to follow the oxidation/reduction procedure. The dimeric construct was obtained in pure form after separating it form the monomer by gel filtration using a Superdex 75 (Amersham Pharmacia) column. All samples were concentrated by using Cenricon concentrators (Amicon).
DNA Bending and Binding Affinities.
All biochemical assays including DNA bending and electrophoretic mobility shift assays were performed essentially as described (6). All experiments were performed at 4°C in 20 μl of reaction buffer containing 10 mM Tris (pH 8.1), 50 mM KCl, 1 mM EDTA, 5% (vol/vol) glycerol, and 0.1 mg/ml BSA.
NMR Sample Preparation.
All lac operator DNA fragments (Fig. 1) were purchased at Carl Roth GmbH (Karlsruhe, Germany) and further purified on a Q-Sepharose (Amersham Pharmacia) column. The following samples were used for NMR measurements: (i) 2 mM 13C- and 15N-labeled HP62-V52C; (ii) 2 mM 13C- and 15N-labeled HP62-V52C, 2 mM 22 bp SymL(−1) operator DNA; (iii) 2 mM 13C- and 15N-labeled HP62-V52C, 2 mM 23 bp wild-type operator DNA; (iv) 2 mM 13C- and 15N-labeled HP62-V52C, 2 mM 24 bp SymR(+1) operator DNA. The free protein sample contained 0.4 M KCl in 0.06 M KPi buffer (pH 5.5). The complexes of HP62-V52C with SymL(−1), SymR(+1), and wild-type operators were dissolved in 0.01 M KPi buffer (pH 6.0) containing 0.02 M KCl. All samples were dissolved in 95% H2O/5% D2O. Trace amounts of NaN3 were added as a preservative.
NMR Spectroscopy and Resonance Assignment.
NMR spectra were recorded on Varian Inova 750 MHz and Bruker DRX-600 spectrometers. Spectra of the free protein were recorded at 300 K, whereas those of HP62-V52C–DNA complexes were recorded at 312 K. Backbone resonance assignments were obtained by a series of 13C- and 15N-edited double and triple resonance experiments (9). The heteronuclear nuclear Overhauser enhancement (NOE) experiments were recorded essentially as described by Dayie and Wagner (10). Amide proton exchange rates were determined at 25°C, pD 6.0, from the time course of the peak intensities in a series of 1H–15N transverse relaxation-optimized spectroscopy (TROSY) spectra (11) after dissolving lyophilized samples in D2O. All spectra were processed by using the nmrpipe software package (12) and analyzed with nmrview (13).
Results and Discussion
Protein Engineering, DNA Affinities, and Bending.
Based on the structure of intact Lac–SymL(−1) DNA complex (3), Falcon et al. (14) designed a mutation at Val52 within the hinge region to generate a protein in which a disulfide bond could be formed between the N-terminal domains. Cross-linking the two hinge helices is expected to strengthen protein–protein interactions that contribute favorably to the stability of the dimer interface as the positive contribution of disulfides to protein stability has been well established (15–17). This had been shown before for lambda repressor by Sauer et al. (18) by engineering a disulfide bond at the dimer interface of the N-terminal domain, which resulted in increase in both protein stability and DNA binding activity. The oxidized LacI-V52C mutant repressor was shown to bind DNA with an approximately 6-fold higher affinity than the wild-type repressor (14). Therefore, we expected that introduction of the same mutation in the Lac headpiece would also result in higher DNA-binding affinity. Further detailed modeling in our lab, based on the high-resolution structure of Lac-HP62–SymL(−1) complex (4), indicated that a disulfide bond of optimal geometry could be accommodated only at the Val-52 position (Fig. 2). In all other positions crosslinking is expected to introduce unfavorable strain in the system.
Figure 2.
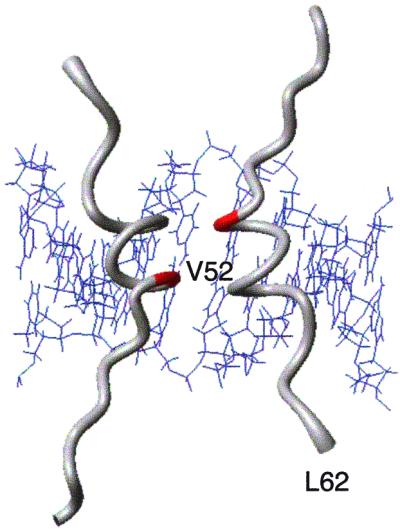
Hinge helices of two units of Lac-HP62 bound to the minor groove of SymL(−1) operator (4). Val-52 (red) was replaced by a cysteine residue.
Table 1 summarizes the affinity of the protein constructs for various operators of DNA. In the reduced form, HP62-V52C protein binds DNA with a 5-fold higher affinity than does wild-type HP62. Remarkably, the disulfide crosslinked HP62-V52C mutant exhibits a 3 × 104-fold higher DNA-binding affinity as compared with the reduced form, reaching an apparent Kd value of 30 pM (Table 1 and Fig. 3 a and b). Under the same conditions, the intact Lac repressor binds DNA with similar affinity (Table 1 and Fig. 3b). The dramatic enhancement of binding strength, which is one of the highest ever reported to arise from simple dimerization, demonstrates that DNA-binding domains can be rationally designed to restore the affinity of the natural repressor. The highest affinity of dimer HP62-V52C is measured for SymL(−1) operator, followed by wild-type (≈2-fold decrease) and SymR(+1) (≈10-fold decrease) operators (Table 1). Moreover, the degree to which DNA bends upon repressor binding appears to follow the same trend [33° in SymL(−1), 24° in wild-type, and 17° in SymR(+1) operator; Table 1 and Fig. 3d]. These biochemical results are very similar to those reported for the intact Lac repressor (6), suggesting that the dimeric HP62-V52C construct behaves in the same way as the intact repressor. The stronger DNA bending determined for intact Lac repressor (Table 1), compared with Lac headpiece, is due to wrapping of the intact repressor around the flanking nonoperator DNA part of the long fragment that was used for the DNA bending assays (19). The structures of intact repressor (3) and Lac headpiece (4) bound to the SymL(−1) operator showed very similar DNA bend angles in the operator site.
Table 1.
Summary of the electrophoretic mobility shift assays with various Lac constructs
| Protein | Operator binding affinity,*
M
|
Bend angle,† °
|
||||
|---|---|---|---|---|---|---|
| Wild type | SymL(−1) | SymR(+1) | Wild type | SymL(−1) | SymR(+1) | |
| LacI‡ | 3 × 10−11 | 2.4 × 10−11 | 9 × 10−11 | 68 | 72 | 56 |
| Wild-type HP62‡ | 1 × 10−6 | 0.8 × 10−6 | n.d.b.‖ | 28 | 35 | n.d.b. |
| HP62-V52C (red)§ | 2 × 10−7 | 23 | 33 | 17 | ||
| HP62-V52C (ox)¶ | 3 × 10−11 | 1.8 × 10−11 | 15 × 10−11 | 25 | 36 | 17 |
The binding affinities for the SymL(−1), wild-type, and SymR(+1) operators were determined at 4°C from four independent experiments and scaled to the value found for the wild-type probe. Values represent apparent Kds and refer to the protein concentration where 50% of the DNA operator is bound to the protein.
Bend angle was determined by circular permutation method and is the average of three independent experiments. The standard deviation in the bending angles determined in repeated experiments is 10%.
Relative affinities and bend angles for LacI and Wt-HP62 were taken from Spronk et al. (6).
Reduced state.
Oxidized state.
No detectable binding.
Figure 3.

(a) Representative binding experiment with a serial dilution of both the reduced and oxidized form of HP62-V52C mutant to the wild-type operator; highest concentration in each experiment is 100 ng/μl. (b) Quantification of a representative binding experiment. (c) Representative experiment of the binding of dimer HP62-V52C to SymR(+1), wt, and SymL(−1) operator sequences. (d) A representative bending experiment using circularly permuted SymL(−1), wt, and SymR(+1) operator sites. (e) Binding of HP62-V52C to the wild-type operator in the presence or absence of 10 mM DTT either reduced or oxidized, or a mixture of 10 mM oxidized and reduced glutathione in the given molar ratio (ox/red).
In Vitro Redox Regulation of DNA Binding.
The binding properties of the HP62-V52C mutant can be exploited in various ways. This protein can be interconverted between two forms, reduced and oxidized, with each of them exhibiting very different affinity for DNA. Our experimental results show that the formation of the disulfide bond is a reversible procedure. Therefore, this construct can be used to promote or suppress protein–DNA complex formation (20) because its DNA-binding affinity can be readily modulated. Simple adjustment of the redox conditions induces a switch between the dimeric (tightly DNA-bound) and monomeric (weakly DNA-bound) form (Fig. 3e). The dimer form binds DNA with pM affinity (similar to intact Lac repressor), whereas the addition of reducing agent decreases the binding by 4 orders of magnitude. Furthermore, different ratios of reducing vs. oxidizing reagents can be used to control the extent of DNA binding (Fig. 3e). An interesting development would be to use the HP62-V52C construct as an artificial control of gene expression or other biological processes (21).
Lac Binding to Wild-Type and SymR(+1) Operators.
Previously it had been shown that the affinity of Lac repressor for left-half and right-site symmetrized operators is highest for SymL(−1) and SymR(+1) operators (6, 22, 23). Symmetric operators with other deletions or insertions bind with very low or negligible affinity. Fig. 4 a and b show the 1H–15N heteronuclear single quantum correlation (HSQC) spectra of free dimer HP62-V52C and its complex with the wild-type operator. Similar to native Lac-HP62 (1), NOE and chemical shift data show that the C-terminal region of dimer HP62-V52C is unstructured when it is free in solution. Upon binding to the wild-type operator, however, characteristic NOE's (Fig. 4 d and e) and chemical shifts of Hα, Cα, and CO resonances unambiguously indicate that the hinge region of both the left and the right half sites of the dimeric HP62-V52C protein form an α-helix. Further evidence for hinge helix formation in both sites was provided by studies of the dynamics of the backbone NH groups (data not shown).
Figure 4.

1H-15N heteronuclear single quantum correlation (HSQC) spectra of dimer HP62-V52C (a) in the free form, (b) in the complex with wild-type DNA operator, and (c) in the complex with SymR(+1) operator. Peaks corresponding to the hinge region residues are labeled. (b) Normal numbers indicate residues of the left site, whereas primed numbers indicate residues of the right site. (c) Peaks corresponding to the different folding states of hinge region residues are indicated in circles. Strip plots of residues 50–58 from (1H-15N) NOESY-HSQC spectra of HP62-V52C–wild-type DNA complex: (d) left site, (e) right site. Hα–HN and HN–HN sequential contacts are indicated with red lines; Hα–HN(i, i + 3) and Hα–HN(i, i + 4) contacts are indicated with blue lines.
From chemical shift and NOE analysis we can conclude that the Lac repressor recognizes and binds the left site of the wild-type operator identically to the SymL(−1) operator. Chemical shift mapping of the dimeric HP62-V52C bound to wild-type operator reveals significant differences between the individual subunits that contact the left and the right site of the operator (Fig. 5). Especially for the recognition helix residues (Y17-N25), which are responsible for sequence-specific contacts, profound chemical shift differences up to 5.3 ppm and 2.5 ppm in the 15N and 1H dimensions, respectively, were measured. These results clearly demonstrate that the inherent differences in the sequence of the two sites influence the mode by which contact is made with the identical N-terminal DNA binding domains (24, 25). Furthermore, a remarkable difference in the backbone 1H and 15N chemical shift of residue Asn-50 between the left and right site was identified. Asn-50 is a highly conserved residue and plays an important role in stabilizing the C-terminal region by making a direct hydrogen bond to DNA backbone (O-1 phosphorus atom of Cyt-9; ref. 4). This contact anchors and orients the hinge helix within the minor groove, and also acts as a “clamp” to stabilize the kinked DNA conformation in the complex. Upon binding to the operator, the HN amide of Asn-50 in the left site of the DNA-complex shifts downfield by 1.1 and 5.4 ppm in the 1H and 15N dimensions, respectively. In contrast, the 1H chemical shift of the backbone amide of Asn-50 that contacts the right site moves 0.3 ppm up-field. Because the 1H chemical shift is very sensitive to, and thus indicative of, the presence of hydrogen bonding (26), it is suggested that this particular H-bond is missing in the right site.
Figure 5.

Chemical shift variations of dimer HP62-V52C induced upon binding to the wild-type DNA operator. The chemical shift mapping is projected on the three-dimensional structure of the HP62-SymL(−1) complex (4). The two sites are depicted independently and have been rotated for easier comparison: (a) left site, (b) right site. (c) The chemical shift difference between bound left and right site is summarized. The color code used is as follows: |Δδav| < 0.3 ppm, gray; 0.3 ppm < |Δδav| < 0.6 ppm, yellow; |Δδav| > 0.6 ppm, red. Δδav is a weighted average of the 15N and 1HN chemical shifts (31): Δδav = 0.5[Δδ(1HN)2 + (0.2 Δδ(15N))2]1/2. Backbone amides that are involved in protein–DNA hydrogen bonding and were shown to be protected in hydrogen exchange experiments are indicated with spheres. The molecular models were drawn with MOLMOL (32).
To obtain additional and independent experimental evidence of differential binding to the left and right site of the wild-type operator, amide proton exchange experiments were performed with the dimeric HP62–V52C complex. Interestingly, unusually high protection factors were obtained for residues participating in intermolecular hydrogen bonding, most likely reflecting the very low dissociation constant of the dimer HP62-V52C–wild-type operator complex. The backbone amide protons of Leu-6 and Ser-31 have been shown to form strong H-bonds to DNA phosphate in the SymL(−1) complex (4). Our data indicate high protection factors for these residues (the NH signal of Leu-6 showed up in the spectrum for ≈2 days and that of Ser-31 for ≈12 h), suggesting that these protein–DNA contacts are present in both sites of the wild-type complex (Fig. 5 a and b). However, in the case of Asn-50, exchange rates are quite different and indicate that its backbone amide is protected only in the left site. Moreover, quantification of signals of hinge region residues demonstrated that the backbone amides of the right hinge helix exchanges with a ≈2-fold higher rate compared with those of the left site. Therefore, the absence of this critical hydrogen bonding is further corroborated by deuterium exchange experiment and may account for the lower stability of the right hinge helix as compared with the left one.
To shed more light on the structural features of the right-site complex, we studied the dimer HP62-V52C complexed with the SymR(+1) operator (Fig. 1). This fragment consists of two symmetric sites, each of them comprising the central base pair and the right site and has been shown to bind DNA relatively tightly. Upon binding to the operator, the hinge region assumes an α-helical structure as judged by chemical shift and NOE analysis (Fig. 4c). Similar to the right site of the wild-type operator, and in contrast to the left site, the backbone of Asn-50 does not hydrogen bond to DNA. Interestingly, NMR analysis indicated slow exchange (of the order of 10 s−1) of hinge region residues between α-helix and random coil form (Fig. 4c). Apparently, the hinge helices are not stable in the complex of Lac headpiece with SymR(+1) operator despite the fact that the rest of the protein appears to be tightly bound to DNA (only one peak was identified for each residue corresponding to the bound state). Therefore, the instability of the hinge helices in the right half-site originates only in the local contact of the protein with the minor groove and is most probably related to the absence of hydrogen bonding between the backbone of Asn-50 and DNA. Our results confirm previous biochemical data indicating that the central base pairs may modulate the binding of the repressor–operator complex by altering the structure and flexibility of DNA (27). Therefore, the DNA phosphate conformation in the minor groove appears to be optimal for hydrogen bonding to Asn-50 only in the left site region. In this context, it is quite interesting that any base pair substitution in the central part of the left site results in significant losses in binding affinity (28). Furthermore, recently reported biochemical data demonstrated that subtle alterations in the central part of DNA sequence have a profound effect on binding affinity and allosteric response for Lac repressor (23).
Formation of Hinge Helices Is an Important Component of the Mechanism of Specific Site Recognition by Lac.
In contrast to its highly homologous Pur repressor (2), the hinge helices of Lac repressor do not make specific contacts to DNA base pairs. However, many biochemical studies have demonstrated that a specific DNA sequence in the central part of the operator is required for formation of hinge helices, which are needed for both increasing binding affinity and DNA bending (23, 28). It becomes now apparent that both protein–protein and protein–DNA interactions are necessary for the stabilization of the dimer interface. This means that optimal spacing of the headpieces, which is needed to provide favorable protein–protein interactions (6), is not sufficient for hinge helix stabilization. As the present results emphasize, “nonspecific” protein–DNA contacts are capable of modulating the stability of the hinge helices and may provide a mechanism for Lac discrimination of different operator binding sites. Only these sequences with both appropriate headpiece spacing and phosphate geometry in the minor groove will give rise to hinge helix formation and ultimately to a stable repressor–operator complex. Although the Lac repressor makes specific contacts in both sites of its cognate wild-type operator, binding to the right site is unable to impose the optimal DNA conformation that is required for formation of the hydrogen bond between Asn-50 and DNA backbone. It thus appears that the weaker binding of repressor to the right site is a consequence of the resulting less perfect fit between protein and DNA. Furthermore, this crucial contact may modulate DNA bending in a similar way as described before for EcoRI endonuclease (29). In the complex with SymL(−1) operator, where this intermolecular hydrogen bond is present in both sites, the protein–DNA interactions in the minor groove are optimal resulting in the stabilization of a severely bent DNA conformation (Table 1 and Fig. 3d). Loss of this contact increases the energetic cost of attaining a highly kinked DNA, thus resulting in lower degree of bending in wild-type and SymR(+1) operators (Table 1 and Fig. 3d).
In conclusion, protein–DNA interactions in the minor groove are capable of modulating hinge helix stability and may provide a structural basis for explaining affinity variations among variant operator sites. Furthermore, they enable the Lac repressor to position hinge helices for indirect readout of DNA sequence resulting in its discrimination. Therefore, the well known notion that sequence-dependent conformational properties of DNA play an integral role in specificity for many sequence-specific proteins (30) applies also to the Lac repressor.
Abbreviations
- HP
headpiece
- NOE(SY)
nuclear Overhauser enhancement (spectroscopy)
References
- 1.Spronk C A E M, Slijper M, van Boom J H, Kaptein R, Boelens R. Nat Struct Biol. 1996;3:916–919. doi: 10.1038/nsb1196-916. [DOI] [PubMed] [Google Scholar]
- 2.Schumacher M A, Choi K Y, Zalkin H, Brennan R G. Science. 1994;266:763–770. doi: 10.1126/science.7973627. [DOI] [PubMed] [Google Scholar]
- 3.Lewis M, Chang G, Horton N C, Kercher M A, Pace H C, Schumacher M A, Brennan R G, Lu P. Science. 1996;271:1247–1254. doi: 10.1126/science.271.5253.1247. [DOI] [PubMed] [Google Scholar]
- 4.Spronk C A E M, Bonvin A, Radha P K, Melacini G, Boelens R, Kaptein R. Structure. 1999;7:1483–1492. doi: 10.1016/s0969-2126(00)88339-2. [DOI] [PubMed] [Google Scholar]
- 5.Bell C E, Lewis M. Nat Struct Biol. 2000;7:209–214. doi: 10.1038/73317. [DOI] [PubMed] [Google Scholar]
- 6.Spronk C A E M, Folkers G E, Noordman A M G W, Wechselberger R, van der Brink N, Boelens R, Kaptein R. EMBO J. 1999;18:6472–6480. doi: 10.1093/emboj/18.22.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suckow J, Markiewicz P, Kleina L G, Miller J H, Kisters-Woike B, Müller-Hill B. J Mol Biol. 1996;261:509–523. doi: 10.1006/jmbi.1996.0479. [DOI] [PubMed] [Google Scholar]
- 8.Chakerian A E, Matthews K S. J Biol Chem. 1991;266:22206–22214. [PubMed] [Google Scholar]
- 9.Cavanagh J, Fairbrother W J, Palmer A G, III, Skelton N J. Protein NMR Spectroscopy: Principles and Practice. London: Academic; 1996. [Google Scholar]
- 10.Dayie K T, Wagner G J. J Magn Reson. 1994;111:121–126. [Google Scholar]
- 11.Pervushin K, Riek R, Wider G, Wüthrich K. Proc Natl Acad Sci USA. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delaglio F, Grzesiek S, Vuister G, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 13.Johnson B A, Blevins R A. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 14.Falcon C M, Swint-Kruse L, Matthews K S. J Biol Chem. 1997;272:26818–26821. doi: 10.1074/jbc.272.43.26818. [DOI] [PubMed] [Google Scholar]
- 15.Robinson C R, Sauer R T. Biochemistry. 2000;39:12494–12502. doi: 10.1021/bi001484e. [DOI] [PubMed] [Google Scholar]
- 16.Matthews B W, Nicholson H, Becktel W J. Proc Natl Acad Sci USA. 1987;84:6663–6667. doi: 10.1073/pnas.84.19.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsumura M, Becktel W J, Levitt M, Matthews B W. Proc Natl Acad Sci USA. 1989;86:6562–6566. doi: 10.1073/pnas.86.17.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sauer R T, Hehir K, Stearman R S, Weiss M A, Jeitler-Nilsson A, Suchanek E G, Pabo C O. Biochemistry. 1986;25:5992–5998. doi: 10.1021/bi00368a024. [DOI] [PubMed] [Google Scholar]
- 19.Tsodikov O V, Saecker R M, Melcher S E, Levandoski M M, Diane E F, Capp M W, Record M T., Jr J Mol Biol. 1999;294:639–655. doi: 10.1006/jmbi.1999.3283. [DOI] [PubMed] [Google Scholar]
- 20.Moore I, Gälveiler L, Grosskopf D, Schell J, Palme K. Proc Natl Acad Sci USA. 1998;95:376–381. doi: 10.1073/pnas.95.1.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beerli R R, Dreier B, Bardas C F., III Proc Natl Acad Sci USA. 2000;97:1495–1500. doi: 10.1073/pnas.040552697. . (First Published January 31, 2000; 10.1073/pnas.040552697) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sasmor H M, Betz J L. Gene. 1990;89:1–6. doi: 10.1016/0378-1119(90)90198-z. [DOI] [PubMed] [Google Scholar]
- 23.Falcon C M, Matthews K S. Biochemistry. 2000;39:11074–11083. doi: 10.1021/bi000924z. [DOI] [PubMed] [Google Scholar]
- 24.Horton N, Lewis M, Lu P. J Mol Biol. 1997;265:1–7. doi: 10.1006/jmbi.1996.0706. [DOI] [PubMed] [Google Scholar]
- 25.Ogata R T, Gilbert W. Proc Natl Acad Sci USA. 1977;74:4973–4976. doi: 10.1073/pnas.74.11.4973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becker E D. In: Encyclopedia of Nuclear Magnetic Resonance. Grant D M, Harris R K, editors. Chichester, U.K.: Wiley; 1996. pp. 2409–2415. [Google Scholar]
- 27.Zhang X, Gottlieb P A. Nucleic Acids Res. 1995;23:1502–1511. doi: 10.1093/nar/23.9.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lehming N, Santorius J, Niemoller M, Genenger G, Wilcken-Bergmann B, Müller-Hill B. EMBO J. 1987;6:3145–3153. doi: 10.1002/j.1460-2075.1987.tb02625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lesser D R, Kurpiewski M R, Jen-Jacobson L. Science. 1990;250:776–786. doi: 10.1126/science.2237428. [DOI] [PubMed] [Google Scholar]
- 30.Jen-Jacobson L. Biopolymers. 1997;44:153–180. doi: 10.1002/(SICI)1097-0282(1997)44:2<153::AID-BIP4>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 31.Foster M P, Wuttke D S, Clemes K R, Jahnke W, Radhakrishnan I, Tennant L, Reymond M, Chung J, Wright P E. J Biomol NMR. 1998;12:51–71. doi: 10.1023/a:1008290631575. [DOI] [PubMed] [Google Scholar]
- 32.Koradi R, Billeter M, Wüthrich K. J Mol Graphics. 1996;14:52–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]