

Abstract
The vast majority of HIV-1 infections in Africa are caused by the A and C viral subtypes rather than the B subtype prevalent in the United States and Western Europe. Genomic differences between subtypes give rise to sequence variations in the encoded proteins, including the HIV-1 protease. Because some amino acid polymorphisms occur at sites that have been associated with drug resistance in the B subtype, it is important to assess the effectiveness of protease inhibitors that have been developed against different subtypes. Here we report the enzymatic characterization of HIV-1 proteases with sequences found in drug-naïve Ugandan adults. The A protease used in these studies differs in seven positions (I13V/E35D/M36I/R41K/R57K/H69K/L89M) in relation to the consensus B subtype protease. Another protease containing a subset of these amino acid polymorphisms (M36I/R41K/H69K/L89M), which are found in subtype C and other HIV subtypes, also was studied. Both proteases were found to have similar catalytic constants, kcat, as the B subtype. The C subtype protease displayed lower Km values against two different substrates resulting in a higher (2.4-fold) catalytic efficiency than the B subtype protease. Indinavir, ritonavir, saquinavir, and nelfinavir inhibit the A and C subtype proteases with 2.5–7-fold and 2–4.5-fold weaker Kis than the B subtype. When all factors are taken into consideration it is found that the C subtype protease has the highest vitality (4–11 higher than the B subtype) whereas the A subtype protease exhibits values ranging between 1.5 and 5. These results point to a higher biochemical fitness of the A and C proteases in the presence of existing inhibitors.
The magnitude of the AIDS epidemic in Africa is well documented (see ref. 1 for a recent review). Of the 34 million people infected with HIV worldwide, 73% or 25 million are in Africa. However, the HIV-1 subtypes prevalent in Africa are not the same that are prevalent in the United States and Western Europe. In the United States and Western Europe the B subtype is responsible for the vast majority of HIV infections whereas in subSaharan Africa the A and C subtypes account for most of the infections. The A subtype predominates in the northern part of subSaharan Africa and the C subtype in southern Africa. This dominance includes areas where the B subtype was previously prevalent. Different factors, including distinct transcriptional regulation, have been proposed to explain differences in transmission rates and pathogenesis among HIV-1 subtypes (2, 3). Since HIV-1 antiretroviral therapy has been developed and tested against the B subtype, an important question relates to the effectiveness of these therapies against different subtypes. At the molecular level this issue is related to the inhibition constants and enzymatic vitalities of these proteases in the presence of existing inhibitors (4, 5). For this reason, we have cloned and expressed proteases with sequences observed in A and C HIV-1 subtypes (6) and characterize their enzymatic behavior and response to four widely used clinical inhibitors (indinavir, saquinavir, nelfinavir, and ritonavir).
Different subtypes of HIV-1 differ from one another anywhere between 10% and 30% along their entire genomes, including the protease gene (6). At the time of this writing, 48 A subtype protease sequences and 53 C subtype protease sequences have been deposited in GenBank (6). Analysis of the HIV-1 sequence database (6) indicates that variations between subtypes do not occur at random within the protease structure and appear to be clustered in certain regions (Fig. 1). A significant number of amino acid polymorphisms exist among subtype A, B, and C sequences (7, 8). Many of these are located in the hinge regions, residues 35, 36, 41, and 57. The polymorphisms E35D/M36I/R41K/R57K are common (red in Fig. 1). Residue 89 in the α-helix and residue 13 in the opposite β-strand are also sites of amino acid polymorphisms with I13V/L89M being predominant (yellow in Fig. 1). Finally, residue 69 in the loop connecting the two β strands is also variable (H69K) (green in Fig. 1). A subset of these polymorphisms (M36I/R41K/H69K/L89M) often is encountered in C subtype proteases.
Figure 1.

(A) Structure of the HIV-1 protease showing the location of amino acid polymorphisms in the A subtype (I13V/E35D/M36I/R41K/R57K/H69K/L89M) (Left). (Right) Shown is the location of a subset of mutations (M36I/R41K/H69K/L89M) found in some C subtype and recombinant HIV-1 isolates. In the hinge of the flap, residues 36 and 41 are affected in A and C subtypes, whereas residue 35 is mostly affected in A (red). Residue 89 in the α helix is affected in A and C whereas residue 13 in the opposite β strand is predominantly affected in A (yellow). Residue 69 in the loop connecting the two β strands is affected in both the A and C proteases (green). (B) The sequences of the proteases used in these studies.
It is noteworthy that some of these differences in sequence occur at sites that are known to be associated with inhibitor resistance in the B subtype (8). Position 36 is occupied by isoleucine in the A and C subtype proteases of drug-naïve patients. In the B subtype M36I is a secondary resistant mutation that has been found in patients treated with ritonavir, indinavir, or nelfinavir (9–11). Although none of the differences found in the A and C proteases are directly located in the catalytic site, some of them are located at sites that are important for the conformational change associated with binding. The hinge region and the α-helix are important for the closing of the flaps and the subunit rotation upon binding (12, 13). Mutations in this region might affect the energetics of these changes and consequently the binding affinity of inhibitors.
In previous papers, we have studied the mechanisms by which specific mutations in the protease molecule cause resistance to inhibitors (13–17). In this paper we have investigated the catalytic efficiency and inhibition of proteases carrying four and seven mutations that are commonly observed in the A and C subtypes.
Materials and Methods
Protease Subtypes.
HIV-1 protease containing the mutations I13V/E35D/M36I/R41K/R57K/H69K/L89M and M36I/R41K/H69K/L89M were generated from the B subtype protease (13, 15, 18) by PCR mutagenesis. The gene encoding the B subtype HIV-1 protease was transferred to the pET24 vector (Novagen), where the expression is under control of the T7 promoter. Mutations at selected positions were introduced by using an in vitro site-directed mutagenesis kit (Stratagene), and mutations were confirmed by DNA sequencing. Proteases were expressed in BL21/DE3 cells by adding isopropyl β-d-thiogalactoside to 1 mM once culture density (as determined by absorbance at 600 nm) was 1.5 or greater.
Protease Purification.
Plasmid-encoded HIV-1 protease was expressed as inclusion bodies in Escherichia coli 1458 (13, 15, 18). Cells were suspended in extraction buffer [20 mM Tris/1 mM EDTA/10 mM 2-mercaptoethanol (2-ME), pH 7.5] and broken with two passes through a French pressure cell (≥16,000 psi, 1 psi = 6.89 kPa). Cell-debris and protease-containing inclusion bodies were collected by centrifugation (20,000 g for 20 min at 4°C). Inclusion bodies were washed with three buffers. Each wash consisted of resuspension (glass homogenizer, sonication) and centrifugation (20,000 g for 20 min at 4°C). In each step a different washing buffer was used: buffer 1 (25 mM Tris/2.5 mM EDTA/0.5 M NaCl/1 mM Gly-Gly/50 mM 2-ME, pH 7.0), buffer 2 (25 mM Tris/2.5 mM EDTA/0.5 M NaCl/1 mM Gly-Gly/50 mM 2-ME/1 M urea, pH 7.0), and buffer 3 (25 mM Tris/1 mM EDTA/1 mM Gly-Gly/50 mM 2-ME, pH 7.0). Protease was solubilized in 25 mM Tris, 1 mM EDTA, 5 mM NaCl, 1 mM Gly-Gly, 50 mM 2-ME, 9 M urea, pH 8.0, clarified by centrifugation, and applied directly to an anion exchange Q-Sepharose column (Q-Sepharose HP, Amersham Pharmacia) previously equilibrated with the same buffer. The protease was passed through the column and then acidified by adding formic acid to 25 mM immediately upon elution from the column. Precipitation of a significant amount of contaminants occurred upon acidification. Protease-containing fractions were pooled, concentrated, and stored at 4°C at 5–10 mg/ml.
The HIV-1 protease was folded by 10-fold stepwise dilution into 10 mM formic acid at 0°C. The pH was gradually increased to 3.8, then the temperature was raised to 30°C. Sodium acetate pH 5.0 was added up to 100 mM and protein was concentrated. Folded protease was desalted into 1 mM sodium acetate at pH 5.0 by using a gel filtration column (PD-10, Amersham Pharmacia) and stored at either 4°C or −20°C (≥2.5 mg/ml) without loss of activity in several weeks. After folding, the protease was estimated to be ≥99% pure.
Clinical Inhibitors Purification.
Clinical inhibitors (indinavir, saquinavir, ritonavir, and nelfinavir) were purified from commercial capsules by HPLC (Waters) using a semipreparative C-18 reversed-phase column developed with 0–100% acetonitrile in 0.05% trifluoroacetic acid. Purified inhibitors were lyophilized and stored at −20°C in the crystalline form (indinavir, nelfinavir) or as suspensions in DMSO (saquinavir, ritonavir).
Determination of Kinetic Parameters.
The catalytic activities of the HIV-1 proteases were monitored by following the hydrolysis of the chromogenic substrate Lys-Ala-Arg-Val-Nle-nPhe-Glu-Ala-Nle-NH2, where Nle stands for norleucine and nPhe stands for p-nitrophenylalanine (California Peptide Research, Napa, CA), and the fluorogenic substrate Arg-Glu(EDANS)-Ser-Gln-Asn-Tyr-Pro-Ile-Val-Gln-Lys(DABCYL)-Arg (Molecular Probes).
In the spectrophotometric assay, protease was added to a 120-μl microcuvette containing substrate at 25°C. Final concentrations in the standard assay were: 30–60 nM active protease, 0–170 μM substrate, 10 mM sodium acetate, and 1 M sodium chloride, pH 5.0. The absorbance was monitored at 6 wavelengths (296–304 nm) by using a HP 8452 diode array spectrophotometer (Hewlett–Packard) and corrected for spectrophotometer drift by subtracting the average absorbance at 446–454 nm. An extinction coefficient for the difference in absorbance upon hydrolysis (1,800 M−1⋅cm−1 at 300 nm) was used to convert absorbance change to reaction rates. Hydrolysis rates were obtained from the initial portion of the data, where at least 80% of the substrate remains unhydrolyzed. The concentration of active protease was determined by performing active site titrations with KNI-272, a very potent inhibitor (at pH 5.0, Ki ≈ 16 pM), using protease concentrations much higher (≈2 μM) than the corresponding Ki.
In the spectrofluorometric assay, protease was added to 200-μl microwells containing substrate at 25°C. Final concentrations in the standard assay were: 25–28 nM active protease, 0–120 μM substrate, 10 mM sodium acetate, and 1 M sodium chloride, pH 5.0. The fluorescence was monitored by using a Cytofluor Microtiter Plate Fluorescence Reader (PerSeptive Biosystems, Framingham, MA) with excitation and emission wavelengths of 360 nm and 508 nm, respectively.
Inhibition constants, Ki, for the inhibitors (indinavir, ritonavir, saquinavir, and nelfinavir) were obtained at 25°C by measuring the rate of chromogenic substrate hydrolysis using 15–30 nM protease in 10 mM sodium acetate, 1 M sodium chloride, pH 5.0, and 42.5 μM substrate plus increasing amounts of inhibitor. Inhibition constants were obtained by fitting the data to standard equations for tight-binding inhibitors and considering the decrease in free inhibitor concentration when a non-negligible portion of the total is bound. In each of the assays, the known experimental concentrations were the total concentration of enzyme (free plus bound) and the total concentration of inhibitor (free plus bound).
Differential Scanning Calorimetry.
The heat capacity function of the HIV-1 proteases was measured as a function of temperature with a high precision differential scanning VP-DSC microcalorimeter (Microcal, Northampton, MA). Protein samples and reference solutions were properly degassed and carefully loaded into the cells to avoid bubble formation. Thermal denaturation scans were performed with freshly prepared buffer-exchanged protease solutions in 10 mM glycine, pH 3.6 as described (18). Data were analyzed by software developed in this laboratory.
Results and Discussion
Protease Sequences.
The studies presented here were performed with recombinant proteases based on sequences of subtype A and C HIV-1 (the complete sequences are given in Fig. 1B). For the subtype A protease our sequence was derived from the consensus sequence of subtype A protease from 14 antiretroviral naïve Ugandan adults (7, 8). This sequence differs from the subtype B consensus sequence (6) at seven positions I13V/E35D/M36I/R41K/R57K/H69K/L89M (Fig. 1A Left) and is identical to the sequences of two Ugandan isolates (nos. 225.706 and 230.706) (8). A second recombinant protease was generated based on a consensus sequence of subtype C proteases [92RW026 (Rwanda, GenBank no. AF009410), C2220 (Ethiopia, GenBank no. U461016), Z1226 (Zimbabwe, GenBank no. AF083603), 96BW01 (Botswana, GenBank no. AF110959) and C11 (Zambia, GenBank no. AF107378)]. The subtype C sequence used in this report is identical to that consensus sequence with the exception of amino acid substitutions V15I and L93I. However, isoleucine is also found at positions 15 and 93 in some subtype C proteases (6). This sequence differs from the subtype B reference sequence at four positions, which are a subset of those described for the subtype A recombinant protein: M36I/R41K/H69K/L89M (Fig. 1A Right). These polymorphisms have been observed in several non-B HIV-1 subtypes (6). Finally, all of the proteases used in these studies carry the mutation Q7K, which eliminates the most significant autocatalytic site and enhances the stability of the preparations for biochemical and biophysical studies without affecting its enzymatic activity (19).
Catalytic Efficiency.
The kinetic constants measured for the A, B, and C HIV-1 subtype proteases with the substrates Lys-Ala-Arg-Val-Nle-nPhe-Glu-Ala-Nle-NH2 and Arg-Glu(EDANS)-Ser-Gln-Asn-Tyr-Pro-Ile-Val-Gln-Lys(DABCYL)-Arg are summarized in Table 1. These substrates resemble two cleavage sites, KARVL/AEAM, between the capsid protein and p2, and SQNY/PIVQ, between the matrix and capsid proteins in the Gag polyprotein precursor. These are two of the most conserved protease cleavage sites in the Gag polyprotein among the different viral subtypes. These two octapeptide sequences comprise more than 94% of the cleavage site sequences corresponding to the viral subtypes A, B, and C (6). As seen in Table 1, for each substrate the catalytic rate constants, kcat, are very similar between subtypes and range between 7 and 9 sec−1. The lack of a significant effect in kcat probably reflects the fact that the sequence variations found in subtypes A and C are located outside the active site and that the chemical environment surrounding the aspartyl diad remains unchanged. However, the situation is not the same with the KM values. The KM for the subtype C protease is 2.6–2.7 times lower than that of the B subtype, whereas the A subtype protease exhibits a somewhat higher KM for both substrates. Accordingly, the catalytic efficiency, defined as kcat/KM, of the C subtype protease is about 2.2–2.5 times higher than that of the B subtype protease, whereas the A subtype protease is only 0.6–0.8 times as efficient as the B subtype.
Table 1.
Catalytic efficiency of HIV-1 protease from different subtypes
| Substrate | Kinetic constants | Subtype B | Subtype A | Subtype C |
|---|---|---|---|---|
| I | KM (μM) | 14.0 ± 1 | 20.0 ± 2 | 5.4 ± 0.4 |
| kcat (s−1) | 8.9 ± 0.2 | 7.8 ± 0.2 | 7.7 ± 0.1 | |
| kcat/KM (s−1⋅μM−1) | 0.64 ± 0.06 | 0.39 ± 0.05 | 1.4 ± 0.1 | |
| II | KM (μM) | 62.0 ± 8 | 65.0 ± 10 | 23.0 ± 3 |
| kcat (s−1) | 8.6 ± 0.5 | 6.9 ± 0.5 | 8.0 ± 0.3 | |
| kcat/KM (s−1⋅μM−1) | 0.14 ± 0.02 | 0.11 ± 0.03 | 0.35 ± 0.05 |
Experiments were performed in 10 mM sodium acetate and 1 M NaCl, pH 5.0 at 25°C using the chromogenic substrate I, Lys-Ala-Arg-Val-Nle-nPhe-Glu-Ala-Nle-NH2, and the fluorogenic substrate II, Arg-Glu(EDANS)-Ser-Gln-Asn-Tyr-Pro-Ile-Val-Gln-Lys(DABCYL)-Arg.
Inhibition of Proteases from Different HIV-1 Subtypes.
Inhibition constants, Ki, for the inhibitors indinavir, ritonavir, saquinavir, and nelfinavir were obtained at 25°C as described in Materials and Methods. The results obtained for all of the inhibitors are summarized in Table 2. It is clear that the Ki values for all inhibitors are higher for the A and C subtype proteases than for the B subtype protease against which they were developed. The relative effects are not the same for each protease or inhibitor as illustrated in Fig. 2. On a relative scale the Kis for ritonavir and indinavir appear to be affected the most, increasing by a factor of 3–4 for the C subtype and 6–7 for the A subtype. It must be noted, however, that despite the higher relative increase observed for ritonavir, the Ki for this inhibitor remains in the subnanomolar range and lower than that of all of the other inhibitors. The Kis for saquinavir and nelfinavir are about 2- to 2.5-fold higher for both the A and C subtype proteases.
Table 2.
Inhibition constants for HIV-1 proteases from different subtypes
| Inhibitor | Ki (nM), subtype B | Ki (nM), subtype A | Ki (nM), subtype C |
|---|---|---|---|
| Indinavir | 0.8 ± 0.06 | 5.1 ± 0.2 | 2.5 ± 0.2 |
| Ritonavir | 0.05 ± 0.005 | 0.36 ± 0.03 | 0.23 ± 0.02 |
| Saquinavir | 0.5 ± 0.03 | 1.2 ± 0.08 | 1.1 ± 0.05 |
| Nelfinavir | 1.0 ± 0.07 | 2.6 ± 0.2 | 2.1 ± 0.1 |
Inhibition constants, Ki, for the inhibitors indinavir, ritonavir, saquinavir, and nelfinavir were obtained at 25°C by measuring the rate of substrate hydrolysis using 15–30 nM Protease in 10 mM sodium acetate, 1 M NaCl, pH 5.0, and 42.5 μM substrate plus increasing amounts of inhibitor.
Figure 2.

Relative increase in Ki for the A and C subtype proteases in relation to the B subtype protease. Solid bars = A subtype; hatched bars = C subtype.
To compare the selective advantage of different protease mutations in the presence of specific inhibitors Gulnik et al. (4, 5) introduced an empirical parameter, called vitality, which is given by the following equation:
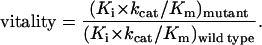 |
The vitality can be considered to be a measure of the enzymatic fitness of a particular mutant in the presence of a given inhibitor. For example, the double resistant mutant in the B subtype V82T/I84V has a reported vitality close to 10 for ritonavir and indinavir. Vitalities as high as 20 have been reported for the single mutants V82F and V82T in the presence of indinavir (5). The vitality also can be used to compare the proteases from other subtypes in relation to the B subtype (wild type). Fig. 3 shows the vitalities obtained for the A and C subtypes in the presence of indinavir, saquinavir, nelfinavir, and ritonavir for the two substrates considered in this study. Even though the experimental errors are amplified in the calculation of vitalities, the A and C proteases exhibit values higher than unity. In general, the vitalities for the A and C proteases are not as high as those reported for primary resistant muations and in some cases they are close to unity indicating a similar biochemical fitness. For the A subtype, vitalities as high as 5 were observed in the presence of indinavir and ritonavir and around 1.5 for nelfinavir and saquinavir. For the C subtype, on the other hand, vitalities around 8–10 were observed in the presence of indinavir and ritonavir and around 5 for nelfinavir and saquinavir.
Figure 3.

Vitality of the A and C proteases in the presence of indinavir, ritonavir, saquinavir, and nelfinavir. For the A protease, the solid bars are for the chromogenic substrate Lys-Ala-Arg-Val-Nle-nPhe-Glu-Ala-Nle-NH2 and the slashed bars for the fluorogenic substrate Arg-Glu(EDANS)-Ser-Gln-Asn-Tyr-Pro-Ile-Val-Gln-Lys(DABCYL)-Arg. For the C protease, the hatched bars are for the chromogenic substrate Lys-Ala-Arg-Val-Nle-nPhe-Glu-Ala-Nle-NH2 and the open bars for the fluorogenic substrate Arg-Glu(EDANS)-Ser-Gln-Asn-Tyr-Pro-Ile-Val-Gln-Lys(DABCYL)-Arg.
Long-Range Effects in Binding Energetics.
It is apparent from the experiments presented here that the amino acid polymorphisms considered in this study are able to affect the Km of the substrates and the Kis of the inhibitors even though they are located outside of the binding site. In all cases, an increase in the Kis is observed whereas the Kms either decrease (C subtype) or remain essentially unchanged (A subtype). This differential effect on Ki and Km has been observed before for inhibitor resistant mutants of the B subtype (20–22). Notably, this effect occurs even for mutations that do not change the chemical nature or polarity of the binding site but are known to change its geometry (21). At the thermodynamic level, the origin of the differential effect appears to be related to the difficulties of conformationally constrained inhibitors to adapt to distortions in the geometry of the binding site (14, 15). The substrates, being linear peptides, adapt well to these changes and their binding affinities are not affected as much. Distortions in the binding site geometry can be elicited by mutations in the binding site itself or by distal mutations that propagate their effects through the protein structure. The sequence differences studied here occur outside of the binding site but at locations that are known to be important to the conformational equilibrium of the protease and the conformational change that occurs upon binding (13, 23).
Fig. 4 shows the results of high-sensitivity differential scanning calorimetry measurements of the structural stabilities of the A, B, and C subtype proteases. These experiments were performed under the same buffer conditions and identical protease concentrations. Because the protease is a dimer, its stability is a function of protein concentration as discussed (18). It is clear that the A and C proteases are about 3.5°C more stable than the B subtype protease (2.7°C for the A protease and 3.7°C for the C protease). Analysis of the calorimetric data indicates that the native states of the A and C proteases are 0.7 and 1.3 kcal/mol more stable than the B subtype protease. Within error, the heat capacity change is the same for the three molecules (1.5 ± 0.1 kcal/K × mol-monomer). When normalized to the same temperature (65°C) the unfolding enthalpies for the three subtypes are identical within the experimental error 59.5 ± 0.5 kcal/mol-monomer, suggesting that the origin of the stability differences are primarily entropic even though the magnitude of the effects (0.7 and 1.3 kcal/mol) are very small for an accurate dissection.
Figure 4.

Structural stability of the A, B, and C subtype HIV-1 proteases as determined by high-sensitivity differential scanning calorimetry. These experiments were performed at pH 3.6, 10 mM glycine conditions under which thermal denaturation is fully reversible. These experiments indicate that the A and C subtype proteases are more stable than B subtype protease.
The sequence differences in the A and C subtype proteases are enough to alter the stability of the protease molecule. Because some of the differences occur at buried locations, they have the potential to induce atomic packing rearrangements within the core of the protease molecule and/or affect the energetics of the conformational change associated with binding. The observed differences in Kis summarized in Table 2 amount to differences in the Gibbs energy of binding ranging between 0.5 and 1.2 kcal/mol. Overall, these are small differences when compared with those observed in drug-resistant mutants of the B subtype protease, which are on the order of 1.2–3.3 kcal/mol (15). It must be noted, however, that the sequence differences of the proteases studied in this paper are outside of the binding site and that they correspond to the viral genomes of naïve patients. At this point, however, little is known about inhibitor-resistant mutations in A and C subtypes (24).
Conclusions
The experiments presented here indicate that current protease inhibitors have higher Kis for the A and C subtype proteases and that the biochemical fitness of these proteases, as defined by their vitality, is higher than that of the B subtype protease. These results are not surprising, considering the fact that current inhibitors were designed against the B subtype protease, and that they are highly constrained molecules preshaped to that binding site. Small geometric distortions in the A and C binding sites or small alterations in the Gibbs energy required to adopt the bound conformation may be sufficient to elicit the observed changes. The differences in stability observed by differential scanning calorimetry are consistent with small free energy differences on the order of 1 kcal/mol. These results demonstrate that small changes in drug targets might have significant effect on binding affinities of conformationally constrained inhibitors without a similar effect on the binding affinity of more relaxed molecules like the peptide substrates. The higher vitality of the A and C enzymes in the presence of existing inhibitors does not necessarily reflect a lower in vivo efficacy because other factors can be important in antiviral potency (25), including the fact that differences between subtypes are not limited to the protease molecule. The entire genomes of A, B, and C HIV-1 subtypes show variations on the order of 10–30% (6). Interestingly, these variations are not uniform throughout the entire genome and seem to be more pronounced in matrix and envelope proteins. An analysis of the HIV-1 sequences in GenBank indicates that relative to the B subtype, the reverse transcriptase enzyme exhibits 7–9% amino acid polymorphisms, the integrase between 7% and 9%, gp120 between 18% and 23%, gp41 between 22% and 27% and the matrix protein p17 between 22% and 27%. Even though systematic long-term clinical studies of the efficacy of existing protease inhibitors on non-B HIV-1 subtypes are not yet available, recent studies have pointed out to the existence of polymorphisms associated with drug resistance (8, 26). At the protease level the studies presented here support those observations. On the other hand, the few existing studies reporting susceptibility of clinical isolates from different HIV-1 subtypes to existing protease inhibitors have shown no difference or only small differences among subtypes (27, 28). Drug-resistance mutations in non-B HIV-1 subtypes are only beginning to be reported (24), and the effects arising from the combination of these mutations with the already existing polymorphisms in non-B subtypes are not yet known.
Acknowledgments
We thank Mr. Matt Merski for his work on the cloning of the A and C subtype proteases and Dr. Susan Eshleman of the Hopkins HIV genotyping Laboratory for providing us with the sequence data and many discussions. This work was supported by National Institutes of Health Grants GM 57144 and GM 51362 and National Science Foundation Grant MCB-9816661. A.V.-C. was partially supported by a postdoctoral fellowship from the Universidad de Granada, Spain (Plan Propio 1,999).
Abbreviation
- 2-ME
2-mercaptoethanol
References
- 1.Cohen J. Science. 2000;288:2150–2170. doi: 10.1126/science.288.5474.2170. [DOI] [PubMed] [Google Scholar]
- 2.Montano M, Novitski V, Blackard J, Cho N, Katzeinstein D, Essex M. J Virol. 1997;71:8657–8665. doi: 10.1128/jvi.71.11.8657-8665.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Essex M. Adv Virus Res. 1999;53:71–88. doi: 10.1016/s0065-3527(08)60343-7. [DOI] [PubMed] [Google Scholar]
- 4.Gulnik S, Suvorov L I, Liu B, Yu B, Anderson B, Mitsuya H, Erickson J W. Biochemistry. 1995;34:9282–9287. doi: 10.1021/bi00029a002. [DOI] [PubMed] [Google Scholar]
- 5.Gulnik S, Erickson J W, Xie D. In: Vitamins and Hormones. Litwack G, editor. New York: Academic; 1999. [Google Scholar]
- 6.Benson D A, Boguski M S, Lipman D J, Ostell J, Francis B F. Nucleic Acids Res. 1998;26:1–7. doi: 10.1093/nar/26.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brennan C A, Lund J K, Golden A, Yamaguchi J, Vallari A S, Phillips J F, Kataaha P K, Jackson J B, Devare S G. AIDS. 1997;11:1823–1832. doi: 10.1097/00002030-199715000-00006. [DOI] [PubMed] [Google Scholar]
- 8.Becker-Pergola G, Kataaha P, Johnston-Dow L, Fung S, Jackson J B, Eshleman S H. AIDS Res Hum Retroviruses. 2000;16:807–813. doi: 10.1089/088922200308800. [DOI] [PubMed] [Google Scholar]
- 9.Patick A K, Duran M, Cao Y, Shugarts D, Keller M R, Mazabel E, Knowles M, Chapman S, Kuritzkes D R, Markowitz M. Antimicrob Agents Chemother. 1998;42:2637–2644. doi: 10.1128/aac.42.10.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boden D, Markowitz M. Antimicrob Agents Chemother. 1998;42:2775–2783. doi: 10.1128/aac.42.11.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirsch M S, Brun-Vezinet F, D'Aquila R T, Hammer S M, Johnson V A, Kuritzkes D R, Loveday C, Mellors J W, Clotet B, Conway B, et al. J Am Med Assoc. 2000;283:2417–2426. doi: 10.1001/jama.283.18.2417. [DOI] [PubMed] [Google Scholar]
- 12.Rose R B, Craik C S, Stroud R M. Biochemistry. 1998;37:2607–2621. doi: 10.1021/bi9716074. [DOI] [PubMed] [Google Scholar]
- 13.Todd M J, Freire E. Proteins. 1999;36:147–156. doi: 10.1002/(sici)1097-0134(19990801)36:2<147::aid-prot2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 14.Luque I, Todd M J, Gomez J, Semo N, Freire E. Biochemistry. 1998;37:5791–5797. doi: 10.1021/bi9802521. [DOI] [PubMed] [Google Scholar]
- 15.Todd M J, Luque I, Velazquez-Campoy A, Freire E. Biochemistry. 2000;39:11876–11883. doi: 10.1021/bi001013s. [DOI] [PubMed] [Google Scholar]
- 16.Velazquez-Campoy A, Todd M J, Freire E. Biochemistry. 2000;39:2201–2207. doi: 10.1021/bi992399d. [DOI] [PubMed] [Google Scholar]
- 17.Velazquez-Campoy A, Luque I, Todd M J, Milutinovich M, Kiso Y, Freire E. Protein Sci. 2000;9:1801–1809. doi: 10.1110/ps.9.9.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Todd M J, Semo N, Freire E. J Mol Biol. 1998;283:475–488. doi: 10.1006/jmbi.1998.2090. [DOI] [PubMed] [Google Scholar]
- 19.Mildner A M, Rothrock D J, Leone J W, Bannow C A, Lull J M, Reardon I M, Sarcich J L, Howe W J, Tomich C C, Smith C W, et al. Biochemistry. 1994;33:9405–9413. doi: 10.1021/bi00198a005. [DOI] [PubMed] [Google Scholar]
- 20.Lin Y, Lin X, Hong L, Foundling S, Heinrikson R L, Thaisrivongs S, Leelamanit W, Raterman D, Shah M, Dunn B D, Tang J. Biochemistry. 1995;34:1143–1152. doi: 10.1021/bi00004a007. [DOI] [PubMed] [Google Scholar]
- 21.Baldwin E T, Bhat T N, Liu B, Pattabiraman N, Erickson J W. Nat Struct Biol. 1995;2:244–249. doi: 10.1038/nsb0395-244. [DOI] [PubMed] [Google Scholar]
- 22.Hong L, Treharne A, Hartsuck J A, Foundling S, Tang J. Biochemistry. 1996;35:10627–10633. doi: 10.1021/bi960481s. [DOI] [PubMed] [Google Scholar]
- 23.Rose R B, Craik C B, Stroud R M. Biochemistry. 1998;37:2607–2621. doi: 10.1021/bi9716074. [DOI] [PubMed] [Google Scholar]
- 24.Holguin A, Rodes B, Soriano V. AIDS Res Hum Retroviruses. 2000;16:1395–1403. doi: 10.1089/08892220050140946. [DOI] [PubMed] [Google Scholar]
- 25.Klabe R M, Bacheler L T, Ala P J, Erickson-Viitanen S, Meek J L. Biochemistry. 1998;37:8735–8742. doi: 10.1021/bi972555l. [DOI] [PubMed] [Google Scholar]
- 26.Cornelissen M, van den Burg R, Zorgdrager F, Lukashov V, Goudsmit J. J Virol. 1997;71:6348–6358. doi: 10.1128/jvi.71.9.6348-6358.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shafer R W, Chuang T K, Hsu P, White C B, Katzenstein D A. AIDS Res Hum Retroviruses. 1999;15:65–69. doi: 10.1089/088922299311727. [DOI] [PubMed] [Google Scholar]
- 28.Palmer S, Alaeus A, Albert J, Cox S. AIDS Res Hum Retroviruses. 1998;14:157–162. doi: 10.1089/aid.1998.14.157. [DOI] [PubMed] [Google Scholar]