

Abstract
Aging increases mitochondrial dysfunction and susceptibility to hypoxia. Previous reports have indicated an association between post-hypoxic hyperoxidation of intra-mitochondrial enzymes and delayed neuronal injury. Therefore we investigated the relationship between NADH fluorescence and neuronal function during and after hypoxia across the lifespan. Hippocampal slices were prepared from adult (1 to >22 months) F344 rats. NADH fluorescence, extracellular voltage and tissue PO2 were recorded from the CA1 region during hypoxia (95% N2) of various lengths following onset of hypoxic spreading depression (hsd). Slices from younger rats recovered evoked neuronal responses to a greater degree and exhibited less hyperoxidation after a hypoxic episode, than slices from older rats. However, the use of Ca2+ free-media in slices from >22 month old rats improved recovery and delayed NADH hyperoxidation (2.5 min hypoxia after hsd). Post-hypoxic decrease of NADH fluorescence (hyperoxidation) was age dependent and correlated with decreased neuronal recovery. Slices exposed to repeated hypoxic episodes yielded data suggesting depletion of the NAD+ pool, which may have contributed to the deterioration of neuronal function.
Keywords: Hypoxic spreading depression, NADH, Hippocampus, Aging, Mitochondria
1. Introduction
Age-related deficiencies in mitochondrial respiratory function may contribute to cellular energy deficits, which reduce the brain's ability to adapt to metabolic stresses, such as hypoxia/ischemia, and as a result increase the brain's vulnerability to subsequent damage (Ames et al., 1995; Zarchin et al., 2002). Stroke is the third leading cause of death in Western civilizations (Bonita, 1992) and may incur a greater amount of injury in the elderly, compared to younger age groups. Brain injury that occurs following a stroke in aged individuals may be due to enhanced reactive oxygen species (ROS) damage to protein (Smith et al., 1991), lipids (Roberts et al., 1998; Yoritaka et al., 1996) and DNA (Hamilton et al., 2001; Nakae et al., 2000); alterations in intracellular calcium handling (Leslie et al., 1985; Murchison et al., 2004); and decreased metabolic activity (Bowling et al., 1993; Fattoretti et al., 1998; Ferrandiz et al., 1994; Harmon et al., 1987; Kwong and Sohal, 2000). Although age is the primary risk factor affecting stroke victims, few experimental hypoxia or stroke studies have been conducted using aged individuals.
During mitochondrial respiration, approximately 2–5% of electrons escape to react directly with oxygen, resulting in the production of ROS at complexes I and III (Boveris and Chance, 1973). Aging has been shown to increase levels of ROS in the brain (Siqueira et al., 2004) which may contribute to progressive mitochondrial damage in aged individuals (Ames et al., 1995; Harman, 2003). Impairment of mitochondrial function or ROS buffering can reduce a cell's ability to adapt to an event such as reperfusion following hypoxia/ischemia, during which considerable amounts of ROS are formed.
Events during reperfusion following a transient ischemic episode have been blamed for so-called delayed brain damage. Recordings made in intact brains as well as in brain slices following either transient ischemic or hypoxic episodes revealed a decrease of NADH fluorescence and cytochrome a,a3 reflectance significantly below control levels, which has been interpreted as hyperoxidation of intra-mitochondrial oxidative enzymes (Dora et al., 1986; Duckrow et al., 1981; Feng et al., 1998; Paschen et al., 1985; Perez-Pinzon et al., 1998a, 1997a, 1998b; Pulsinelli et al., 1982; Rosenthal et al., 1995, 1997; Siesjo, 1981; Tanaka et al., 1986; Welsh et al., 1991; Welsh et al., 1982). The alternative explanation of a depletion of the electron carrier pool has been rejected by other authors (Rosenthal et al., 1997). Some of our present observations suggest, however, that a decrease in pool size may be an additional factor besides hyperoxidation. An association between hyperoxidation and cell damage was supported in studies which have shown that free radical scavengers lessened hyperoxidation and at the same time augmented functional recovery following hypoxia (Perez-Pinzon et al., 1997), while hyperoxic gas mixtures enhanced hyperoxidation and hindered recovery (Feng et al., 1998). Another potential influencing factor in hyperoxidation and tissue recovery is calcium. It has been shown that low concentrations of extracellular calcium limit hyperoxidation and improve neuronal recovery in adult rat hippocampal slices and that high concentrations have the opposite effect (Perez-Pinzon et al., 1998a).
Previous studies have shown hyperoxidation to be inversely related to adenosine 5′-triphosphate (ATP) recovery but directly correlated with histopathology (Welsh et al., 1991). Since the aged brain has shown increased vulnerability to hypoxic/ischemic damage (Roberts and Chih, 1995, Roberts and Chih, 1998; Roberts et al., 1990), it is conceivable that injury to the aged brain will be exacerbated by the combined effects of changes in calcium buffering, prolonged hsd and resulting NADH hyperoxidation. To answer this question, a hippocampal tissue slice preparation of rats across the lifespan was used to test the following hypotheses: (1) NADH hyperoxidation following hypoxia is exacerbated in aged animals and is associated with diminished neuronal recovery compared to other age groups; (2) the removal of calcium during hypoxia in aged rats will decrease NADH hyperoxidation and improve neuronal recovery.
2. Materials and methods
2.1. Tissue slice preparation
Hippocampal tissue slices were prepared from F344 rats (Harlan, Indianapolis, Indiana, USA) of the following ages: 1–2 months old (mo) (young adult), 3–6 months (adult), 12–20 months (mature adult) and 22 months (senescent). The Institutional Animal Care Committees at Duke University and the Durham Veterans Affairs Medical Center approved all experimental protocols. The rats were anesthetized with halothane (Abbott Laboratories, North Chicago, IL, USA) in an anesthesia induction chamber until respirations ceased, and decapitated. The brain was rapidly removed from the skull and placed in chilled artificial cerebrospinal fluid (ACSF, for composition, refer to Section 2.2) for 2 min. The brain was then cut in half sagitally and one hippocampus was isolated at a time. Transverse hippocampal slices of 400 μm thickness were cut using a manual tissue chopper with a micrometer. Slices were immediately placed in an oxygenated holding chamber consisting of ACSF at room temperature. The slices were maintained for 60–90 min to allow for recovery and stabilization. Individual slices were then transferred to an “Oslo-style” interface-recording chamber contained within a physiology and imaging workstation. The chamber was perfused with ACSF (maintained at 35–36 °C) at a flow rate of 1.5 ml/min and continuously aerated with a 95% O2–5% CO2 humidified gas mixture (400 ml/min).
2.2. Solutions
The ACSF was composed of the following (in mM): NaCl, 130; KCl, 3.5; NaH2PO4, 1.25; NaHCO3, 24; CaCl2, 1.2; MgSO4, 1.2; dextrose, 10. The pH was 7.4 when saturated with a gas mixture containing 5% CO2. All chemicals were analytical grade. Calcium-free medium was identical to ACSF except that no CaCl2 was added. The concentration of MgSO4 was increased to 4.8 mM and 1 mM ethylene glycolbis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) was added to chelate any residual Ca2+ remaining in the extracellular space.
2.3. Synaptic activation
A bipolar stimulating electrode made of insulated stainless steel wire was placed in the stratum radiatum (SR) of the Schaffer collaterals of the CA1 region. Stimulus current was adjusted, using single pulses (0.1ms, up to 0.2 mA at 10 s intervals) to produce a field excitatory post-synaptic potential (fEPSP) of 50% of the maximum amplitude. Borosilicate glass microelectrodes were pulled to produce a tip resistance of ∼5MΩ. The electrodes were filled with 150 mM NaCl and were used to measure extracellular evoked and dc potentials. The recording electrode was placed as close to the stimulating electrode as possible at a depth of ∼110 μm (for maximal field potential).
2.4. Tissue oxygen tension (PO2) measurement
A Clark-style glass oxygen microelectrode (737gc, tip diameter 8–10 μm, Diamond General, Ann Arbor, MI, USA) was used to measure brain tissue PO2 during all experiments. Details regarding dimensions, oxygen consumption, electrode drift and volume of tissue measurement of this electrode have been described previously (Foster et al., 2005). The electrode was connected to a polarographic amplifier (Chemical Microsensor II, Diamond General, Ann Arbor, MI, USA) and the cathode (noble metal type) was polarized at −800 mV for up to 12 h in normal saline at 36 °C. Following polarization, a two-point calibration (in amperes) was performed by inserting the electrode in normal saline (at 36 °C) equilibrated (up to 2h) with 95% O2–5% CO2–0% N2 (medical grade) until the ampere values stabilized. The electrode was then inserted into normal saline (at 36 °C) equilibrated with 95% N2–5% CO2–0% O2 (medical grade). Calibrations were repeated before and after each slice to account for any drift in PO2 values. The final PO2 values were corrected for electrode drift and also took into account the accuracy of the amplifier (range ±2%).
Following calibration, the oxygen electrode was inserted in the SR in close proximity to the recording electrode. The oxygen electrode was then slowly advanced using a micrometer through the tissue in 50 μm increments to the depth at which the PO2 was at a minimum (nadir). Since the oxygen electrode was positioned in the micro-manipulator at a 36° angle to the surface of the slice, the nominal values of 50 μm were subsequently corrected to yield actual tissue depth values of 29 μm. All subsequent references to depth reflect actual tissue depth values. Oxygen tension (PO2) was measured at every 29 μm in the SR of 400 μm thick hippocampal slices from rats of each age group. The minimal PO2 (nadir) was determined in each slice and did not vary significantly between the four age groups: 1–2 months (254 ± 64 mmHg), 3–6 months (294 ± 73 mmHg), 12–20 months (279 ±70 mmHg) and 22–24 months (252 ±118 mmHg). The oxygen electrode was kept at the nadir depth of each slice for subsequent measurements during hypoxia.
2.5. Hypoxia
Hippocampal slices were deprived of oxygen following 10 min of baseline recording by replacing the 95% O2–5% CO2 humidified gas mixture above the slices with 95% N2–5% CO2. As soon as hsd occurred (typically within 2–3 min), hypoxia was continued for a set amount of time (0.25, 2.5,5, or 10 min) followedby reoxygenation for 60 min (refer to Fig. 1A). The percentage of neuronal recovery, i.e. the difference in fEPSP peak amplitude measured at 50 min following reoxygenation compared to pre-hypoxia, was assessed. These calculations were derived from 5 min of data recording averaged at each time point.
Fig. 1.
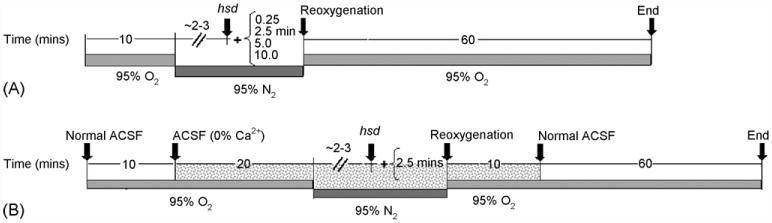
(A) Timeline for hypoxia experiments. The arrow labeled hsd indicates the approximate time after the initiation of hypoxia at which the hsd occurred. The times (in min) shown in brackets indicate the time remaining before reoxygenation. (B) Timeline for hypoxia experiments with 0% Ca2+. Normal ACSF is replaced with Ca2+-free ACSF at 10 min following the start of the experiment (as indicated by the grey shaded area). This is continued for 20 min before the start of hypoxia, during hypoxia and for 10 min following reoxygenation.
2.6. Removal of calcium
In separate experiments, calcium was removed from the ACSF in slices from >22 months old ratsinorderto determine the effect of calcium removal on neuronal recovery. Slices were exposed to 10min of normal ACSF, 20min of ACSF lacking calcium followed by hypoxia for 2.5 min post-hsd (see Fig. 1B). During reoxygenation, slices were perfused with ACSF lacking calcium for an additional 10 min before switching to normal ACSF for the remainder of the experiment (60 min). In these experiments, recovery of fEPSP was calculated as the difference between the peak amplitude at baseline (i.e. prior to both calcium removal and hypoxia) and at 70 min following reoxygenation (average of 5 min of experimental recording at each time point).
2.7. NADH imaging
NADH fluorescence was imaged in slices by epi-illuminating the tissue using a Sutter DG-2 Xenon light source with a 330WB80 filter from Omega Filters (Brattleboro, VT, USA). Excitation light was at wavelengths of 300–370 nm while emitted light was restricted to 400 nm or above. Slices in the interface chamber were imaged through a Nikon upright microscope (UM-2) with a compound lens (4×), using a Cooke Instruments Sensicam QE (Auburn Hills, MI, USA), with 1280 × 1040 digital spatial resolution (12-bit; interline charge-coupled device, cooled to −15 °C). Images were taken once every 5 s with 1000 ms exposure. The images were acquired as 8 × 8 binned images, reducing the effective spatial resolution to 160 × 130 pixels. The digital images were analyzed by selecting an anatomically based region such as the SR. A series of 12-bit intensity numbers were generated from this region, which were then plotted versus time. The NADH signal drifted over time (∼5% over 1–2 h), which was likely to have resulted from irreversible photolysis of NADH to NAD+ by some means other than direct photo-oxidation. It has been suggested that this ‘irreversible behavior’ could occur in situations in which the NAD pool that is available for replenishing the lost NADH is small (Joubert et al., 2004). To account for NADH drift over time, a regression line was created for each set of experimental data from the baseline prior to hypoxia. The line was then extrapolated to the end of the data in order to correct for the drift.
The NADH fluorescence measured in the following experiments is representative of both cytosolic and mitochondrial NADH. NADPH levels within the brain are low and are not directly involved with metabolism (Chance et al., 1962; Kaplan, 1985; Klaidman et al., 2001). The depth of NADH fluorescence was confirmed in previous studies by placing a slice on a net shown to be fluorescent in ultraviolet light (Foster et al., 2005). The fluorescence of the net imaged through the slice was 82.7% of the fluorescence imaged from the net alone. Therefore, one can surmise that in a 400 μm thick slice, the fluorescence originates from the entire depth of the slice.
In order to establish a redox baseline for NADH, we determined the maximum percentage of oxidation and reduction. Hypoxia (10min post-hsd) resulted in a 45% increase in NADH fluorescence intensity, while the use of ACSF without glucose (hypoglycemia) resulted in a 40% decrease in NADH fluorescence, representing maximal oxidation. The percentage of NADH hyperoxidation (i.e. % change in NADH below baseline following hypoxia) was calculated relative to the baseline at 15min following reoxygenation using linear regression. This time interval was selected since the peak of hyperoxidation in the majority of slices occurred approximately 15min following reoxygenation.
2.8. Experimental design and statistical analyses
Experiments were conducted in a randomized fashion. Each hypoxic time interval (0.25, 2.5, 5, or 10min) following hsd was examined only once in slices of the same rat. All data values (n = number of slices) are presented as the mean ± standard deviation. Statistical significance (p < 0.05) was determined using t-tests (SigmaPlot 2002, Version 8.02) or one-way ANOVA (GraphPad Prism, Version 3.0) where needed. Tukey's post-test was used to compare pairs of group means.
3. Results
3.1. Short-term hypoxia
Hippocampal slices from each age group were subjected to 15 s of hypoxia post-hsd. Fig. 2A shows a series of NADH fluorescence images during the progression of hypoxia (hsd + 15 s) in a hippocampal slice from a 20 months old rat with times relative to hsd onset (indicated to the right of the images). The top image in each sequence shows a control image of a hippocampal slice (CA1 region)inan interface chamber setup. The subsequent frames are difference images of NADH fluorescence prior to, during and following hypoxia (diffuse gray indicating no change).
Fig. 2.
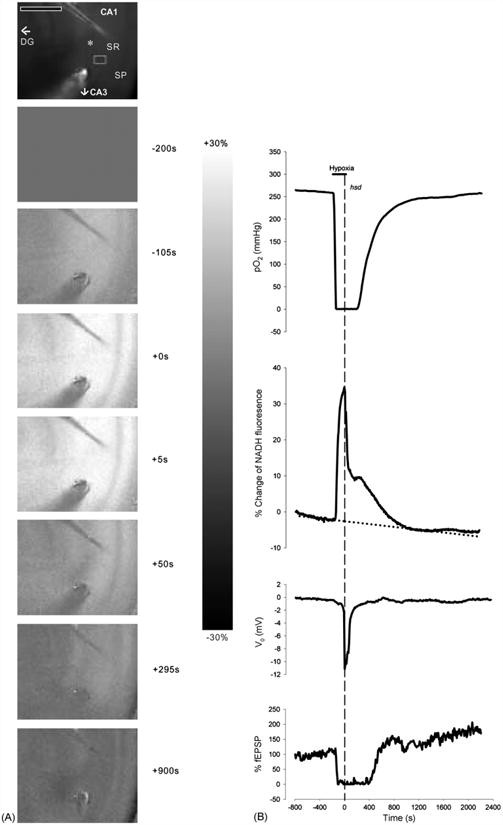
(A) Series of NADH fluorescence images during the progression of reversible hypoxia (hsd + 15 s) in a hippocampal slice from a 20 months old rat. The top image shows an unsubtracted raw camera picture with the region of interest indicated by a gray rectangle in the SR of the CA1 region. The asterisk indicates the position of the recording electrode. The oxygen and stimulating electrodes are above and below the recording electrode, respectively. Scale bar indicates 500 μm. Hypoxia was initiated at −180 s (as shown in the right margin). Zero time marks the onset of hsd. The scale bar in the right margin indicates the scale for the difference images, with white representing a 30% increase above baseline and black, a 30% decrease. Note that the CA1 region is preferentially affected by hypoxia, with a marked increase in NADH signal. (B) Corresponding responses of NADH as well as PO2, % fEPSP and dc voltage to reversible hypoxia (hsd + 15 s). The horizontal line indicates the hypoxic interval, while the dotted vertical line shows the onset of hsd. The PO2 was measured at the nadir of the slice. To account for the drift in NADH over time, a regression line (dotted line) was created from the baseline data prior to hypoxia.
Fig.2B illustrates the corresponding time course of NADH fluorescence (from a SR region of interest) as well as tissue PO2 (measured at the slice nadir within the SR), dc voltage level and evoked fEPSP from the hippocampal slice (20 months old rat) in response to 15 s of hypoxia post-hsd. PO2 levels started to rapidly decrease several seconds after the initiation of hypoxia. The PO2 reached values close to 0 mmHg after approximately 70 s. The PO2 remained at minimal values for approximately 4 min after which hsd occurred. Within 15 s of hsd initiation, oxygen was reintroduced. The PO2 values started to increase slowly from 0 mmHg approximately 1 min following reoxygenation, and reached baseline values after 20 min.
The NADH response to hypoxia can be separated into a number of sequential phases. The first phase (pre-hsd phase) consisted of a prominent increase (NADH reduction) at the end of which the initiation of hsd occurred (hsd phase). Reoxygenation was performed within 15 s of hsd initiation by switching the ambient gas supply from 95% N2 to 95% O2. The NADH levels abruptly decreasedasaresultofthe reintroduction of oxygen to the slice (initial reoxygenation phase). However, following reoxygenation, NADH levels decreased at a slower rate toward baseline levels (late reoxygenation phase). During this time, NADH levels increased temporarily (represented by a ‘shoulder’) while the V0 was already recovering. The increase in NADH continued while the V0 reached baseline.Inthe majorityofcases (i.e. slices from 1–2, 3–6 and 12–20 months old rats), the NADH responses were completely reversible following 15 s of hsd and reoxygenation. However, in slices from >22 months old rats, NADH levels continued to decrease slightly below baseline over the remaining 60 min, a phenomenon known as ‘hyperoxidation’. The percentage change below baseline measured after 15 min of reoxygenation was −1.8 ± 2.8% in this older age group.
3.2. Prolonged hypoxia
To determine whether factors such as age or the duration of hypoxia following hsd alter neuronal (fEPSP) recovery or the responses of NADH and PO2, the hypoxic period was then extended to 2.5, 5 or 10 min and assessed in hippocampal slices from rats across the lifespan. Changes in NADH fluorescence as a result of prolonged hypoxia (5 min post-hsd) are illustrated in a series of fluorescence images produced during the progression of hypoxia and following reoxygenation in a hippocampal slice from a 25-mo rat (Fig. 3A). Similar to shorter periods of hypoxia, NADH fluorescence increased to peak values following hsd, represented by the increase in fluorescence across the CA1 area of the slice (NADH reduction). Following reoxygenation, however, the images became increasingly darker indicating a decrease in NADH fluorescence below baseline. The greatest decrease in NADH is represented in the last image and was measured at 15 min following reoxygenation (peak of NADH hyperoxidation).
Fig. 3.
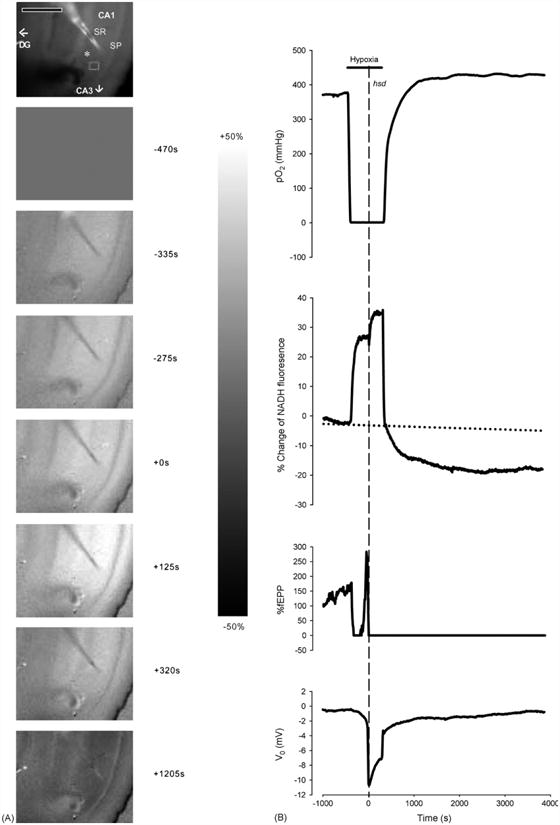
(A) Series of NADH fluorescence images during the progression of irreversible hypoxia (hsd + 5 min) in a hippocampal slice from a 25 months rat. The top image shows an unsubtracted raw camera picture with the region of interest indicated by a gray rectangle in the SR of the CA1 region. The asterisk indicates the position of the recording electrode. The oxygen and stimulating electrodes are above and below the recording electrode, respectively. Scale bar indicates 500 μm. Image times are shown in the right margin. Hypoxia was initiated at −450 s. Zero time marks the onset of hsd. The scale bar in the right margin indicates the scale for the difference images, with white representing a 50% increase above baseline and black, a 50% decrease. Note that the CA1 region is preferentially affected by hypoxia, with a marked increase in NADH signal. (B) Responses of PO2, NADH (% change), fEPSP (% change from baseline) and dc voltage to hypoxia (hsd + 5 min) in a 25 months rat hippocampal slice. The horizontal line indicates the hypoxic interval, while the dotted vertical line shows the onset of hsd. The PO2 was measured at the nadir of the slice. A regression line (dotted line) was created from the baseline data to account for the drift in NADH over time.
Fig. 3B shows the corresponding plots of PO2 (measured at the slice nadir within the SR), NADH values (from a SR region of interest), fEPSP and the dc voltage level in a hippocampal slice from a 25 months old rat following 5 min of hypoxia post-hsd. NADH levels increased rapidly from baseline following the initiation of hypoxia (pre-hsd phase). This phase, which ended in an inflection point, was then followed by a further increase in NADH levels in response to the initiation of hsd (hsd phase). The extension of the hypoxic period to 5 min slowed the incline of NADH levels to a plateau-like elevation (hypoxic plateau phase), which continued until reoxygenation. Following reoxygenation, NADH levels rapidly decreased to baseline but in contrast to reversible hypoxia, continued to decrease below baseline over the next 60 min, indicating NADH hyperoxidation.
Following the initiation of hypoxia, the PO2 started to decrease from baseline values within several seconds and reached values close to 0 mmHg in approximately 50 s. Hypoxic spreading depression occurred after another 4 min. The PO2 increased from minimal values following reoxygenation to baseline at faster rates (∼8 min) compared to 15 s of hypoxia post-hsd. In addition, the PO2 continued to increase to values higher than baseline (PO2 overshoot) and remained at these elevated values for the remainder of the experimental period.
Ten minutes of hypoxia post-hsd caused a maximal NADH fluorescence increase in all age groups. The peak percentage of NADH measured during 10 min hypoxia post-hsd in slices from 3–6 and >22 months old rats was significantly greater than the NADH peak during 15 s hypoxia (Fig. 4). However, no significant differences were found when the NADH reduction peaks (during either 15 s or 10 min hypoxia) were compared between age groups.
Fig. 4.
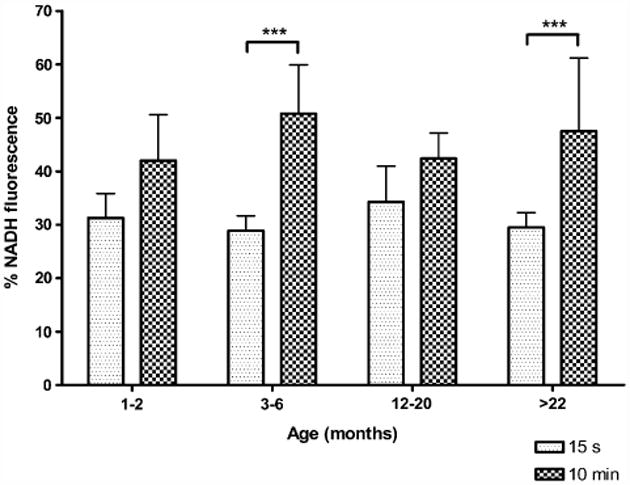
Maximal % increase of NADH fluorescence (i.e. reduction) during hypoxia 15 s or 10 min after hsd onset in slices from 1–2, 3–6, 12–20 and >22 months old rats. Significant differences were found between the NADH reduction during 15 s and 10 min hypoxia post-hsd in slices from 3–6 and >22 months old rats (***p < 0.001).
3.3. Time to hsd
The time from when the PO2 reached 0mmHg (following the initiation of hypoxia) to the onset of the hsd-related negative V0 shift was determined in slices from each age group. The mean time to hsd increased with age, but the differences between the age groups were not statistically significant: 1–2 months (128±55 s, n=27), 3–6 months (162±126 s, n= 24), 12–20 months (168±120 s, n= 25), and >22 months (174 ± 87 s, n = 30).
The time taken for the fEPSP to recover to 50% of baseline was measured in slices from across the lifespan following 15 s of hypoxia post-hsd (Fig. 5). The fEPSPs of slices from >22 months old rats were significantly slower to recover (677 ± 298 s) compared to all younger age groups (1–2 months, 397±133 s; 3–6 months, 356±98 s; 12–20 months, 413 ± 114 s).
Fig. 5.
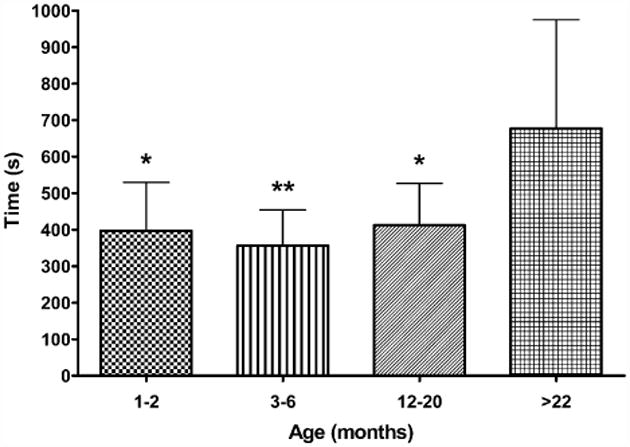
Time taken for fEPSP amplitude to recover to 50% of control in hippocampal slices from 1–2 months old rats (n = 7), 3–6 months old rats (n = 8), 12–20 months old rats (n = 8) and >22 months old rats (n = 7) following 15 s hypoxia post-hsd. The fEPSPs of slices from >22 months old rats were significantly slower to recover compared to slices from the younger age groups (1–2 and 12–20 months *p < 0.05; 3–6 months **p < 0.01).
3.4. Neuronal (fEPSP) recovery
The effect of hypoxia, of varying durations, was tested in four age groups across the lifespan to assess the limits of neuronal recovery. Fig. 6 shows the recovery of the fEPSP amplitudes (%) from baseline in hippocampal slices from rats aged 1–2 to >22 months following varying periods of hypoxia (15 s–10 min following hsd). At all ages tested, 15 s of hypoxia was a reversible event with the majority of slice fEPSPs recovering to greater than 100%. A 2.5 min hypoxic interval following hsd initiation, however, caused a decrease in fEPSP recovery in slices from 12 to 20 months old (50±57%, n=6) and >22 months old (40±49%, n= 9) animals but had no adverse effects on fEPSP recovery in slices from younger rats (1–2 months, 95 ± 58% (n = 8); 3–6 months, 116 ± 47%; (n = 8)). Neuronal recovery in slices from >22 months old rats was significantly lower compared to slices from 3 to 6 months old rats following 2.5 min hypoxia.
Fig. 6.
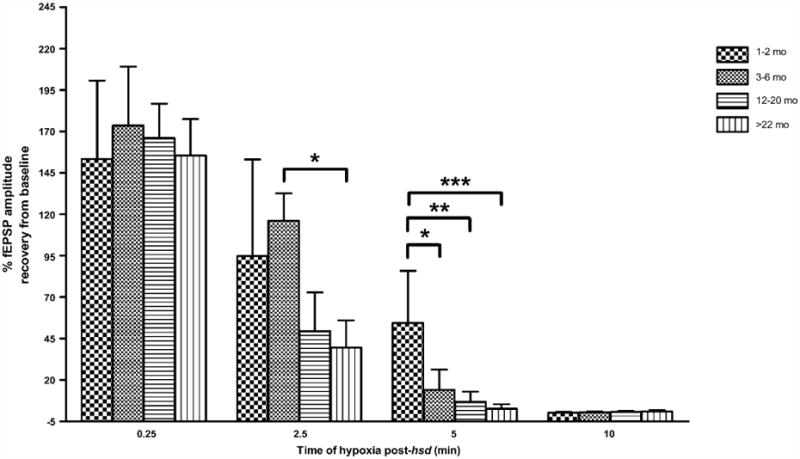
Recovery of fEPSP amplitude in hippocampal slices from 1–2, 3–6, 12–20, >22 months old rats following varying lengths of hypoxia post-hsd (0.25, 2.5, 5 and 10 min). Following 2.5 min of hypoxia post-hsd, fEPSP recovery differed significantly between slices from 3–6 months old (116 ± 47%, n = 8) and >22 months old rats (40 ± 49%, n = 9) (*p < 0.05) while following 5 min hypoxia post-hsd, the recovery of fEPSP in slices from 1–2 months old rats (54 ± 32%, n = 8) was significantly greater than the recoveries in slices from all older age groups [(3–6 months, 14 ± 32%, n = 7, *p < 0.05); (12–20 months, 7 ± 15%, n = 6, **p < 0.01); (>22 months, 3 ± 8%, n = 9, ***p < 0.001)].
Five minutes of hypoxia following hsd onset led to irreversible neuronal impairment in all age groups with the exception of slices from 1 to 2 months old rats, whose neuronal responses recovered to 54 ± 32%. The percentage of neuronal recovery in this age group was significantly higher than the recovery in slices from all older age groups, i.e. 3–6 months old rats(14 ± 32%), 12–20months old rats(7 ± 15%) and >22 months old rats (3 ±8%). A 10 min interval of hypoxia post-hsd resulted in 0% recovery across the lifespan.
3.5. NADH hyperoxidation
Hyperoxidation was evident (shown in Fig. 7) following 15 s of hypoxia post-hsd in slices from >22 months old rats (−1.9 ± 2.8%), and following 2.5 min of hypoxia in slices from both 12–20 months old rats (−0.1 ± 2.8%) and >22 months old rats (−1.8 ± 3.1%). Hyperoxidation was present in the younger age groups only after 5 min of hypoxia post-hsd (1–2 months, −0.8 ± 4.1% and 3–6 months old, −0.6 ± 4.9%). The extent of hyperoxidation following 5 min hypoxia in these groups was significantly less than the hyperoxidation noted in slices from >22 months old rats. Hyperoxidation was most pronounced following 10 min of hypoxia post-hsd in all age groups. However, a significantly greater degree of hyperoxidation occurred in slices from 1 to 2 months old rats (−16.4 ± 2.5%) compared to the 3–6 months old (−6.1±1.5%) and >22 months old (−7.3±6.3%) age groups.
Fig. 7.
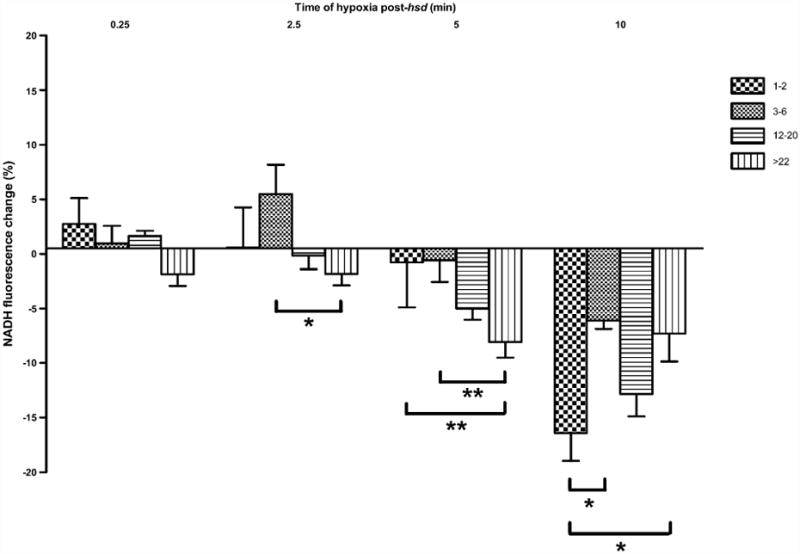
Change in NADH fluorescence (%) in hippocampal slices from 1–2, 3–6, 12–20, >22 months old rats following varying lengths of hypoxia post-hsd (0.25, 2.5, 5 and 10 min). Following 2.5 min hypoxia post-hsd, NADH hyperoxidation was significantly greater in slices from >22 months old rats (n = 9) compared to slices from 3 to 6 months old rats (n = 7; *p < 0.05). This effect was amplified between these age groups at 5 min hypoxia (**p < 0.01). In addition, the percentage of hyperoxidation was greater following 5 min hypoxia in slices from >22 months old rats (n = 6) compared to 1–2 months old rats (n = 7; **p < 0.01). Ten minutes of hypoxia post-hsd resulted in the greatest amount of hyperoxidation in all age groups with slices from 1 to 2 months old rats (n = 4) exhibiting significantly higher amounts of hyperoxidation compared to slices from 3 to 6 months old rats (n = 4) and >22 months old rats (n = 6; *p < 0.05).
Correlation analysis revealed a significant relationship between decreased fEPSP recovery and increased NADH hyperoxidation across all age groups following longer periods of hypoxia (p < 0.0001, R2 = 0.28).
3.6. Repeated episodes of prolonged hypoxia
The residual reduction response of NADH was investigated, along with the PO2 response, following both an initial prolonged period of hypoxia (10 min post-hsd), and then a second 10 min period of hypoxia at 50 min following the first episode. Fig. 8 shows typical PO2 and NADH traces following the repeated episodes of hypoxia in a hippocampal slice from a 1–2 months old rat. Reoxygenation following the first episode of prolonged hypoxia resulted in a PO2 overshoot and NADH hyperoxidation. The PO2 continued to increase until the second episode of hypoxia was induced, while NADH levels stabilized below baseline during this time. The second episode resulted in an even more pronounced PO2 overshoot and a smaller NADH reduction peak compared to the first hypoxic episode (i.e. a decrease by 8 ± 11%). Note also the absence of any further NADH hyperoxidation following the second episode of hypoxia.
Fig. 8.
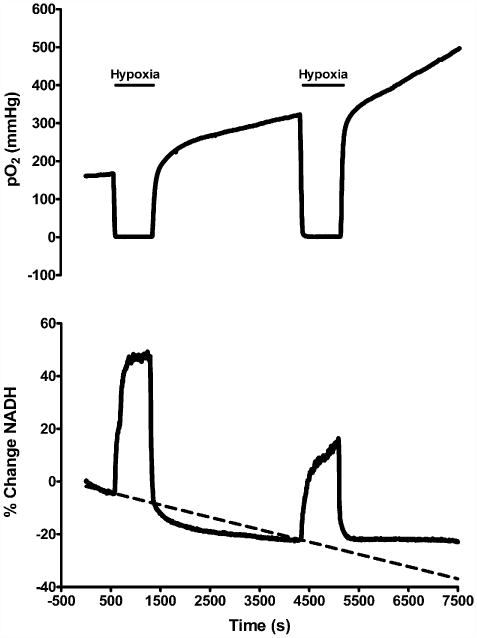
Graphs of PO2 and NADH following repeated episodes of hypoxia (10 min post-hsd) in slices from 1 to 2 months old rats (n = 4). Reoxygenation following the first episode of hypoxia resulted in a PO2 overshoot (increase above baseline) and NADH hyperoxidation (decrease below baseline). During the second episode of hypoxia the NADH fluorescence increase was less than during the first, and the second event was not followed by hyperoxidation. The PO2 overshoot was enhanced after the second episode.
3.7. Effect of calcium removal
Following 10 min perfusion with normal ACSF, hippocampal slices from >22 months old rats were perfused with ACSF lacking calcium for 20 min and then exposed to hypoxia for 2.5 min post-hsd. The removal of calcium resulted in a complete loss of the evoked fEPSP within ∼7 min. Slices perfused without calcium experienced hsd at 100 ± 46 s on average, compared to a slightly more prolonged time in slices perfused with calcium, 150 ± 94 s. At 10 min following reoxygenation, calcium was reintroduced to the perfusate. Slices in which the calcium had been removed generally took longer to recover baseline synaptic transmission than slices perfused with calcium. Fig. 9A shows the recovery of fEPSP amplitude from slices perfused with and without calcium. The removal of Ca2+ from the ACSF showed a trend towards increased neuronal (fEPSP) recovery in the aged slices following hypoxia (2.5 min post-hsd) from 40 ± 49% (n = 9) to 76 ± 72% (n = 8), although the change was not significant due to the large variability in neuronal recovery in both control and treatment groups.
Fig. 9.
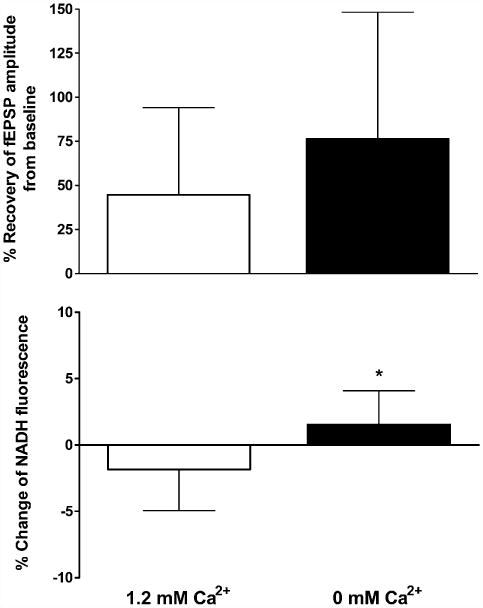
(A) Recovery of fEPSP amplitude following 2.5 min hypoxia post-hsd in 1.2 and 0 mM calcium conditions in slices from >22 months old rats. The removal of calcium from the ACSF improved neuronal recovery following hypoxia from 40 ±49% (n= 9) to 76 ±72% (n=8) however the effect was not significant. (B) NADH fluorescence change (%) measured at 15 min following 2.5 min hypoxia post-hsd and reoxygenation in 1.2 and 0 mM calcium conditions in slices from>22months old rats. Hyperoxidation was absent 15 min after reoxygenation in slices perfused with 0% calcium (+1.5 ± 0.9%) compared to slices with calcium (−1.8 ± 1%, *p < 0.05).
The absence of Ca2+ prevented the occurrence of NADH hyperoxidation at 15 min following reoxygenation (+1.5 ± 0.9%) in comparison with slices perfused with Ca2+ (−1.8 ± 1%, p < 0.05) (Fig. 9B). However, at 45 min post-reoxygenation, NADH hyperoxidation was present in slices without Ca2+ and had increased (to −6 ± 4.4%) compared to slices with Ca2+ (+3.0 ± 3.9%, p < 0.001). In contrast to the early (15 min post-hsd) measure of hyperoxidation, this later occurrence of hyperoxidation did not correlate with decreased recovery of the fEPSP.
Control experiments (no hypoxia) were also conducted in the absence of calcium to determine whether the removal of calcium itself or the length of calcium deprivation compromised neuronal recovery. The recovery of the fEPSP in these experiments was 120 ± 34% (n = 5) and was not significantly different to the recovery noted in the hypoxia experiments conducted in the absence of calcium (76 ± 72% (n = 8)). In addition, no NADH hyperoxidation was evident in the control experiments (absence of hypoxia and calcium) when measured at 15 min following the time at which reoxygenation would have occurred (+0.24 ± 0.53%).
4. Discussion
The major goal of this study was to identify whether alterations in oxidative metabolism following hypoxia, such as NADH hyperoxidation, are exacerbated with aging and as a result contribute to a greater degree of neuronal injury. The results presented in this study demonstrate significant alterations in mitochondrial function and enhanced susceptibility to hypoxia as a result of aging. The major findings of this study were: (1) hippocampal slices from aged rats are more susceptible to shorter periods of hypoxia compared to younger age groups; (2) the degree of hyperoxidation increases both with the length of hypoxia (post-hsd) and with age and is inversely correlated with neuronal recovery; and (3) the removal of calcium provides neuroprotection in slices from aged rats by delaying NADH hyperoxidation.
4.1. Duration of hypoxia (pre- and post-hsd) versus outcome
Neuronal recovery in the CA1 region of the hippocampus has been shown to be more sensitive to the length of time following the onset of hsd than to the total time exposed to hypoxia (Perez-Pinzon et al., 1998b). We have shown that slices from all ages are tolerant to 15 s of further hypoxia following hsd. In fact, the fEPSP responses in all age groups recovered to greater than control values, which suggest that mild synaptic potentiation occurred. A similar result was obtained in which 30 s of hypoxia post-hsd in hippocampal slices resulted in higher fEPSP amplitudes compared to control slices (Perez-Pinzon et al., 1998b).
A number of previous studies in brain slices have provided evidence that neuronal recovery is worsened as the duration of hypoxia following hsd is increased (Balestrino et al., 1989, 1999; Balestrino and Somjen, 1986; Jing et al., 1991; Kawasaki et al., 1988; Lee and Lowenkopf, 1993; Perez-Pinzon et al., 1998b; Roberts and Sick, 1988; Tombaugh, 1994; Watson and Lanthorn, 1995). Extending the length of post-hsd hypoxia to 2.5 min in this study resulted in depressed synaptic activity and therefore reduced neuronal recovery in slices from the oldest age groups (12–20 and >22 months) while slices from 1 to 2 and 3 to 6 months old rats were more resistant (and recovered to ≥95%). In a previous study, shorter periods of anoxic depolarization (1 min) caused greater than 50% suppression of synaptic activity in adult hippocampal slices while 2 min resulted in <20% recovery of the evoked potential (Perez-Pinzon et al., 1998b). The reasons for the differences in susceptibility between these two studies are not directly apparent since slice preparation conditions and the types of chambers used were very similar. However, factors such as oxygen flow rates, slice thickness, rat strain used in the experiments and temperature may influence the specific period of hypoxia, which leads to various levels of neuronal recovery.
The threshold for irreversible neuronal damage in the two oldest age groups in the current study was 5 min hypoxia post-hsd while some recovery still occurred in slices from 3 to 6 months animals (14%). Slices from the 1–2 months old rats were the most resistant and recovered to 54%. Again, these results were in contrast to those obtained in the previous study, in which complete suppression of synaptic activity in adult slices occurred following 5 min hypoxia post-hsd (Perez-Pinzon et al., 1998b).
No significant differences were found in this study between the various age groups in terms of time to hsd onset following initiation of hypoxia, although slices from the youngest age group (1–2 months old) underwent hsd slightly earlier than the three older age groups (2.1 min compared to 2.5–3.0 min, respectively; NS). These results are in contrast to previous studies in which slices from aged rats (26–27 months) lost homeostasis at a significantly faster rate and the delay to hsd was shorter following hypoxia compared to slices from younger rats (6–7 months) (Robertset al., 1990; Roberts and Chih, 1995). Anoxic depolarization (or hsd as we have defined the event) has been significantly correlated with a decrease in ATP concentration (generated by glycolysis (Allen et al., 2005; Chih et al., 1989; Kass and Lipton, 1989; Rosenthal and Sick, 1992)). Also, aging decreases the levels of ATP maintained during anoxia (Kass and Lipton, 1989). Therefore, it was suggested that slices from aged rats in these previous studies were unable to enhance anaerobic metabolism in response to hypoxia in order to maintain ionic pump function (Roberts and Chih, 1995).
Slices from >22 months old rats in the current study were, however, slower to recover to 50% of baseline fEPSP following 15 s of hypoxia post-hsd compared to all younger age groups (11 min versus 6–7 min). A similar result was obtained in which recovery of orthodromic spike activity and homeostasis following hypoxia in slices from aged rats was delayed, compared to slices from 6 to 7 months old rats (Roberts et al., 1990). The delays in recovery of synaptic transmission in both studies may have been the result of age-related decreases in energy metabolism, particularly glycolysis, as opposed to oxidative phosphorylation and/or less effective pH buffering (Roberts and Chih, 1998).
4.2. NADH reduction and hyperoxidation
The NADH peak or plateau prior to reoxygenation was indicative of maximal reduction during hypoxia. Exposure of slices from different age groups to 15 s hypoxia post-hsd did not cause significant differences in peak magnitude. However, an in vivo study demonstrated that the NADH peak in response to anoxia (30 s of 100% N2) was significantly diminished in the cortex of aged rats (9.6%, 25–30 months) compared to adult rats (36%, 2–3 months) (Zarchin et al., 2002) suggesting a progressive reduction of the NADH redox range with age. The differences in these results may have been due to various experimental factors in the in vivo preparation. For example, an increased sensitivity of the older animals to the anesthesia used in the in vivo study may have caused a differential effect. In addition, cerebral atrophy and an increase in cerebral spinal fluid volume, which occur with age, may have increased the light scattering across the meninges in the intact aged brain compared to the adult brain.
The ability of neurons to recover from a hypoxic/ischemic event is dependent upon the function of mitochondrial enzymes as well as the electron transport chain to produce adequate levels of ATP. Reperfusion following hypoxia/ ischemia triggers a number of events, which contribute to subsequent hypoxic/ischemic cell damage. One such event is mitochondrial hyperoxidation, which includes hyperoxidation of NADH and cytochrome a,a3. Not only is NADH hyperoxidation associated with decreased neuronal recovery following prolonged periods of hypoxia post-hsd (Perez-Pinzon et al., 1998b), but as we have shown for the first time in this study, NADH hyperoxidation is also exacerbated with aging. Hyperoxidation was present in slices from aged rats (12–20 and >22 months) even following short periods of hsd (15 s and 2.5 min). The hyperoxidation was accompanied by a decrease in neuronal recovery of ≥50% compared to the youngest age groups, in which 5 min of hypoxia post-hsd was required to decrease neuronal recovery to ≤54% and to produce hyperoxidation. The correlation between increased hyperoxidation and reduced neuronal recovery across the lifespan (R2 = 0.28) does not necessarily suggest a causal relationship between the two events. Instead we suggest that NADH hyperoxidation and the loss of electrical activity are two parallel outcomes that both occur as a result of cell damage following increasing lengths of hypoxia.
The mechanisms by which mitochondrial hyperoxidation occurs remain to be fully elucidated. However past studies suggest that hyperoxidation is modulated by intracellular derangements during and following reperfusion and may result from the combined effects of ROS production, activation of poly-ADP ribose polymerase (PARP), inhibition of the pyruvate dehydrogenase complex (PDHC) and accumulation of intracellular calcium. The proposed link between ROS and mitochondrial hyperoxidation is reinforced by a study in which the use of antioxidants ascorbate and glutathione lessened the severity of NADH hyperoxidation and improved the recovery of field potentials following 2 min of hypoxia post-hsd in adult rat hippocampal slices (Perez-Pinzon et al., 1997). In addition, it has been shown that the use of hyperoxia (30–100% O2) at reperfusion following 30 min global cerebral ischemia in adult rats produced hyperoxidation of cytochrome a,a3, while 15% O2 prevented hyperoxidation (Feng et al., 1998).
NADH hyperoxidation could in part result from a decreased NAD pool available for metabolism due to its binding by PARP. PARP is a nuclear enzyme, which scans for deoxyribonucleic acid (DNA) damage (Skaper, 2003) caused by free radicals (Yabuki et al., 1997). Following activation (as early as 15min after transient ischemia (Narasimhan et al., 2003)), PARP hydrolyses its substrate, nicotinamide adenine dinucleotide (NAD+) to nicotinamide and transfers poly(ADP-ribose) chains to a variety of other nuclear proteins. In the presence of significant injury, PARP can be activated up to several hundred fold (Benjamin and Gill, 1980) and can lead to NAD+ depletion, particularly in the cytosol (Gaal et al., 1987), resulting in severe impairment of glycolysis.
Decreased NADH turnover in the tricarboxylic acid (TCA) cycle due to inhibition of the PDHC could also contribute to hyperoxidation. Following ischemia and reperfusion, alterations in mitochondrial activity occur, among them the inhibition of the PDHC, an enzyme required for the facilitation of substrates for oxidative phosphorylation (specifically for the conversion of pyruvate to acetyl CoA). The inhibition of this enzyme is suggested to occur as a result of free radical mediated protein oxidation and may be potentiated by increased calcium concentrations (Bogaert et al., 1994). Inhibition of the PDHC (to 50% of control levels) takes place as early as 15 min following reperfusion (Cardell et al., 1989) consistent with the occurrence of NADH hyperoxidation following hypoxia (as shown in this study) and ischemia (Feng et al., 1998). Inhibition may, as a result, restrict the supply of reducing equivalents and substrates to mitochondria thereby decreasing aerobic metabolism. In addition, the dramatic increase in oxygen levels observed during reoxygenation could potentially accelerate the rate of oxidative phosphorylation causing the already limited NADH supply from the TCA cycle to be depleted.
Mitochondrial uncoupling has been shown to cause the acceleration of electron transport and as a result, NADH oxidation. For example, a previous study demonstrated that the exogenous application of the uncoupler carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) in rat hippocampal slices caused a reversible decrease in NADH levels (i.e. oxidation) due to the occurrence of mitochondrial depolarization (as shown by an increase in R123) and the activation of mitochondrial respiration (Gerich et al., 2006).
The endogenous uncoupling protein UCP-2 has been shown to play a role in ischemia through the modulation of mitochondrial ROS release and caspase-3 activation (Mattiasson et al., 2003). Sublethal oxygen-glucose deprivation (OGD) in cell culture causes UCP-2 mRNA induction, which is suggested to contribute to the subsequent lack of neuronal damage following another longer period of OGD (10 min). However, mRNA induction of UCP-2 is upregulated only several hours (24–96 h) after the period of OGD. Due to this delayed activation, endogenous uncoupling proteins are not likely to play a role during the reoxygenation period in the acute hippocampal slice preparation used in our study (Mattiasson et al., 2003).
The exacerbation of NADH hyperoxidation in older rats may have been attributed to compromised mitochondrial electron transport as a result of aging as well as an increased production of ROS due to a partially reduced tissue oxygen fraction (LeBel and Bondy, 1992). In addition, aging-induced perturbations in calcium homeostasis may have caused greater intracellular calcium accumulation during hypoxia.
During periods of oxidative stress, the levels of pyridine nucleotides, including NADPH, NADP+, NADH and NAD+, determine a cell's ability to continue functioning, since the levels of pyridine nucleotides determine the balance of reductive power and ATP production rate (Klaidman et al., 2001). In an oxygen-depleted environment, a decline in NAD+ levels determines the onset of cell death since NAD+ is required in the production of ATP. In order to further elucidate the phenomenon of NADH hyperoxidation and to determine the residual NADH cycling capacity of the slice following the occurrence of hyperoxidation, we performed repeated episodes of prolonged hypoxia (10 min post-hsd) in young adult hippocampal slices. Ten minutes of hypoxia was chosen since this interval is known to cause irreversible neuronal injury and the greatest amount of hyperoxidation.
Both the smaller NADH peak (by 8 ± 11%) during the second episode of prolonged hypoxia and the absence of hyperoxidation (refer to Fig. 8) indicate an absolute decline in the nucleotide pool and therefore less NAD+/NADH available for metabolic turnover. The corresponding PO2 overshoot following both episodes of hypoxia suggest decreased oxygen utilization and therefore neuronal impairment. The decline in NADH is corroborated by a previous study conducted in vivo, which showed a 43% decrease in the total pool of NAD (NADH+NAD+) in ATP depleted areas of cortex compared to control following 30 min of ischemia and reperfusion. In areas of the cortex in which restoration of ATP was more complete, the NAD pool was only slightly decreased (Welsh et al., 1982). The present results, however, are in contrast to another in vivo study in which the maximal levels of NAD and cytochrome a,a3 reduction did not differ when measured either prior to cerebral anoxia or during NADH hyperoxidation (Rosenthal et al., 1997). The differences in the results may have been attributed to the limitations of NADH detection in the previous study since NADH was measured at single time points and averaged across a larger tissue volume as opposed to imaging in real time.
A mechanism for NAD pool depletion may be the actual loss of NAD+/NADH from mitochondria. In the presence of oxidative stress and calcium overload, mitochondria swell and become depolarized, a process referred to as mitochondrial permeability transition (MPT). A previous study has shown that prolonged (≤10 min) and repeated episodes of hypoxia (equivalent to the periods of hypoxia used in this study) in the hippocampal slice caused MPT (shown by an increase in R123 fluorescence indicating mitochondrial depolarization) (Bahar et al., 2000). The cause of MPT is the opening of the mitochondrial permeability transition pore (MPTP) within the inner mitochondrial membrane, which during hypoxia or ischemia results in the release of small molecules such as NAD+/NADH (Halestrap et al., 2002) and the pro-apoptotic factor cytochrome c from the mitochondria (Galeffi et al., 2000; Perez-Pinzon et al., 1999).
Although mitochondria contain a significant fraction of the cellular NAD+ pool (approximately 72% in cardiac myocytes) (Di Lisa et al., 2001), the contribution of NAD+ loss from the mitochondria versus the cytosol remains controversial. Recent efforts to discriminate NADH fluorescence originating from the cytosol versus the mitochondria have involved techniques such as two-photon imaging in neuronal cell cultures. For example, changes in NADH levels between mitochondria and cytosol were monitored in cultured Purkinje cells following the separate applications of FCCP, rotenone and potassium chloride (Hayakawa et al., 2005). The successful application of these techniques is at present limited to cell culture preparations. However, future advancements in imaging technology and dual labeling of neurons and glia will allow the use of these techniques in more complex systems such as acutely isolated slice preparations.
4.3. Influence of calcium
Under normal physiological conditions, calcium is tightly regulated by mitochondria, which buffer cytosolic calcium levels during synaptic transmission (David et al., 1998). In the case of hypoxia/ischemia, mitochondria sequester the calcium, but become damaged once the levels of Ca2+ are in excess of their buffering capacity. The reduction or removal of calcium from the experimental media in adult rat hippocampal slice preparations during both brief and prolonged hsd improved the recovery of rat hippocampal slices compared to slices perfused with normal concentrations of calcium (Amagasa et al., 1990; Balestrino and Somjen, 1986; Roberts and Sick, 1988; Young et al., 1991). In addition, a relationship has been demonstrated between the mitochondrial redox status (i.e. NADH levels), synaptic activity and cytosolic calcium levels in adult hippocampal slices. For example, lowering calcium levels to 0.5 or 1 mmol/L lessened the subsequent amount of NADH hyperoxidation (measured at 60 min post-reoxygenation) and improved neuronal recovery following 2 min of hypoxia post-hsd, whereas 2 and 4 mM/L of calcium exacerbated the hyperoxidation and worsened neuronal recovery (Perez-Pinzon et al., 1998a).
Aging causes perturbations in cellular calcium homeostasis, which include elevated voltage-gated calcium influx (Campbell et al., 1996; Pitler and Landfield, 1990) and decreases in the mitochondrial buffering of calcium influx through voltage-gated channels (Murchison et al., 2004). In addition, it has been demonstrated that during anoxia, the percentage increase in calcium uptake is higher in adult hippocampal slices compared to young slices (Kass and Lipton, 1989). Therefore, due to the relationship between calcium and NADH hyperoxidation during hypoxia/ischemia and the fact that calcium function changes as a result of aging, we chose to remove the calcium from the extracellular media of slices from <22 months old rats during hypoxia, in order to determine whether neuronal recovery could be improved and NADH hyperoxidation lessened. We examined the responses of slices following 2.5 min of hypoxia post-hsd since this length of hypoxia resulted in partial but not complete synaptic suppression in this age group.
The elimination of calcium improved neuronal recovery (NS) and prevented the occurrence of NADH hyperoxidation at 15 min following reoxygenation. The fact that most slices were better able to recover synaptic transmission when hyperoxidation was delayed (to 45 min post-reoxygenation) suggests that biochemical events occurring during reoxygenation in the presence of calcium are critical for causing subsequent neuronal injury. We observed in control experiments (i.e. hypoxia in the presence of calcium) that optimal neuronal recovery following hypoxia is associated with decreased NADH hyperoxidation. In addition, it was demonstrated that reducing the concentration of calcium in slices during hypoxia lessens the amount of NADH hyperoxidation observed following reoxygenation (Perez-Pinzon et al., 1998a). However, it is not clear whether the protective effect in the current study was primarily due to the delay in hyperoxidation or the removal of calcium. The differences in the ability of the slices to recover synaptic activity may have been partly due to aging induced changes in calcium regulation, such as decreased buffering of calcium by mitochondria, which may have reduced the benefits of calcium removal.
4.4. Conclusions
The results of this study demonstrate the critical role of NADH hyperoxidation in the pathophysiology of hypoxia/ischemia and its relationship with calcium signaling and neuronal survival. The results also implicate increased NADH hyperoxidation in aged rat slices as an intrinsic contributing factor in the increased vulnerability of aged individuals to pathological events such as hypoxia/ischemia and its clinical equivalent, stroke (Williams, 2001).
Acknowledgments
This study was funded by the American Heart Association, National Institutes of Aging (AG13165) and the Veterans Affairs Medical Center (Merit Review Grant). We are grateful to Professor George Somjen for his critical reading of the manuscript and his helpful suggestions.
Abbreviations
- ACSF
artificial cerebral spinal fluid
- ATP
adenosine 5′-triphosphate
- CA1
cornu ammonis, region 1
- CA3
cornu ammonis, region 3
- DG
dentate gyrus
- DNA
deoxyribonucleic acid
- FCCP
carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone
- fEPSP
field excitatory post-synaptic potential
- hsd
hypoxic spreading depression
- MPT
mitochondrial permeability transition
- MPTP
mitochondrial permeability transition pore
- NAD(P)H
nicotinamide adenine dinucleotide (phosphate)
- OGD
oxygen glucose deprivation
- PARP
poly-ADP ribose polymerase
- PDHC
pyruvate dehydrogenase complex
- PO2
tissue oxygen tension (mmHg)
- ROS
reactive oxygen species
- SP
stratum pyramidale
- SR
stratum radiatum
- TCA
tricarboxylic acid
Footnotes
Disclosure statement: There are no potential or actual conflicts of interest. Approval for the use of F344 rats in this study was granted by the Institutional Animal Care Committees at Duke University and the Durham Veterans Affairs Medical Center.
References
- Allen NJ, Karadottir R, Attwell D. A preferential role for glycolysis in preventing the anoxic depolarization of rat hippocampal area CA1 pyramidal cells. J Neurosci. 2005;25:848–859. doi: 10.1523/JNEUROSCI.4157-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amagasa M, Ogawa A, Yoshimoto T. Effects of calcium and calcium antagonists against deprivation of glucose and oxygen in guinea pig hippocampal slices. Brain Res. 1990;526(1):1–7. doi: 10.1016/0006-8993(90)90242-4. [DOI] [PubMed] [Google Scholar]
- Ames BN, Shigenaga MK, Hagen TM. Mitochondrial decay in aging. Biochim Biophys Acta. 1995;1271(1):165–170. doi: 10.1016/0925-4439(95)00024-x. [DOI] [PubMed] [Google Scholar]
- Bahar S, Fayuk D, Somjen GG, Aitken PG, Turner DA. Mitochondrial and intrinsic optical signals imaged during hypoxia and spreading depression in rat hippocampal slices. J Neurophysiol. 2000;84(1):311–324. doi: 10.1152/jn.2000.84.1.311. [DOI] [PubMed] [Google Scholar]
- Balestrino M, Aitken PG, Somjen GG. Spreading depression-like hypoxic depolarization in CA1 and fascia dentata of hippocampal slices: relationship to selective vulnerability. Brain Res. 1989;497(1):102–107. doi: 10.1016/0006-8993(89)90975-x. [DOI] [PubMed] [Google Scholar]
- Balestrino M, Rebaudo R, Lunardi G. Exogenous creatine delays anoxic depolarization and protects from hypoxic damage: dose–effect relationship. Brain Res. 1999;816(1):124–130. doi: 10.1016/s0006-8993(98)01131-7. [DOI] [PubMed] [Google Scholar]
- Balestrino M, Somjen GG. Chlorpromazine protects brain tissue in hypoxia by delaying spreading depression-mediated calcium influx. Brain Res. 1986;385(2):219–226. doi: 10.1016/0006-8993(86)91067-x. [DOI] [PubMed] [Google Scholar]
- Benjamin RC, Gill DM. ADP-ribosylation in mammalian cell ghosts Dependence of poly(ADP-ribose) synthesis on strand breakage in DNA. J Biol Chem. 1980;255(21):10493–10501. [PubMed] [Google Scholar]
- Bogaert YE, Rosenthal RE, Fiskum G. Postischemic inhibition of cerebral cortex pyruvate dehydrogenase. Free Radic Biol Med. 1994;16(6):811–820. doi: 10.1016/0891-5849(94)90197-x. [DOI] [PubMed] [Google Scholar]
- Bonita R. Epidemiology of stroke. Lancet. 1992;339(8789):342–344. doi: 10.1016/0140-6736(92)91658-u. comment. [DOI] [PubMed] [Google Scholar]
- Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowling AC, Mutisya EM, Walker LC, Price DL, Cork LC, Beal MF. Age-dependent impairment of mitochondrial function in primate brain. J Neurochem. 1993;60(5):1964–1967. doi: 10.1111/j.1471-4159.1993.tb13430.x. [DOI] [PubMed] [Google Scholar]
- Campbell LW, Hao SY, Thibault O, Blalock EM, Landfield PW. Aging changes in voltage-gated calcium currents in hippocampal CA1 neurons. J Neurosci. 1996;16(19):6286–6295. doi: 10.1523/JNEUROSCI.16-19-06286.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardell M, Koide T, Wieloch T. Pyruvate dehydrogenase activity in the rat cerebral cortex following cerebral ischemia. J Cereb Blood Flow Metab. 1989;9(3):350–357. doi: 10.1038/jcbfm.1989.53. [DOI] [PubMed] [Google Scholar]
- Chance B, Cohen P, Jobsis F, Schoener B. Intracellular oxidation–reduction states in vivo. Science. 1962;137(3529):499–508. doi: 10.1126/science.137.3529.499. [DOI] [PubMed] [Google Scholar]
- Chih CP, Feng ZC, Rosenthal M, Lutz PL, Sick TJ. Energy metabolism, ion homeostasis, and evoked potentials in anoxic turtle brain. Am J Physiol. 1989;257(4 Pt 2):R854–R860. doi: 10.1152/ajpregu.1989.257.4.R854. [DOI] [PubMed] [Google Scholar]
- David G, Barrett JN, Barrett EF. Evidence that mitochondria buffer physiological Ca2+ loads in lizard motor nerve terminals. J Physiol (London) 1998;509(Pt 1):59–65. doi: 10.1111/j.1469-7793.1998.059bo.x. see comment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276(4):2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- Dora E, Tanaka K, Greenberg JH, Gonatas NH, Reivich M. Kinetics of microcirculatory, NAD/NADH, and electrocorticographic changes in cat brain cortex during ischemia and recirculation. Ann Neurol. 1986;19(6):536–544. doi: 10.1002/ana.410190604. [DOI] [PubMed] [Google Scholar]
- Duckrow RB, LaManna JS, Rosenthal M. Disparate recovery of resting and stimulated oxidative metabolism following transient ischemia. Stroke. 1981;12(5):677–686. doi: 10.1161/01.str.12.5.677. [DOI] [PubMed] [Google Scholar]
- Fattoretti P, Bertoni-Freddari C, Caselli U, Paoloni R, Meier-Ruge W. Impaired succinic dehydrogenase activity of rat Purkinje cell mitochondria during aging. Mech Ageing Dev. 1998;101(1–2):175–182. doi: 10.1016/s0047-6374(97)00176-0. [DOI] [PubMed] [Google Scholar]
- Feng ZC, Sick TJ, Rosenthal M. Oxygen sensitivity of mitochondrial redox status and evoked potential recovery early during reperfusion in post-ischemic rat brain. Resuscitation. 1998;37(1):33–41. doi: 10.1016/s0300-9572(98)00031-8. [DOI] [PubMed] [Google Scholar]
- Ferrandiz ML, Martinez M, De Juan E, Diez A, Bustos G, Miquel J. Impairment of mitochondrial oxidative phosphorylation in the brain of aged mice. Brain Res. 1994;644(2):335. doi: 10.1016/0006-8993(94)91699-3. [DOI] [PubMed] [Google Scholar]
- Foster KA, Beaver CJ, Turner DA. Interaction between tissue oxygen tension and NADH imaging during synaptic stimulation and hypoxia in rat hippocampal slices. Neuroscience. 2005;132:645–657. doi: 10.1016/j.neuroscience.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Gaal JC, Smith KR, Pearson CK. Cellular euthanasia mediated by a nuclear enzyme: a central role for nuclear ADP-ribosylation in cellular metabolism. Trends Biochem Sci. 1987;12:129–130. [Google Scholar]
- Galeffi F, Sinnar S, Schwartz-Bloom RD. Diazepam promotes ATP recovery and prevents cytochrome c release in hippocampal slices after in vitro ischemia. J Neurochem. 2000;75(3):1242–1249. doi: 10.1046/j.1471-4159.2000.0751242.x. [DOI] [PubMed] [Google Scholar]
- Gerich FJ, Hepp S, Probst I, Muller M. Mitochondrial inhibition prior to oxygen-withdrawal facilitates the occurrence of hypoxia-induced spreading depression in rat hippocampal slices. J Neurophysiol. 2006;96(1):492–504. doi: 10.1152/jn.01015.2005. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, McStay GP, Clarke SJ. The permeability transition pore complex: another view. Biochimie. 2002;84(2–3):153–166. doi: 10.1016/s0300-9084(02)01375-5. [DOI] [PubMed] [Google Scholar]
- Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, Walter CA, Richardson A. Does oxidative damage to DNA increase with age? Proc Natl Acad Sci U S A. 2001;98(18):10469–10474. doi: 10.1073/pnas.171202698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. The free radical theory of aging. Antioxid Redox Signal. 2003;5(5):557–561. doi: 10.1089/152308603770310202. [DOI] [PubMed] [Google Scholar]
- Harmon HJ, Nank S, Floyd RA. Age-dependent changes in rat brain mitochondria of synaptic and non-synaptic origins. Mech Ageing Dev. 1987;38(2):167–177. doi: 10.1016/0047-6374(87)90076-5. [DOI] [PubMed] [Google Scholar]
- Hayakawa Y, Nemoto T, Iino M, Kasai H. Rapid Ca2+-dependent increase in oxygen consumption by mitochondria in single mammalian central neurons. Cell Calcium. 2005;37(4):359–370. doi: 10.1016/j.ceca.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Jing J, Aitken PG, Somjen GG. Lasting neuron depression induced byhigh potassium and its prevention by low calcium and NMDA receptor blockade. Brain Res. 1991;557(1–2):177–183. doi: 10.1016/0006-8993(91)90132-f. [DOI] [PubMed] [Google Scholar]
- Joubert F, Fales HM, Wen H, Combs CA, Balaban RS. NADH enzyme-dependent fluorescence recovery after photobleaching (ED-FRAP): applications to enzyme and mitochondrial reaction kinetics, in vitro. Biophys J. 2004;86(1 Pt 1):629–645. doi: 10.1016/S0006-3495(04)74141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan NO. The role of pyridine nucleotides in regulating cellular metabolism. Curr Top Cell Regul. 1985;26:371–381. doi: 10.1016/b978-0-12-152826-3.50035-8. [DOI] [PubMed] [Google Scholar]
- Kass IS, Lipton P. Protection of hippocampal slices from young rats against anoxic transmission damage is due to better maintenance of ATP. J Physiol (London) 1989;413:1–11. doi: 10.1113/jphysiol.1989.sp017638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki K, Czeh G, Somjen GG. Prolonged exposure to high potassium concentration results in irreversible loss of synaptic transmission in hippocampal tissue slices. Brain Res. 1988;457(2):322–329. doi: 10.1016/0006-8993(88)90702-0. [DOI] [PubMed] [Google Scholar]
- Klaidman LK, Mukherjee SK, Adams JD., Jr Oxidative changes in brain pyridine nucleotides and neuroprotection using nicotinamide. Biochim Biophys Acta. 2001;1525(1–2):136–148. doi: 10.1016/s0304-4165(00)00181-1. [DOI] [PubMed] [Google Scholar]
- Kwong LK, Sohal RS. Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Arch Biochem Biophys. 2000;373(1):16–22. doi: 10.1006/abbi.1999.1495. [DOI] [PubMed] [Google Scholar]
- LeBel CP, Bondy SC. Oxidative damage and cerebral aging. Prog Neurobiol. 1992;38(6):601–609. doi: 10.1016/0301-0082(92)90043-e. [DOI] [PubMed] [Google Scholar]
- Lee KS, Lowenkopf T. Endogenous adenosine delays the onset of hypoxic depolarization in the rat hippocampus in vitro via an action at A1 receptors. Brain Res. 1993;609(1–2):313–315. doi: 10.1016/0006-8993(93)90888-t. [DOI] [PubMed] [Google Scholar]
- Leslie SW, Chandler LJ, Barr EM, Farrar RP. Reduced calcium uptake by rat brain mitochondria and synaptosomes in response to aging. Brain Res. 1985;329(1–2):177–183. doi: 10.1016/0006-8993(85)90523-2. [DOI] [PubMed] [Google Scholar]
- Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T, Gonzalez-Zulueta M, Nikolich K, Wieloch T. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat Med. 2003;9(8):1062–1068. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- Murchison D, Zawieja DC, Griffith WH. Reduced mitochondrial buffering of voltage-gated calcium influx in aged rat basal forebrain neurons. Cell Calcium. 2004;36(1):61–75. doi: 10.1016/j.ceca.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Nakae D, Akai H, Kishida H, Kusuoka O, Tsutsumi M, Konishi Y. Age and organ dependent spontaneous generation of nuclear 8-hydroxydeoxyguanosine in male Fischer 344 rats. Lab Invest. 2000;80(2):249–261. doi: 10.1038/labinvest.3780028. [DOI] [PubMed] [Google Scholar]
- Narasimhan P, Fujimara M, Noshita N, Chan PH. Role of superoxide in poly(ADP-ribose) polymerase upregulation after transient cerebral ischemia. Brain Res Mol Brain Res. 2003;113(1–2):28–36. doi: 10.1016/s0169-328x(03)00062-7. [DOI] [PubMed] [Google Scholar]
- Paschen W, Sato M, Pawlik G, Umbach C, Heiss WD. Neurologic deficit, blood flow and biochemical sequelae of reversible focal cerebral ischemia in cats. J Neurol Sci. 1985;68(2–3):119–134. doi: 10.1016/0022-510x(85)90094-2. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Mumford PL, Carranza V, Sick TJ. Calcium influx from the extracellular space promotes NADH hyperoxidation and electrical dysfunction after anoxia in hippocampal slices. J Cereb Blood Flow Metab. 1998a;18(2):215–221. doi: 10.1097/00004647-199802000-00013. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Mumford PL, Rosenthal M, Sick TJ. Antioxidants, mitochondrial hyperoxidation and electrical recovery after anoxia in hippocampal slices. Brain Res. 1997;754(1–2):163–170. doi: 10.1016/s0006-8993(97)00066-8. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Mumford PL, Sick TJ. Prolonged anoxic depolarization exacerbates NADH hyperoxidation and promotes poor electrical recovery after anoxia in hippocampal slices. Brain Res. 1998b;786(1–2):165–170. doi: 10.1016/s0006-8993(97)01438-8. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA, Xu GP, Born J, Lorenzo J, Busto R, Rosenthal M, Sick TJ. Cytochrome C is released from mitochondria into the cytosol after cerebral anoxia or ischemia. J Cereb Blood Flow Metab. 1999;19(1):39–43. doi: 10.1097/00004647-199901000-00004. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Landfield PW. Aging-related prolongation of calcium spike duration in rat hippocampal slice neurons. Brain Res. 1990;508(1):1–6. doi: 10.1016/0006-8993(90)91109-t. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Levy DE, Duffy TE. Regional cerebral blood flow and glucose metabolism following transient forebrain ischemia. Ann Neurol. 1982;11(5):499–502. doi: 10.1002/ana.410110510. [DOI] [PubMed] [Google Scholar]
- Roberts EL, Jr, Chih CP. The pH buffering capacity of hippocampal slices from young adult and aged rats. Brain Res. 1998;779(1–2):271–275. doi: 10.1016/s0006-8993(97)01120-7. [DOI] [PubMed] [Google Scholar]
- Roberts EL, Jr, Rosenthal M, Sick TJ. Age-related modifications of potassium homeostasis and synaptic transmission during and after anoxia in rat hippocampal slices. Brain Res. 1990;514(1):111–118. doi: 10.1016/0006-8993(90)90441-d. [DOI] [PubMed] [Google Scholar]
- Roberts EL, Jr, Sick TJ. Calcium-sensitive recovery of extracellular potassium and synaptic transmission in rat hippocampal slices exposed to brief anoxia. Brain Res. 1988;456(1):113–119. doi: 10.1016/0006-8993(88)90352-6. [DOI] [PubMed] [Google Scholar]
- Roberts ELJ, Chih CP. Age-related alterations in energy metabolism contribute to the increased vulnerability of the aging brain to anoxic damage. Brain Res. 1995;678(1–2):83–90. doi: 10.1016/0006-8993(95)00168-p. [DOI] [PubMed] [Google Scholar]
- Roberts LJ, II, Montine TJ, Markesbery WR, Tapper AR, Hardyr P, Chemtob S, Dettbarn WD, Morrow JD. Formation of isoprostane-like compounds (neuroprostanes) in vivo from docosahexaenoic acid. J Biol Chem. 1998;273(22):13605–13612. doi: 10.1074/jbc.273.22.13605. [DOI] [PubMed] [Google Scholar]
- Rosenthal M, Feng ZC, Raffin CN, Harrison M, Sick TJ. Mitochondrial hyperoxidation signals residual intracellular dysfunction after global ischemia in rat neocortex. J Cereb Blood Flow Metab. 1995;15(4):655–665. doi: 10.1038/jcbfm.1995.81. [DOI] [PubMed] [Google Scholar]
- Rosenthal M, Mumford PL, Sick TJ, Perez-Pinzon MA. Mitochondrial hyperoxidation after cerebral anoxia/ischemia Epiphenomenon or precursor to residual damage? Adv Exp Med Biol. 1997;428:189–195. [PubMed] [Google Scholar]
- Rosenthal M, Sick TJ. Glycolytic and oxidative metabolic contributions to potassium ion transport in rat cerebral cortex. Can J Physiol Pharmacol. 1992;70(Suppl. l):S165–S169. doi: 10.1139/y92-258. [DOI] [PubMed] [Google Scholar]
- Siesjo BK. Cell damage in the brain: a speculative synthesis. J Cereb Blood Flow Metab. 1981;1(2):155–185. doi: 10.1038/jcbfm.1981.18. [DOI] [PubMed] [Google Scholar]
- Siqueira IR, Cimarosti H, Fochesatto C, Salbego C, Netto CA. Age-related susceptibility to oxygen and glucose deprivation damage in rat hippocampal slices. Brain Res. 2004;1025(1–2):226–230. doi: 10.1016/j.brainres.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Skaper SD. Poly(ADP-ribose) polymerase-1 in acute neuronal death and inflammation: a strategy for neuroprotection. Ann N Y Acad Sci. 2003;993:217–228. doi: 10.1111/j.1749-6632.2003.tb07532.x. [DOI] [PubMed] [Google Scholar]
- Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci U S A. 1991;88(23):10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Dora E, Greenberg JH, Reivich M. Cerebral glucose metabolism during the recovery period after ischemia—its relationship to NADH-fluorescence, blood flow EcoG and histology. Stroke. 1986;17(5):994–1004. doi: 10.1161/01.str.17.5.994. [DOI] [PubMed] [Google Scholar]
- Tombaugh GC. Mild acidosis delays hypoxic spreading depression and improves neuronal recovery in hippocampal slices. J Neurosci. 1994;14(9):5635–5643. doi: 10.1523/JNEUROSCI.14-09-05635.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson GB, Lanthorn TH. Phenytoin delays ischemic depolarization, but cannot block its long-term consequences, in the rat hippocampal slice. Neuropharmacology. 1995;34(5):553–558. doi: 10.1016/0028-3908(95)00005-q. [DOI] [PubMed] [Google Scholar]
- Welsh FA, Marcy VR, Sims RE. NADH fluorescence and regional energy metabolites during focal ischemia and reperfusion of rat brain. J Cereb Blood Flow Metab. 1991;11(3):459–465. doi: 10.1038/jcbfm.1991.88. [DOI] [PubMed] [Google Scholar]
- Welsh FA, O'Connor MJ, Marcy VR, Spatacco AJ, Johns RL. Factors limiting regeneration of ATP following temporary ischemia in cat brain. Stroke. 1982;13(2):234–242. doi: 10.1161/01.str.13.2.234. [DOI] [PubMed] [Google Scholar]
- Williams GR. Incidence and characteristics of total stroke in the United States. BMC Neurol. 2001;1:2. doi: 10.1186/1471-2377-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabuki M, Inai Y, Yoshioka T, Hamazaki K, Yasuda T, Inoue M, Utsumi K. Oxygen dependent fragmentation of cellular DNA by nitric oxide. Free Radic Res. 1997;26(3):245–255. doi: 10.3109/10715769709097803. [DOI] [PubMed] [Google Scholar]
- Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci U S A. 1996;93(7):2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JN, Aitken PG, Somjen GG. Calcium, magnesium, and long-term recovery from hypoxia in hippocampal tissue slices. Brain Res. 1991;548(1–2):343–345. doi: 10.1016/0006-8993(91)91146-r. [DOI] [PubMed] [Google Scholar]
- Zarchin N, Meilin S, Rifkind J, Mayevsky A. Effect of aging on brain energy-metabolism. Comp Biochem Physiol A: Mol Integr Physiol. 2002;132(1):117–120. doi: 10.1016/s1095-6433(01)00537-2. [DOI] [PubMed] [Google Scholar]