

Abstract
In order to directly compare the reactivity of positively charged carbon-centered aromatic σ-radicals toward methanol in solution and in the gas phase, the 2-, 3-, and 4-dehydropyridinium cations (distonic isomers of the pyridine radical cation) were generated by ultraviolet photolysis of the corresponding iodo-precursors in a mixture of water and methanol at varying pH. The reaction mixtures were analyzed by using liquid chromatography/mass spectrometry. Hydrogen atom abstraction was the only reaction observed for the 3- and 4-dehydropyridinium cations (and –pyridines) in solution. This also was the major reaction observed earlier in the gas phase. Depending on the pH, the hydrogen atom can be abstracted from different molecules (i.e., methanol or water) and from different sites (in methanol) by the 3- and 4-dehydropyridinium cations/-pyridines in solution. In the pH range of 1 – 4, the methyl group of methanol is the main hydrogen atom donor site for both 3- and 4-dehydropyridinium cations (just like in the gas phase). At higher pH, the hydroxyl groups of water and methanol also act as hydrogen atom donors. This finding is rationalized by a greater abundance of the unprotonated radicals that preferentially abstract hydrogen atoms from the polar hydroxyl groups. The percentage yield of hydrogen atom abstraction by these radicals was found to increase with lowering the pH in the pH range of 1.0-3.2. This pH effect is rationalized by polar effects: the lower the pH, the greater the fraction of protonated (more polar) radicals in the solution. This finding is consistent with previous results obtained in the gas phase and suggests that gas-phase studies can be used to predict solution reactivity, but only as long as the same reactive species is studied in both experiments. This was found not to be the case for the 2-iodopyridinium cation. Photolysis of this precursor in solution resulted in the formation of two major addition products, 2-hydroxy- and 2-methoxypyridinium cations, in addition to the hydrogen atom abstraction product. These addition products were not observed in the earlier gas-phase studies on 2-dehydropyridinium cation. Their observation in solution is explained by the formation of another reactive intermediate, the 2-pyridyl cation, upon photolysis of 2-iodopyridinium cation (and 2-iodopyridine). The same intermediate was observed in the gas phase but it was removed before examining the reactions of the desired radical, 2-dehydropyridinium cation (which cannot be done in solution).
Introduction
Chemical reactivity of radicals toward simple hydrogen atom donors has been of great interest for decades.1-7 However, the factors that control the efficiency of hydrogen atom abstraction are still not well understood, in spite of the importance of this knowledge on many research areas, such as radical-induced cleavage of nucleic acids initiated by hydrogen atom abstraction from the sugar-phosphate backbone.8,9 For example, the enediyne anti-tumor antibiotics, such as neocarzinostatin10,11 and calicheamicin,12 undergo rearrangement and aromatization to form carbon-centered σ,σ-biradical intermediates, which abstract hydrogen atoms from sugar-moieties in DNA and lead to double-strand scission. Although these enediyne compounds are potent drugs, they are also highly toxic to normal cells.13,14 Hence, a better understanding of the factors that control the reactivity and selectivity of σ-radicals and σ,σ-biradicals toward hydrogen atom abstraction could facilitate the rational design of antitumor drugs.
Substituent effects on the reactivity of phenyl radicals toward various hydrogen atom donors have been studied extensively.15,16 For example, electron-withdrawing groups in phenyl radicals (that increase the electron affinity (EA) of the radical site) have been found to increase the rate of hydrogen atom abstraction by these radicals from toluene, cyclohexane and acetone.15 This effect was rationalized by the ability of the substituents to polarize the transition state and hence decrease the activation energy of the reactions.17 Polar effects are known to be a key factor in controlling the reactivities of phenyl radicals both in solution15,16,18 and in the gas phase.19-21 For electrophilic radicals, such as phenyl radicals, the transition state of a radical reaction can be represented by the resonance structures shown in Scheme 1.1,17,22 The barrier heights of these reactions can be lowered by increasing the electron affinity (EA) of the radical (R• in Scheme 1), which increases its ability to polarize the transition state.1,23
Scheme 1.

Gas-phase experiments allow the delineation of structure/reactivity relationships for radicals in a solvent-free environment. The reactivity of aromatic monoradicals,19-21,24,25 biradicals,26-28 and triradicals29-31 have been studied in the gas phase. Based on these studies, the hydrogen atom abstraction ability of basic aromatic carbon-centered σ-radicals in solution may be tuned by controlling the pH of the system since protonation increases the EA and hence the reactivity of these radicals.19,25,32,33 However, to the best of our knowledge, the effect of pH on the reactivity of monoradicals in solution has not been studied. On the other hand, a related species, the 2,5-didehydropyridine biradical, has been reported to abstract hydrogen atoms more readily in the presence of a protic acid (HBF4; solvent and hydrogen atom donor: diisopropyl ether).34 This observation was attributed to a smaller singlet-triplet gap (S-T gap: the energy difference between the singlet ground state and the lowest energy triplet state) of 2,5-didehydropyridinium cation than 2,5-didehydropyridine. However, polar effects would provide an alternative (or additional) explanation.
In order to learn more about the effects of pH on the reactivity of aromatic carbon-centered σ-radicals, the reactions of isomeric 2-, 3-, and 4-dehydropyridines with methanol were studied in acidic aqueous solution. These radicals were selected since their reactivity has been already examined in the gas phase.19,21,32,33
Experimental
3-Iodopyridine (98%), 4-iodopyridine (97%), 2-iodopyridine (98%), 2-hydroxypyridine (97%), 2-methoxypyridine (98%), methanol (99.9%), and partially labeled methanol (CD3OH, 99%) were purchased from Sigma-Aldrich and used as received. Photolysis experiments were carried out in a Rayonet chamber reactor (Model RPR-200, Southern New England Ultraviolet Company) equipped with 16 UV lamps (300 nm) and a nitrogen manifold, which was used to cool the reaction chamber. Iodopyridine (10 mM) was dissolved in a mixture of methanol and triply distilled water (1:1, v/v). The pH of the solutions was adjusted by using either hydrochloric acid/potassium chloride or phosphoric acid/monosodium phosphate solutions. UV irradiation time was 30 minutes. Cold nitrogen gas from vaporization of liquid nitrogen was used to cool the reaction chamber in order to maintain the temperature at room temperature. In all cases, “dark control” experiments showed no reactivity in the absence of light. Photolysis experiments were also performed in the presence of air and under an Ar atmosphere. Because no difference was found, all experiments were performed in the presence of air. The concentrations of unreacted iodopyridinium cation, and its reaction products, were determined by using liquid chromatography/mass spectrometry.
The HPLC system employed was a Surveyor Plus instrument (Thermo Scientific) equipped with an autosampler and a photodiode array detector. The HPLC was coupled with a linear quadrupole ion trap mass spectrometer (LQIT; specifically, Thermo Scientific LTQ), which was equipped with an atmospheric pressure chemical ionization (APCI) source. The reaction mixtures were diluted 100-fold in water (HPLC grade, Sigma) and spiked with d5-pyridine (purity, 99.5%; from Sigma-Aldrich) used as the internal standard. Typically, 3 μL of the diluted and spiked reaction mixture was injected into the LC-MS for analysis. The column used was an Aquasil C18 column (dimensions 100×2.1 mm, particle size 5 μm, Thermo Scientific). The mobile phase (flow rate of 0.2 mL/min) was a mixture of two solutions: solution A (water, 0.1% formic acid) and solution B (methanol, 0.1% formic acid). The detailed elution conditions are given in Table 1. The APCI heater temperature was 450°C, and the MS scan range was m/z 65-500.
Table 1.
Gradient Elutiona Timetable for HPLC/MS Analysis
| Time (min) | A (%) | B (%) |
|---|---|---|
| 0.0 | 90 | 10 |
| 7.0 | 10 | 90 |
| 7.9 | 10 | 90 |
| 8.0 | 90 | 10 |
| 10.0 | 90 | 10 |
A: Water containing 0.1% (v/v) formic acid; B: Methanol containing 0.1% (v/v) formic acid.
Each reactant and reaction product was quantified by using d5-pyridine as an internal standard and a calibration curve obtained by using five concentrations of the analyte (spiked with the same amount of d5-pyridine) ranging from 30 to 300 pmol. The ratio of the HPLC peak area of d5-pyridine and the HPLC peak area of the respective standard solution was plotted against the concentration of the standard solution to generate the calibration curve. The concentration of each compound was then determined by using the ratio of the HPLC peak area of d5-pyridine to the HPLC peak area of the analyte and the calibration curve.
The identities of reaction products were verified by retention time and mass spectrometric fragmentation patterns created by collisional activation. Collision-activated dissociation (CAD) was performed by resonance excitation for 30 ms. Collisions with the helium bath gas converted the ion’s kinetic energy into internal energy. The hot precursor ions underwent unimolecular dissociation to form fragment ions.
Geometries for the neutral and positively-charged aryl radicals were computed by using density functional theory (DFT) with the correlation-consistent polarized valence-double-ζ (cc-pVDZ35) basis set. The DFT calculations were of two types. Both used the gradient-corrected exchange functional of Becke,36 which was combined either with the gradient-corrected correlation functional of Lee, Yang and Parr37 (BLYP) or that of Perdew et al.38 (BPW91). All DFT geometries were verified to be local minima by computation of analytic vibrational frequencies. DFT calculations for doublet states employed an unrestricted formalism and total spin expectation values for Slater determinants formed from the optimized Kohn-Sham orbitals did not exceed 0.76.
Atomic charges were calculated at the BPW91/cc-pVDZ level of theory by using the BPW91/cc-pVDZ optimized geometries and the CHELPG procedure.39 For these calculations, the atomic charges were fitted in order to reproduce the overall molecular dipole moment.
In order to compute vertical electron affinities (EA) for the neutral and positively charged aryl radicals, single-point calculations (BLYP/aug-cc-pVDZ40) using the BLYP/cc-pVDZ optimized geometry for each aryl radical were also carried out for the states that are produced when a single electron is added to the nonbonding σ-orbital of each molecule.41 For the positively charged aryl radicals studied here, these calculations involve (zwitterionic) singlet states.42 The vertical electron affinities of the aryl radicals were computed as [E0(monoradical; doublet state)] − [E0(monoradical + electron; singlet state)]. Note that because these are vertical electron affinities, zero-point vibrational energies (ZPVEs) and 298K thermal contributions to the enthalpy are not included.
Proton affinities were calculated at the RHF-UCCSD(T)/cc-pVTZ level of theory by using the BPW91/cc-pVDZ optimized geometries. The proton affinities include zero-point vibrational energies and 198 K thermal contributions calculated at the BPW91/cc-pVDZ level of theory.
All DFT and coupled-cluster calculations were carried out with Gaussian 0343 and Mopro44 electronic structure program suite.
Results and Discussion
The 2-, 3-, and 4-dehydropyridinium cations (in equilibrium with the 2-, 3- and 4-dehydropyridines; Table 2) were generated at varying pH in a mixture of water and methanol. The ratio of the protonated to unprotonated 2-, 3-, and 4-iodopyridine molecules at the different pH conditions used in this work is given in Table 2. After ultraviolet photolysis for 30 minutes, the reaction mixtures were analyzed by using liquid chromatography (Table 1) coupled with an ion trap mass spectrometer. For the 3- and 4-dehydropyridinium cations (and -pyridines), only hydrogen atom abstraction products were observed. The methyl group of methanol was found to be the main hydrogen atom donor in the pH range of 1 - 4, as discussed below. The percentage yields of hydrogen atom abstraction were found to increase with a decrease in pH in the range of 1 - 3 (Figure 1). For the 3- and 4-dehydropyridinium cations, the trend was found to be linear (R2 = 0.9946 and 0.8520, respectively). These findings are in agreement with previous gas-phase studies that indicated enhanced reactivity when the EA of the radical site increases.19,21,33 For the 2-dehydropyridinium cation, the percentage yields of hydrogen atom abstraction were found to be unaffected by pH. This is because of formation of a second reaction intermediate, the 2-pyridyl cation, which complicates the experiment in solution. The results are shown in Tables 3-7 and discussed in detail below.
Table 2.
Ratio of Protonated to Unprotonated 2-, 3-, and 4-Iodopyridine Molecules at Different pH in a Mixture of Methanol and Water Obtained Using the Henderson-Hasselbach equation.
 pKa 2.9a pKa 2.9a
|
 pKa 2.3a pKa 2.3a
|
 pKa 1.1a pKa 1.1a
|
|
|---|---|---|---|
| Ratio of protonated to unprotonated molecules | pH 1.1b | pH 1.0b | pH 1.0b |
| 63 | 20 | 1.3 | |
| pH 2.0b | pH 2.3b | pH 1.7b | |
| 7.9 | 1 | 0.25 | |
| pH 2.8b | pH 2.8b | pH 2.2b | |
| 1.3 | 0.32 | 0.079 | |
| pH 3.0b | pH 3.1b | pH 2.6b | |
| 0.79 | 0.16 | 0.032 | |
| pH 3.7c | pH 3.5c | pH 3.5c | |
| 0.16 | 0.063 | 0.004 | |
| pH 5.5c | pH 5.6c | pH 5.6c | |
| 0.0025 | 0.0005 | 0.000032 |
The pKa values were measured previously in a mixture of water and ethanol (1:1, v/v). See reference 37.
The pH of the solution was adjusted by using a solution containing hydrochloric acid and potassium chloride.
The pH of the solution was adjusted by using a solution containing phosphoric acid and monosodium phosphate.
Figure 1.

Yields of the hydrogen atom abstraction product for 2-dehydropyridinium cation (
 ), 3-dehydropyridinium cation (
), 3-dehydropyridinium cation (
 ) and 4-dehydropyridinium cation (
) and 4-dehydropyridinium cation (
 ) as a function of pH. Error bars reflect standard deviations.
) as a function of pH. Error bars reflect standard deviations.
Table 3.
Branching Ratios for Deuterium and Hydrogen Atom Abstraction at Varying pH in a Mixture of Partially Labeled Methanol (CD3OH) and Water
 |
 |
 |
||||
|---|---|---|---|---|---|---|
| Branching ratios of reaction products | pH 1.1a | pH 1.0a | pH 1.0a | |||
| D abs | 99% | D abs | 97% | D abs | 65% | |
| H abs | 1% | H abs | 3% | H abs | 35% | |
| pH 2.0a | pH 2.3a | pH 1.7a | ||||
| D abs | 98% | D abs | 93% | D abs | 29% | |
| H abs | 2% | H abs | 7% | H abs | 71% | |
| pH 2.8a | pH 2.8a | pH 2.2a | ||||
| D abs | 99% | D abs | 89% | D abs | 26% | |
| H abs | 1% | H abs | 11% | H abs | 74% | |
| pH 3.0a | pH 3.1a | pH 2.6a | ||||
| D abs | 98% | D abs | 79% | D abs | 25% | |
| H abs | 2% | H abs | 21% | H abs | 75% | |
| pH 3.7b | pH 3.5b | pH 3.5b | ||||
| D abs | 97% | D abs | 71% | D abs | 20% | |
| H abs | 3% | H abs | 29% | H abs | 80% | |
| pH 5.5b | pH 5.6b | pH 5.6b | ||||
| D abs | 92% | D abs | 50% | D abs | 25% | |
| H abs | 8% | H abs | 50% | H abs | 75% | |
The pH of the solution was adjusted by using a solution containing a mixture of hydrochloric acid and potassium chloride.
The pH of the solution was adjusted by using a mixture containing phosphoric acid and monosodium phosphate.
Table 7.
Total Yields (Percent of Starting Material Converted into Products) and Product Branching Ratios in Methanol/Water After Ultraviolet Photolysis for 30 minutes
|
|
|
||||
|---|---|---|---|---|---|---|
| EA of radical sitea(eV) | 0.99 | 0.93 | 0.32 | |||
| PA (kcal/mol)b | 219.6 | 218.5 | 212.2 | |||
| EA (eV) of radical site after protonationa | 5.84 | 6.08 | 6.59 | |||
| Estimated pKacof the iodopyridine precursor | 2.9 | 2.3 | 1.1 | |||
| Branching ratios of reaction products and reaction yields (%) | pH 1.0d | pH 1.1 | pH 1.0 | |||
| H abs. | 100% | H abs. | 100% | H abs. | 33% | |
| Hydroxyl abs. | 36% | |||||
| Methoxyl abs. | 31% | |||||
| Yield | 44% | Yield | 43% | Yield | 23% | |
| pH 1.7 | pH 1.7 | pH 1.6 | ||||
| H abs. | 100% | H abs. | 100% | H abs. | 28% | |
| Hydroxyl abs. | 39% | |||||
| Methoxyl abs. | 33% | |||||
| Yield | 40% | Yield | 38% | Yield | 19% | |
| pH 2.2 | pH 2.4 | pH 2.2 | ||||
| H abs. | 100% | H abs. | 100% | H abs. | 31% | |
| Hydroxyl abs. | 34% | |||||
| Methoxyl abs. | 35% | |||||
| Yield | 40% | Yield | 31% | Yield | 22% | |
| pH 3.1 | pH 3.1 | pH 2.7 | ||||
| H abs. | 100% | H abs. | 100% | H abs. | 29% | |
| Hydroxyl abs. | 36% | |||||
| Methoxyl abs. | 35% | |||||
| Yield | 38% | Yield | 25% | Yield | 19% | |
Calculated at the (U)BLYP/aug-cc-pVDZ//(U)BLYP/cc-pVDZ level of theory.
Calculated at the RHF-UCCSD(T)/cc-pVTZ//UBPW91/cc-pVDZ level of theory by using an isodesmic reaction involving pyridine as the reference base; corrected for zero-point vibrational energy differences at 298 K by using the (unscaled) UBPW91/cc-pVDZ frequencies.
The pKa values were reported previously as measured in a mixture of water and ethanol (1:1, v/v). See Reference 37.
The pH of the reaction mixtures was adjusted by using a solution containing a mixture of HCl and KCl.
3- and 4-Dehydropyridines
When 3- and 4-iodopyridines were dissolved in acidic mixtures of methanol and water and exposed to UV irradiation for 30 minutes, homolytic cleavages of the carbon-iodine bonds yielded (charged and uncharged) monoradicals that abstracted a hydrogen atom, forming pyridine. This was the exclusive product observed (Tables 4, 5 and 7). Pyridine was not formed in the absence of UV irradiation under identical conditions. The reactions were not affected by the presence or absence of oxygen in the solutions. Similar results have been reported earlier for these two iodopyridines in pure methanol.45 Further experiments were carried out to explore the details of the reactions.
Table 4.
Results (Errors Are Standard Deviations) Obtained Upon HPLC/MS Analysis of the Reaction Mixtures of 3-Iodopyridine in Methanol/Water
| Unreacted 3-Iodopyridine | Pyridine | Sum of Pyridine and 3-Iodopyridine | ||
|---|---|---|---|---|
| pH 1.1 | Dark Control | (10.5 ± 0.9) mM | (0.10 ± 0.01) mM | - |
| Sample | (5.9 ± 0.1) mM | (4.5 ± 0.3) mM | (10.4 ± 0.3) mM | |
| Yield | (43 ± 2)% | |||
| pH 1.7 | Dark Control | (10.7 ± 0.8) mM | (0.16 ± 0.01) mM | - |
| Sample | (6.7 ± 0.1) mM | (4.1 ± 0.4) mM | (10.8 ± 0.4) mM | |
| Yield | (38 ± 2)% | |||
| pH 2.4 | Dark Control | (10.7 ± 0.5) mM | (0.09 ± 0.02) mM | - |
| Sample | (7.3 ± 0.6) mM | (3.2 ± 0.2) mM | (10.5 ± 0.6) mM | |
| Yield | (31 ± 2)% | |||
| pH 3.1 | Dark Control | (10.5 ± 0.9) mM | 0.14 ± 0.01 mM | - |
| Sample | (7.5 ± 0.6) mM | (2.5 ± 0.1) mM | (10.1 ± 0.6) mM | |
| Yield | (25 ±1)% |
Table 5.
Results (Errors Are Standard Deviations) Obtained Upon HPLC/MS Analysis of the Reaction Mixtures of 4-Iodopyridine in Methanol/Water
| Unreacted 4-Iodopyridine | Pyridine | Sum of Pyridine and 4-Iodopyridine | ||
|---|---|---|---|---|
| pH 1.0 | Dark Control | (11.0 ± 0.4) mM | 0.0 mM | - |
| Sample | (5.7 ± 0.1) mM | (4.5 ± 0.1) mM | (10.2 ± 0.1) mM | |
| Yield | (44 ± 1)% | |||
| pH 1.7 | Dark Control | (10.3 ± 0.3) mM | 0.0 mM | - |
| Sample | (6.5 ± 0.8) mM | (4.4 ± 0.3) mM | (10.9 ± 0.8) mM | |
| Yield | (40 ± 2)% | |||
| pH 2.2 | Dark Control | (9.8 ± 0.3) mM | 0.0 mM | - |
| Sample | (6.2 ± 0.2) mM | (4.2 ± 0.1) mM | (10.4 ± 0.2) mM | |
| Yield | (40 ± 1)% | |||
| pH 3.1 | Dark Control | (10.2 ± 0.1) mM | 0.0 mM | - |
| Sample | (6.6 ± 0.5) mM | (4.0 ± 0.1) mM | (10.6 ±0.5) mM | |
| Yield | (38 ± 2)% |
Hydrogen atom donor sites in a mixture of MeOH and H2O
To identify the hydrogen atom donor site(s) in the methanol/water mixture, a photolysis experiment was performed in partially labeled methanol (CD3OH) and water (1:1, v/v) at pH 5.5 for 3- and 4-iodopyridine. The pH of the solution was adjusted by adding a mixture of phosphoric acid and monosodium phosphate. After UV photolysis for 30 minutes, both 3- and 4-deuteropyridine and pyridine were observed in the reaction mixture. For 4-dehydropyridine, the branching ratio of deuteron abstraction was 92%, while for 3-dehydropyridine, it was 50% (Table 3). This finding indicates that the methyl group of methanol acts as the main atom donor site for 4-dehydropyridine. However, for 3-dehydropyridine, both the methyl group of methanol and the hydroxyl groups of methanol and/or water act as atom donor sites under these conditions. While the hydroxyl groups in phosphoric acid and monosodium phosphate could potentially also act as hydrogen atom donor sites, their concentrations are about six orders of magnitude lower than those of water and methanol, and it is therefore unlikely that they play a significant role.
In order to learn more, photolysis experiments of 3-iodopyridine were performed in pure, partially labeled methanol (CD3OH) and in a mixture of methanol and D2O (1:1, v/v). In labeled methanol (CD3OH), both deuteron (70%) and hydrogen atom abstraction products (30%) were observed. In the mixture of methanol and D2O (1:1, v/v), both deuteron (30%) and hydrogen atom abstraction products (70%) were detected. These results demonstrate that the hydroxyl groups of both methanol and water also act as atom donor sites for 3-dehydropyridine.
The different selectivies observed for the 3- and 4-dehydropyridinium cations may be explained by the presence of different amounts of the protonated and unprotonated radicals. These radicals are expected to show quite different reactivities due to their different EA values. The calculated ((U)BLYP/aug-cc-pVDZ//(U)BLYP/cc-pVDZ) EAs of 3- and 4-dehydropyridines are 0.93 eV and 0.99 eV, respectively. In sharp contrast, the calculated (same level of theory) EAs of the 3- and 4-dehydropyridinium cations are 6.08 eV and 5.84 eV, respectively. The latter values are substantially greater than those of their neutral precursors, and higher reactivity is expected (and was observed, as discussed below) for the 3- and 4-dehydropyridinium cations. Further, the protonated 3-isomer is expected to be more reactive than the protonated 4-isomer, while the opposite should be observed for the unprotonated radicals.
To examine the influence of pH on the hydrogen atom donor site, photolysis of 3-iodopyridine and 4-iodopyridine was examined (in separate experiments) in a mixture (1:1, v/v) of water and partially labeled methanol (CD3OH). The pH of the mixture was adjusted by using hydrochloric acid/potassium chloride or phosphoric acid/monosodium phosphate. The pKa values of 3- and 4-iodopyridines in aqueous solution containing 50% ethanol have been reported to be 2.3 and 2.9, respectively.46 The pKa values of 3- and 4-iodopyridines in aqueous solution containing 50% methanol are likely to be similar to these values. When the pH of the reaction mixture is lower than their pKa values, the majority of 3- and 4-iodopyridine molecules are protonated (Table 2), and upon photolysis should predominantly yield 3- and 4-dehydropyridinum cations (Scheme 2). When the pH is higher than the pKa values, the majority of 3- and 4-iodopyridine molecules are not protonated (Table 2) and, upon photolysis, yield 3- and 4-dehydropyridines (Scheme 2).
Scheme 2.

For 3-dehydropyridine, both deuteron and hydrogen atom abstraction products were observed, and the branching ratio of hydrogen atom abstraction was found to increase with pH (Table 3). Hence, the hydrogen atom donor site is related to the pH of the reaction mixture, and as the pH increases (less of the radicals are protonated), more hydrogen atoms are abstracted from the hydroxyl groups of water and/or methanol. However, in the pH range of 1.0-3.5, mainly the deuteron atom abstraction product was observed for 3-iodopyridine (79-97%; Table 3). Similarly, deuteron atom abstraction dominated (>98%) for 4-dehydropyridine in this pH range. The observation of a greater selectivity toward the thermodynamically favored hydrogen atom donor site (methyl group in methanol) for 4-dehydropyridine than for 3-dehydropyridine under these acidic conditions may be explained by the lower reactivity of the protonated 4-isomer due to its lower EA. Previous studies of positively charged phenyl radicals in the gas phase have shown that the selectivity of hydrogen atom abstraction from ethanol is related to the EAs of the radicals.47 Radicals with a lower EA (indicating lower reactivity) tend to attack the CH2-group rather than the CH3-group of ethanol because the CH2-group has a lower homolytic C-H bond dissociation energy. Indeed, in the gas phase, the hydrogen atom abstraction efficiency from methanol is 1.7 % and 0.4 % for the protonated 3- and 4-isomers, respectively.33 The lowered selectivity of both radicals upon increase in pH suggests that the unprotonated radicals have the opposite polarity from the protonated radicals, and act as nucleophilic radicals – hence, preferring abstraction of a hydrogen atom bound to an electron-withdrawing oxygen atom rather than the α-carbon atom. The calculated (UBPW91/cc-pVDZ//UBPW91/cc-pVDZ; Figure 2) heavy-atom charges for the 3- and 4-isomers indicate a partial negative charge on the radical site, which supports the above hypothesis of them behaving like nucleophilic radicals. In sharp contrast, the 2-isomer has a partial positive charge on the radical site and hence should behave like the protonated radicals (i.e., as an electrophilic radical).
Figure 2.
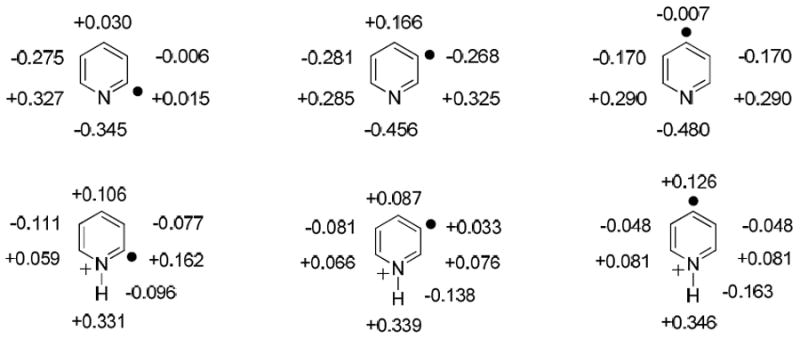
The 2-, 3- and 4-dehydropyridines (top) and their protonated forms (bottom) shown with atomic charges (calculated at the UBPW91/cc-pVDZ//UBPW91/cc-pVDZ level of theory by using the CHELPG procedure and by fitting to the molecular dipole moment).
Effect of pH on the efficiency of hydrogen atom abstraction by 3- and 4-dehydropyridine
The percentage yields for hydrogen atom abstraction at different pH values were also determined (Tables 4 and 5). For 3-dehydropyridine, the percentage yield of hydrogen atom abstraction increased from 25% to 43% as pH was decreased from 3.1 to 1.1 (Table 4 and Figure 1). A similar but weaker trend was observed for 4-iodopyridine (from 38% to 44%, Table 5). The weaker trend for 4-iodopyridine can be explained based on the smaller decrease in the ratio of protonated to unprotonated molecules as the pH increases, which is related to its pKa (Table 2). For example, when pH is increased from 1.1 to 2.0, the ratio of protonated to unprotonated 4-iodopyridine molecules decreases by about a factor of eight. The analogous decrease (factor of twenty from pH 1.0 to 2.3) for 3-iodopyridine is much more pronounced.
A decrease in the reactivity of the radicals at higher pH is in an excellent agreement with the above prediction that the protonated radicals (dominating at low pH) are more reactive than the unprotonated radicals (dominating at high pH), which can be explained based on their calculated gas-phase EAs. However, they are not in agreement with the expectation of the protonated 3-isomer to react faster due to its greater EA than the protonated 4-isomer. This observation is likely explained by the greater pKa of 4-iodopyridine in comparison to the 3-isomer (Table 7). Since the pKa of 4-iodopyridine is greater than that of 3-iodopyridine, more protonated 4-iodopyridine molecules exist in solution at a given pH than for 3-iodopyridine (Table 2). Hence, photolysis of 4-iodopyridine yields more protonated radicals than photolysis of 3-iodopyridine. After formation and before reactions, the protonated and unprotonated radicals may be able to reach a new equilibrium controlled by their basicities. Since 4-dehydropyridine has a PA greater by about one kcal/mol than its 3-isomer (Table 7), it almost certainly also has a greater pKa (e.g., for 2-bromopyridine, PA = 216.3 kcal/mol48a and pKa = 0.9,48b while for 3-bromopyridine, the corresponding values are 217.5 kcal/mol and 2.84, respectively; the same situation is also true for 2- and 3-chloropyridines48). Hence, even in this situation, more protonated 4-dehydropydidines should remain.
In order to explore the influence of pH on the yields of radicals formed under photolysis, the UV absorption (at 300 nm) of the 3- and 4-dehydropyridine in water/methanol solution was measured as a function of pH. No significant changes were observed in the pH range 1-3 (Table 8). Therefore, photolysis of the 3- and 4-iodopyridine under different pH should give the same yields of radicals.
Table 8.
UV Absorption (at 300 nm) of 3- and 4-Iodopyridine in 10 mM Methanol/Water Solutions at Various pHa
| pH | UV absorption at 300 nm | |
|---|---|---|
| 3-iodopyridine | 4-iodopyridine | |
| 1.1 | 0.3116 | 0.3214 |
| 1.7 | 0.3102 | 0.3223 |
| 2.3 | 0.3080 | 0.3260 |
| 3.1 | 0.3120 | 0.3184 |
The pH was adjusted by using a mixture of HCl and KCl in water.
Furthermore, the dependence of the bond dissociation energies of the iodopyridines on their protonation state was also explored. The homolytic C-I bond dissociation energies of the 2-, 3-, and 4-iodopyridinium cations were calculated ((U)B3LYP/6-311G(d,p)//(U)B3LYP/6-311G(d,p)) to be 68.7, 67.2, and 67.2 kcal/mol, respectively. The homolytic C-I bond dissociation energies of 3- and 4-iodopyridines have been shown49 to be similar to that of iodobenzene. The calculated C-I bond dissociation energy of iodobenzene (at the same level of theory as the other calculated values) is 65.7 kcal/mol, which is in very good agreement with the experimentally determined50 bond dissociation energy (66.7 kcal/mol). Hence, the difference in the C-I bond dissociation energies of the 3- and 4-iodopyridines before and after protonation is likely to be insignificant, and should not play an important role here.
2-Dehydropyridine
The photochemistry of 2-iodopyridine has been studied previously in pure methanol.36,50 In these experiments, 2-methoxypyridine was the major product (branching ratio, 64%). The hydrogen atom abstraction product, pyridine, was also formed (branching ratio, 36%). These results suggested the formation of a cationic intermediate, 2-pyridyl cation, through electron transfer within the initially formed radical pair (2-dehydropyridine and iodine atom; Scheme 3). Molecular orbital calculations provided support for this mechanism.51 The energy of the singly occupied molecular orbital (SOMO) of 2-dehydropyridine is higher than the SOMOs of 3- and 4-dehydropyridines. Therefore, it is energetically easier for 2-dehydropyridine to transfer an electron to an iodine atom to form the 2-pyridyl cation.
Scheme 3.

In this study, photolysis of 2-iodopyridine in a mixture of water and methanol produced pyridine, 2-methoxypyridine, and 2-hydroxypyridine. The identities of the 2-methoxypyridine and 2-hydroxypyridine products were confirmed via collision-activated dissociation (CAD) of the protonated reaction products and comparison of their fragmentation patterns with those of the authentic compounds. The same fragment ions were observed: both the protonated 2-hydroxypyridine and 2-methoxypyridine fragment to give the 2-pyridyl cation by loss of water or methanol, respectively.
As discussed above, 2-methoxypyridine is a product of the reaction between 2-pyridyl cation and methanol. As expected, the use of partially labeled methanol (CD3OH) instead of methanol (CH3OH) resulted in the formation of 2-d3-methoxypyridine. 2-Hydroxypyridine is probably formed in an analogous manner upon nucleophilic addition of water to the 2-pyridyl cation (Scheme 3). Indeed, photolysis of 2-iodopyridine in water yielded 2-hydroxypyridine.
The concentration and percent yield of each product formed upon photolysis of 2-iodopyridine in the different solvents studied are listed in Tables 6 and 7. Examination of the photoreaction in a mixture of water and partially labeled methanol (CD3OH) revealed that this radical, as opposed to its isomers, abstracts hydrogen/deuterium atom from both methyl and hydroxyl groups in the pH range 1.0-5.6. For example, at pH 1.0, both deuteron atom abstraction (65%) and hydrogen atom abstraction (35%) products were observed (Table 3). This finding may be explained by the substantially lower PA (and hence pKa) of 2-dehydropyridine than its 3- and 4-isomers (Table 7), which means that the fraction of protonated 2-dehydropyridine molecules is always less than for its isomers (Table 2). Indeed, at greater pH values, the 3- and 4-isomers also show major hydrogen atom abstraction products in water and partially labeled methanol (CD3OH) (Table 3), which is indicative of the presence of unprotonated radicals.
Table 6.
Results (Errors Are Standard Deviations) Obtained Upon HPLC/MS Analysis of the Reaction Mixtures of 2-Iodopyridine in Methanol/Water
| Unreacted 2-Iodopyridine | Pyridine |  |
 |
Sum of All Products and 2-Iodopyridine | ||
|---|---|---|---|---|---|---|
| pH 1.0 | Dark Control | (12 ± 2) mM | 0.0 mM | 0.0 mM | 0.0 mM | - |
| Sample | (9.3 ± 0.3) mM | (0.90 ± 0.04) mM | (1.03 ± 0.04) mM | (0.86 ± 0.01) mM | (12.2 ± 0.3) mM | |
| Yield | (7.4 ± 0.5)% | (8.5 ± 0.5)% | (7.1 ± 0.1)% | |||
| pH 1.6 | Dark Control | (12 ± 1) mM | 0.0 mM | 0.0 mM | 0.0 mM | - |
| Sample | (8.9 ± 0.3) mM | (0.61 ± 0.02) mM | (0.82 ± 0.05) mM | (0.72 ± 0.05) mM | (11.0 ± 0.3) mM | |
| Yield | (5.5 ± 0.1)% | (7.4 ± 0.2)% | (6.5 ± 0.6)% | |||
| pH 2.2 | Dark Control | (11 ± 2) mM | 0.0 mM | 0.0 mM | 0.0 mM | - |
| Sample | (8.3 ± 0.7) mM | (0.71 ± 0.01) mM | (0.77 ± 0.02) mM | (0.8 ± 0.2) mM | (10.6 ± 0.7) mM | |
| Yield | (6.7 ± 0.5)% | (7.3 ± 0.5)% | (8 ± 1)% | |||
| pH 2.7 | Dark Control | (11 ± 2) mM | 0.0 mM | 0.0 mM | 0.0 mM | - |
| Sample | (8.3 ± 0.9) mM | (0.58 ± 0.01) mM | (0.72 ± 0.04) mM | (0.69 ± 0.09) mM | (10.3 ± 0.9) mM | |
| Yield | (5.7 ± 0.7)% | (7.1 ± 0.3)% | (6.7 ± 0.3)% |
An earlier gas-phase study showed that 2-dehydropyridinium cation reacts with methanol only by hydrogen atom abstraction.33 This result provides further support for the intermediacy of the 2-pyridyl cation in the formation of the methoxy- and hydroxypyridines in solution since this ion was removed before examination of the reactions of the 2-dehydropyridinium cation in the gas phase.
The pKa of 2-iodopyridine in a mixture of water and ethanol is 1.1.46 The pKa of 2-iodopyridine in a mixture of water and methanol is likely to be similar. Hence, at pH 1.0, both 2-iodopyridine and its protonated form, 2-iodopyridinium, are present in solution (Table 1). Upon photolysis, 2-dehydropyridine and 2-dehydropyridinium cations, as well as 2-pyridyl cation, are likely formed. Both 2-dehydropyridine and 2-dehydropyridinium cation can undergo hydrogen atom abstraction. However, because the hydrogen atom abstraction product is a minor reaction product, the pH effect is not obvious in the photolysis of 2-iodopyridine. 2-Methoxypyridine and 2-hydroxypyridine were observed as the major products. The yields of these products were not affected by pH (Table 6), possibly because the 2-pyridyl cation may be formed from both 2-dehydropyridine and 2-dehydropyridinium cation. Earlier gas-phase experiments have revealed that 2-pyridyl cation is formed from 2-iodopyridinium cation upon collisional activation.52 Methanol was found to react with the 2-pyridyl cation exclusively by nucleophilic addition, as observed in solution.
Conclusions
The isomeric 2-, 3-, and 4-dehydropyridinium cations were generated by using UV photolysis in an acidic mixture of water and methanol in order to compare their solution reactivity to previously published33 gas-phase reactivity. Liquid chromatography-mass spectrometric analysis of the reaction mixtures revealed hydrogen atom abstraction from the solvent as the only reaction for the 3- and 4-isomers. The same observation has been made previously for these radicals in the gas phase.33 For 2-dehydropyridinium cation, however, nucleophilic addition products, 2-hydroxypyridine and 2-methoxypyridine, were also observed in solution (formation of 2-methoxypyridine has been reported previously36,51) but not in the gas phase. These products are rationalized by the formation of a second intermediate, 2-pyridyl cation, under both conditions. However, in the gas-phase studies, it was possible to eject the 2-pyridyl cation from the reaction chamber before examination of the reactions of the 2-dehydropyridinium cation, and hence, only hydrogen atom abstraction was observed.
Isotope labeling experiments were employed to investigate the identity of the hydrogen atom donor sites in methanol and water. In less acidic conditions, both the methyl group of methanol as well as the hydroxyl groups in methanol and water can act as hydrogen atom donors to the radicals studied. The abstraction of hydrogen atoms from the hydroxyl groups is attributed to unprotonated radicals that dominate at high pH. However, in the pH range of 1 - 4, the methyl group of methanol was found to be the main hydrogen atom donor site for 3-dehydropyridinium and 4-dehydropyridinium cations (as observed for these radicals in the gas phase). Hence, this pH range was chosen for a study on the effects of pH on the efficiency of hydrogen atom abstraction by the radicals from methanol. For 3-dehydropyridinium cation, the yield of the hydrogen atom abstraction product, pyridine, increased from 25% to 43% as the pH decreased from 3.1 to 1.0. A similar but weaker pH effect was observed for 4-dehydropyridinium cation. These results demonstrate that the efficiency of hydrogen atom abstraction can be increased by lowering the pH of the system, as predicted in previous gas-phase studies.19,25,33 This finding is explained by the higher polarity (and hence greater gas-phase electron affinity) of the radical site after protonation of the molecule. For example, the gas-phase electron affinity of the radical site for 3-dehydropyridine increases from 0.93 eV to 6.08 eV upon protonation of the nitrogen atom, which enhances its ability to polarize (and hence stabilize) the transition states of its reactions in the gas phase.17 Based on the results presented here, this is also true for aqueous solutions. Hence, these results suggest that the reactivity of charged radicals in solution may be predicted based on gas-phase studies as long as exactly the same species is studied in both environments.
The finding that the reactivity of some radicals may be enhanced by lowering the pH of the solution is interesting in its own right. Although the conditions used here are more acidic than in tissues, these results may facilitate the design of drugs that are more selective for tumor than normal tissues due to the lower pH in tumor tissues.53
Finally, the attribution of the enhancement in reactivity upon protonation of 2,5-didehydropyridine in solution34 to a reduction in the singlet-triplet gap now warrants reconsideration. Based on the results presented here, the different polarities of these two biradicals should cause a major difference in reactivity, with that of the protonated species being greater. The influence of different polarities (EAs) and singlet-triplet gaps on biradicals’ reactivity in the gas phase and in solution is currently under investigation.
Supplementary Material
Acknowledgments
The project described was supported by Grant Number R01GM052418 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. F.W. also thanks the Frederick N. Andrews endowment for a Graduate Fellowship.
Footnotes
Supporting Information Available. Complete ref. 43 and 44. The absolute energies (in Hartrees) and the coordinates of the atoms in all the molecules whose geometries were optimized. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Donahue NM. Chem Rev. 2003;103:4593–4604. doi: 10.1021/cr020650g. [DOI] [PubMed] [Google Scholar]
- 2.Blowers P, Masel R. AlChE Journal. 2000;46:2041–2052. [Google Scholar]
- 3.Chen YH, Tschuikowroux E. J Phys Chem. 1993;97:3742–3749. [Google Scholar]
- 4.Galano A, Alvarez-Idaboy JR, Bravo-Perez G, Ruiz-Santoyo ME. Phys Chem Chem Phys. 2002;4:4648–4662. [Google Scholar]
- 5.Pardo L, Banfelder JR, Osman R. J Am Chem Soc. 1992;114:2382–2390. [Google Scholar]
- 6.Mebel AM, Lin MC, Yu T, Morokuma K. J Phys Chem A. 1997;101:3189–3196. [Google Scholar]
- 7.Roberts BP. Chem Soc Rev. 1999;28:25–35. [Google Scholar]
- 8.Pogozelski WK, Tullius TD. Chem Rev. 1998;98:1089–1108. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 9.Nicolaou KC, Smith AL, Yue EW. PNAS. 1993;90:5881–5888. doi: 10.1073/pnas.90.13.5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldberg IH. Acc Chem Res. 1991;24:191–198. [Google Scholar]
- 11.Sugiyama H, Fujiwara T, Kawabata H, Yoda N, Hirayama N, Saito I. J Am Chem Soc. 1992;114:5573–5578. [Google Scholar]
- 12.Lee MD, Ellestad GA, Borders DB. Acc Chem Res. 1991;24:235–243. [Google Scholar]
- 13.Nicolaou KC, Dai WM, Tsay SC, Estevez VA, Wrasidlo W. Science. 1992;256:1172–1178. doi: 10.1126/science.256.5060.1172. [DOI] [PubMed] [Google Scholar]
- 14.Guanti G, Banfi L, Basso A, Riva R. Polynes Synth Prop App. 2006:453–491. [Google Scholar]
- 15.Pryor WA, Echols JT, Smith K. J Am Chem Soc. 1966;88:1189–1199. [Google Scholar]
- 16.Takayama K, Kosugi M, Migita T. Chem Lett. 1973:193–195. [Google Scholar]
- 17.Fossey J, Lefort D, Sorba J. Free Radicals in Organic Chemistry. John Wiley & Sons; New York: 1995. [Google Scholar]
- 18.Takayama K, Kosugi M, Migita T. Chem Lett. 1973:215–218. [Google Scholar]
- 19.Heidbrink JL, Ramirez-Arizmendi LE, Thoen KK, Guler L, Kenttämaa HI. J Phys Chem A. 2001;105:7875–7884. [Google Scholar]
- 20.Li R, Smith RL, Kenttämaa HI. J Am Chem Soc. 1996;118:5056–5061. [Google Scholar]
- 21.Petucci C, Nyman M, Guler L, Kenttämaa H. J Am Chem Soc. 2002;124:4108–4115. doi: 10.1021/ja012243c. [DOI] [PubMed] [Google Scholar]
- 22.Donahue NM, Clarke JS, Anderson JG. J Phys Chem A. 1998;102:3923–3933. [Google Scholar]
- 23.Clarke JS, Rypkema HA, Kroll JH, Donahue NM, Anderson JG. J Phys Chem A. 2000;104:4458–4468. [Google Scholar]
- 24.Liu J, Petzold CJ, Ramirez-Arizmendi LE, Perez J, Kenttämaa HI. J Am Chem Soc. 2005;127:12758–12759. doi: 10.1021/ja052766a. [DOI] [PubMed] [Google Scholar]
- 25.Ramirez-Arizmendi LE, Heidbrink JL, Guler LP, Kenttämaa HI. J Am Chem Soc. 2003;125:2272–2281. doi: 10.1021/ja020632g. [DOI] [PubMed] [Google Scholar]
- 26.Nash JJ, Nizzi KE, Adeuya A, Yurkovich MJ, Cramer CJ, Kenttämaa HI. J Am Chem Soc. 2005;127:5760–5761. doi: 10.1021/ja0449473. [DOI] [PubMed] [Google Scholar]
- 27.Adeuya A, Yang L, Amegayibor FS, Nash JJ, Kenttämaa HI. J Am Soc Mass Spectrom. 2006;17:1325–1334. doi: 10.1016/j.jasms.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Nash JJ, Yurkovich MJ, Jin Z, Vinueza NR, Kenttämaa HI. Org Lett. 2008;10:1889–1892. doi: 10.1021/ol800312g. [DOI] [PubMed] [Google Scholar]
- 29.Nizzi KE, Amegayibor FS, Nash JJ, Kenttämaa HI. J Am Chem Soc. 2005;127:13152–13153. doi: 10.1021/ja054514f. [DOI] [PubMed] [Google Scholar]
- 30.Jankiewicz BJ, Reece JN, Vinueza NR, Nash JJ, Kenttämaa HI. Angew Chem Int Ed. 2008;47:9860–9865. doi: 10.1002/anie.200802714. [DOI] [PubMed] [Google Scholar]
- 31.Jankiewicz BJ, Adeuya A, Yurkovich MJ, Vinueza NR, Gardner SJ, Zhou M, Nash JJ, Kenttämaa HI. Angew Chem Int Ed. 2007;46:9198–9201. doi: 10.1002/anie.200701732. [DOI] [PubMed] [Google Scholar]
- 32.Jing LH, Nash JJ, Kenttämaa HI. J Am Chem Soc. 2008;130:17697–17709. doi: 10.1021/ja801707p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adeuya A, Price JM, Jankiewicz BJ, Nash JJ, Kenttämaa HI. J Phys Chem A. 2009;113:13663–13674. doi: 10.1021/jp901380y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoffner J, Schottelius MJ, Feichtinger D, Chen P. J Am Chem Soc. 1998;120:376–385. [Google Scholar]
- 35.Dunning TH., Jr J Chem Phys. 1989;90:1007–1023. [Google Scholar]
- 36.Becke AD. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 37.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 38.Perdew JP, Burke K, Wang Y. Phys Chem B. 1996;54:16533–16539. doi: 10.1103/physrevb.54.16533. [DOI] [PubMed] [Google Scholar]
- 39.Breneman CM, Wiberg KB. J Comput Chem. 1990;11:361–373. [Google Scholar]
- 40.Kendall RA, Dunning TH, Harrison RJ. J Chem Phys. 1992;96:6796–6806. [Google Scholar]
- 41.Note that, for these calculations, we are computing the vertical electron affinity of the radical site, not the vertical electron affinity of the molecule.
- 42.Because the positively chargeed aryl radicals studied here contain the formal positive charge on the nitrogen atom, the state that is produced when an electron is added to the nonbonding orbital is formally zwitterionic; that is, it contains localized positive (π) and negative (σ) charges.
- 43.Frisch MJ, et al. Gaussian 98, revision A.11.3. Gaussian, Inc.; Pittsburgh, PA: 2002. for complete reference, see the Supporting Information. [Google Scholar]
- 44.MOPRO. Wener H-J, Knowles PJ, Lindh R, Manby FR, Schütz M, Celani P, Korona T, Mitrushenkov A, Rauhut G, Adler TB, Amos RD, Bernhardsson A, Berning A, Cooper DL, Deegan MJO, Dobbyn AJ, Eckert F, Goll E, Hampel C, Hetzer G, Hrenar T, Knizia G, Köppl C, Liu Y, Lloyd AW, Mata RA, May AJ, McNicholas SJ, Meyer W, Mura ME, Nicklass A, Palmieri P, Pflüger K, Pitzer R, Reiher M, Schumann U, Stoll H, Stone AJ, Tarroni R, Thorsteinsson T, Wang M, Wolf A. version 2008.1, a package of ab initio programs. See http://www.molpro.net.
- 45.Ohkura K, Seki K, Terashima M, Kanaoka Y. Tet Lett. 1989;30:3433–3436. [Google Scholar]
- 46.Holubek J, Volke J. Collect Czech Chem C. 1962;27:680–692. [Google Scholar]
- 47.Jing L, Guler LG, Nash JJ, Kenttämaa HI. J Am Soc Mass Spectrom. 2004;15:913–919. doi: 10.1016/j.jasms.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 48.(a) Hunter EP, Lias SG. J Phys Chem Ref Data. 1998;27:413–656. [Google Scholar]; (b) Brown HC, McDaniel DH. J Am Chem Soc. 1955;77:3732–3755. [Google Scholar]
- 49.Danen WC, Saunders DG, Rose KA. J Am Chem Soc. 1974;96:4558–4562. [Google Scholar]
- 50.Kumaran SS, Su MC, Michael JV. Chem Phys Lett. 1997;269:99–106. [Google Scholar]
- 51.Ohkura K, Terashima M, Kanaoka Y, Seki K. Chem Pharm Bull. 1993;41:1920–1924. [Google Scholar]
- 52.Kirkpatrick L. Ph.D. Thesis. Purdue University; 2010. [Google Scholar]
- 53.Tannock IF, Rotin D. Cancer Res. 1989;49:4373–4384. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.