

Abstract
A model system for biological Rieske clusters that incorporates bis-benzimidazolate ligands (Prbbim)2- has been developed (PrbbimH2 = 4,4-bis(benzimidazol-2-yl)heptane). The di-ferric and mixed-valence clusters have been prepared and characterized in both their protonated and deprotonated states. The thermochemistry of interconversions of these species has been measured, and the effect of protonation on the reduction potential is in good agreement to that observed in the biological systems. The mixed-valence and protonated congener [Fe2S2(Prbbim)(PrbbimH)](Et4N)2 (4) reacts rapidly with TEMPO or p-benzoquinones to generate di-ferric and deprotonated [Fe2S2(Prbbim)2](Et4N)2 (1) and 1 equiv of TEMPOH or 0.5 equiv p-benzohydroquinones, respectively. The reaction with TEMPO is the first well-defined example of a concerted proton-electron transfer (CPET) at a synthetic ferric/ferrous [Fe-S] cluster.
Iron-sulfur clusters are ubiquitous electron-transfer (ET) cofactors and they play an integral role in many enzymes that catalyze multi-e—/H+ redox events.1 In some cases, the [Fe-S] clusters are known to undergo proton-coupled electron transfer (PCET).2 Biochemical and model studies are providing insight into the PCET reactivity of low-valent CO-ligated [Fe-S] clusters in hydrogenases,3 but much less is known about the involvement of protons in the more widespread high-valent clusters.2 The [2Fe-2S] Rieske cluster, in which one Fe is ligated by two Cys residues and the other by two His residues, is perhaps the best characterized biological [Fe-S] cluster that undergoes PCET.2a,4 This PCET reactivity enables Rieske clusters to act as structural gates in oxygenase enzymes (reduction/protonation of the cluster induces structural changes that affect the active site),5 and PCET is their primary function in the Q-cycle of the mitochondrial and photosynthetic electron transport chains.4b,6 In the latter role, the di-ferric Rieske cluster accepts a net H-atom (e–+H+) from a hydroquinone (H2Q), reducing the cluster to the mixed-valence state and protonating the imidazolate (His) ligand. The mechanism of H2Q oxidation by the Rieske cluster is a topic of debate, with both stepwise proton transfer/electron transfer (PT-ET) and concerted proton-electron transfers (CPET) having been invoked.4b,7 This issue is important because the ET steps within the Q-cycle are tightly regulated to avoid energetically wasteful short-circuits and formation of reactive oxygen species.6a,8
Reported here are studies of the PCET reactivity of a model Rieske cluster, including the first example of CPET reactivity of a non-carbonyl [Fe-S] cluster.3b Four congeners of a family of bisbenzimidazolate ligated [2Fe-2S] clusters have been prepared and interconverted, thus defining the full PCET square scheme (1-4, Scheme 1). The protonated congeners are the first characterized examples of protonated high-valent [Fe-S] clusters.9
Scheme 1.

Square Scheme for [Fe2S2(Prbbim)x(PrbbimH)2-x]n– Clusters
Di-ferric and mixed-valence bis-benzimidazolate ligated clusters were first prepared and characterized by Gibson (including 1 and 2),10 and recent detailed spectroscopic studies have been done by Franc Meyer and coworkers.11 Di-ferric [FeIII2S2(Prbbim)2](Et4N)2 (1) and mixed-valence [FeIII/II2S2(Prbbim)2](Et4N)3 (2) are readily prepared by minor modification of the literature procedures.10a Clusters 1 and 2 are comprised of high-spin iron centers that are antiferromagnetically coupled to give S = 0 and S = ½ ground-states, respectively.10b,10c,111H NMR spectra of 1 and 2 each display 9 well-resolved resonances close to the diamagnetic region (δ 0.9–12.2 ppm; Figure 1a,c and Supporting Information Figures S2–S5), indicating D2h symmetry in solution. The (Prbbim)2- to Et4N+ ratios of 1:1 for 1 and 2:3 for 2 confirm the stoichiometries. The latter spectrum also indicates valence delocalization on the NMR timescale for 2.10b
Figure 1.

1H NMR spectra (d7-DMF) of: (a) mixed-valence 2 (-20 °C); (b) reaction of 2 with DMAP-H+ to give mixed-valence and protonated 4 (-20 °C); (c) reaction of 4 with 0.5 equiv of tBu2Q to give 1 and 0.5 equiv tBu2H2Q. Legend:  = 2;
= 2;  = 1;
= 1;  = tBu2H2Q; ● = DMAP; ○ = Et4N+; * = DMF.
= tBu2H2Q; ● = DMAP; ○ = Et4N+; * = DMF.
The solid-state structures of both 1 and 2 have been obtained. The structures are very similar and that of 2 is shown in Figure 2. The metrical parameters of 1, d(Fe-Nave) = 1.978 Å and d(Fe-Save) = 2.197 Å, are as expected for di-ferric clusters.11-12 Upon reduction, the Fe-Nave and Fe-Save bond distances elongate by ca. 0.07 Å and 0.03 Å, respectively. The iron centers in 2 are crystallographically unique (unlike the one other mixed-valence [2Fe-2S] structure11), with one iron having Fe-Save and Fe-Nave distances that are statistically longer than the other. This is consistent with Mössbauer studies on related compounds indicating that the valence may be partially localized in the crystal at 110K.10c,11
Figure 2.

Thermal ellipsoid (50%) representation of mixed-valence cluster 2. Hydrogen atoms, Et4N+, and minor components of disorder have been omitted for clarity; see the SI for details and metrical comparisons of 1 and 2.
Di-ferric 1 is converted to the protonated derivative 3 by 1 equiv of [pyH]OTf in MeCN. By analogy with the biological Rieske clusters, we tentatively assign the site of protonation as a benzimidazolate nitrogen, [FeIII2S2(Prbbim)(PrbbimH)](Et4N). The IR spectrum of 3 shows no discernible N-H or S-H stretch and there are no noticeable differences between the IR spectra of 3 and 3-D (Figure S14).
When the protonation is monitored by optical spectroscopy, isosbestic points are observed with ≤ 1 equiv [pyH]OTf, and addition of DBU regenerates 1 (DBU = 1,8-diazabicycloundec-7-ene; Figure S11). Reversible protonation is observed by 1H NMR spectroscopy as well. 1H NMR spectra of 3 show a single set of ligand resonances, indicating that proton transfer among the four benzimidazolates is rapid on the NMR timescale (Figure S6). NMR spectra of mixtures of 1 and 3 also show a single set of ligand resonances that shift according to the ratio of 1 and 3, indicating that intermolecular PT is fast between di-ferric clusters (Figure S7). No decoalescence of the resonances for 3 is observed upon cooling samples in CD2Cl2 (-70 °C), d8-THF (-50 °C), d7-DMF (-50 °C), or CD3CN (-30 °C).
Complex 3 is unstable in MeCN solution, decomposing over the course of hours to PrbbimH2, 1, and unidentified iron-containing species (t1/2 ca. 4 h at 22 °C; 3.8 mM solution). The formation of both PrbbimH2 and 1 suggests that the decomposition involves a second protonation. Protonation has been previously shown to facilitate ligand exchange and decomposition in synthetic [Fe-S] clusters.9b,12
Mixed-valence 2 can likewise be protonated, with [DMAP-H]OTf or [pyH]OTf, to generate [FeIII/II2S2(Prbbim)(PrbbimH)]-(Et4N)2 (4) (4-DMAP = 4-dimethylaminopyidine). Again, this protonation is reversible, with 1H NMR spectra showing regeneration of 2 upon addition of the strong non-coordinating base tbutylimino-tri(pyrrolidino)phosphorane (tBuNP(pyrr)3; Figure S9). Solutions of mixed-valence 4 in MeCN show no degradation over at least 30 min at –24 °C, but decompose at 22 °C with a t1/2 of ca. 20 min. Complex 4 is thus less stable than di-ferric 3. The decomposition products have not been identified, but their 1H NMR spectra suggest formation of a monomeric iron(II) species (a similar species forms upon mixing FeCl2 and Prbbim2Tl2, Figure S25), indicating that the Fe2S2 core does not stay intact. NMR experiments show that 3 and 4 can be interconverted with [Cp2Fe]PF6 and Cp2Co, although not without some decomposition.
Mixed-valence and protonated 4 has a solution magnetic moment of 2.4 B.M. in DMF-d7 at -25 °C. The EPR spectrum of 4 was obtained, and is shown in Figure 3a. The spectrum can be fit to a rhombic g tensor with g = [1.983, 1.943, 1.860] and corresponding linewidths W = [61, 22, 84 G]. For comparison, the EPR spectrum of 2 obtained under similar conditions has g = [2.012, 1.940, 1.835] and W = [40, 70, 145 G] (Figure S13). Thus 4 has an S = ½ ground-state like 2.
Figure 3.

(a) X-band EPR spectrum of 4 collected at 120 K in a 2:3 MeCN:toluene glass (solid black) and the calculated fit (dotted red; displaced vertically from the experimental). (b) CVs of 2 in the absence (yellow) and presence (purple) of 1 equiv [pyH]OTf at -20 °C in 0.25 M [nBu4N]PF6 in MeCN, with scan-rates of 100 mV s-1.
Protonated 4 is essentially NMR silent (Figures 1b and S8), in contrast to the sharp signals observed for 2. Only resonances ascribed to the Et4N+ cations are readily observed within ±170 ppm, in both CD3CN (-20 to 25 °C) and d7-DMF (-50 to 25 °C). NMR spectra of mixtures of 2 and 4 show a single set of resonances that is not shifted from that of 2 and integrate according to the mole fraction of 2 in solution (relative to the conjugate base or Et4N+). Thus PT between 2 and 4 is slow on the NMR timescale, and the lack of an observable spectrum for 4 cannot be due to exchange with 2. Most likely the larger NMR linewidths for 4 are due to a change in the electronic relaxation upon protonation of 2.13 The electronic relaxation of mixed-valence [2Fe-2S] clusters is dominated by an Orbach mechanism at these temperatures,14 and hence perturbation of the excited-state energies will affect NMR linewidths.
The ability to generate the protonated congeners 3 and 4 allows for the direct measurement of their pKas. Solutions of diferric 3, generated in situ from 1 and [pyH]OTf, were titrated with quinuclidine and monitored by optical spectroscopy. Using the quinuclidine pKa of 19.51 in MeCN,15 the titrations give a pKa of 20.6 ± 0.2. For comparison, the protonated congener of [Fe2S2Cl4]2- has a pKa of 18.1 in MeCN (determined kinetically by Henderson because the protonated species is too unstable to observe).16 That 1 is 2.5 units more basic than [Fe2S2Cl4]2- despite having 0.2 V more positive redox potential17 supports the suggestion that the site of protonation is different in the two clusters, N in 1 and S in [Fe2S2Cl4]2-.
Titrations of CD3CN solutions of 4 (generated in situ from 2 and [DMAP-H]OTf or [pyH]OTf) with DBU (pKa = 24.31 in MeCN)18 were monitored by 1H NMR spectroscopy (see SI). Assuming mass balance, integration of the ArH resonance of 2 at 12.16 ppm gives the relative amounts of 2 and (NMR silent) 4. A pKa of 24.7 ± 0.4 was obtained for 4.
Cyclic voltammograms (CVs) of 1 or 2 show two electro-chemically reversible couples at -1.225 (± 0.003) and -2.198 (± 0.008) V vs. Fc/Fc+ (Figures 3b and S18). These correspond to the FeIII2/FeIII/II2 and FeIII/II2/FeII2 couples, respectively. The large 0.973 V separation between the two couples is consistent with the observed stability of 2 towards redox disproportionation.
In contrast, the CVs of 4 and 3 show several redox events (Figures 3b and S20). This behavior is not due to species degradation, as addition of base regenerates the CVs of 2 and 1. Additionally, CVs recorded at -20 °C are qualitatively similar to those obtained at room temperature. In the region of the FeIII2/FeIII/II2 couple, the CV of 4 features two broad redox events that are anodically shifted relative to that of 2. The first wave, assigned to the 3/4 couple, is estimated to be -0.98 ± 0.02 V (from the shift in the oxidation peak of 2 in the presence of 1 equiv [pyH]OTf, see SI). The more anodic wave, at ~–0.6 V, likely corresponds to the couple of a doubly protonated cluster (see SI).
The thermochemistry of interconversion of the cluster congeners is summarized in Scheme 1. The measured change in pKa upon reduction corresponds to a free energy change of 5.6 ± 0.4 kcal mol-1, and is in excellent agreement with the measured change in E1/2 upon protonation (5.65 ± 0.10 kcal mol-1), which must be equal by Hess's Law.19 These changes are also in accord with those observed in biological Rieske clusters. For example, a change in pKa of 6.6 ± 0.4 kcal mol-1 is observed upon reduction of the Rieske cluster from the cytochrome bc1 complex of Rhodobacter sphaeroides (RsRp).4a This similar extent of thermodynamic coupling in the two systems suggests that the site of protonation is similar in both clusters.
The bond dissociation free energy (BDFE) of the NH bond of 4 is 60.5 ± 1.0 kcal mol-1, as defined by the pKa and E1/2 values (BDFE = 23.06E1/2 + 1.37pKa + CG; CG = 54.9 ± 1.0 kcal mol-1 in MeCN).19 This BDFE is 14.6 kcal mol-1 less than that of RsRp (75.1 ± 1.0 kcal mol-1).4a Compounds 1-4 are thus a reasonable model for Rieske clusters in terms of the thermodynamic coupling mentioned above, but are substantially more reducing than the biological systems (4 is a better H-atom donor). This is perhaps due to the presence of three anionic ligands in 4 vs. two anionic ligands in the mixed-valence biological Rieske congener (both His ligands are protonated in the mixed-valence state). This discrepancy could also be due to the differences in the ligand type: benzimidazolate in 4 and imidazolate/thiolate in Rieske clusters.
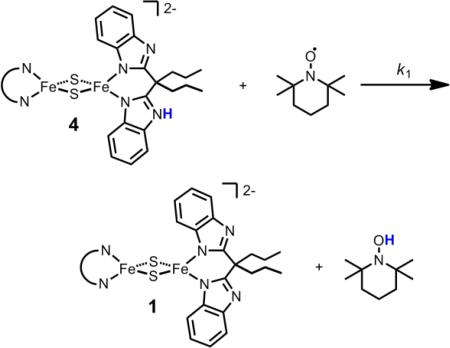
The relatively weak BDFE indicates that H-atom abstraction from 4 should be facile. Indeed, in situ generated 4 reacts quantitatively with 1 equiv of the nitroxyl radical TEMPO to generate oxidized and deprotonated 1 plus 1 equiv TEMPO-H, as ascertained by NMR spectroscopy (eq 1;  = Prbbim2-; Figure S15).
= Prbbim2-; Figure S15).
 |
(1) |
To gain insight into the mechanism of reaction 1, double-mixing stopped-flow kinetic measurements in MeCN were undertaken, under pseudo-first-order conditions of excess TEMPO (Figures S22-S23). Solutions of 2 (0.74 mM) and [DMAP-H]OTf (0.69 mM) were mixed to generate 4 in situ, and after a delay time of 10 s, 4 was mixed with TEMPO (3.2 – 8.5 mM). The rate constant at 25 °C is 2200 ± 350 M-1 s-1, equivalent to ΔG‡ = 12.9 ± 0.3 kcal mol-1. The temperature dependence of k1 (-25 to 50 °C) gives ΔS‡1 = -38 ± 2.4 cal K-1 mol-1 and ΔH‡1 = 1.6 ± 0.3 kcal mol-1. These values are similar to those obtained for the PCET reaction between monomeric and high-spin Fe(H2bim)32+ and TEMPO (H2bim = 2,2'-bi-2-imidazoline).20
The rate constant and thermochemical data indicate that the TEMPO reaction proceeds by concerted transfer of the e– and H+ (called CPET or H-atom transfer, HAT).19 The free energy of this step, from the BDFEs of 4 (see above) and of TEMPO-H,19 is ΔG°CPET = –6.0 kcal mol-1. The alternative stepwise pathways of initial ET followed by PT (with 3 + TEMPO– as intermediates) or PT followed by ET (via 2 + TEMPO-H·+), have very unfavorable free energies associated with the initial steps: ΔG°ET = +22.4 kcal mol-1; ΔG°PT = +22.3 kcal mol-1. Since these free energies are both much larger than the measured free energy barrier (ΔG‡1 = 12.9 ± 0.3 kcal mol-1), these pathways are ruled out. As further evidence against initial ET, TEMPO is unreactive with 2 in CD3CN, even though 2 is a better outer-sphere reductant than 4 (ΔG°ET(2+TEMPO) = +16.7 kcal mol-1).
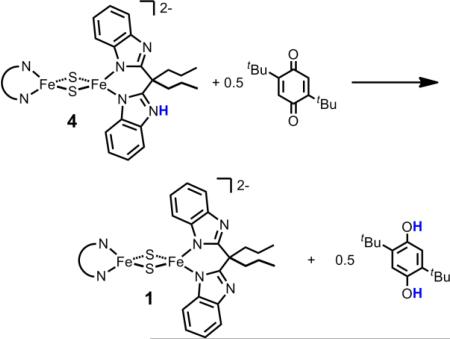
Complex 4 also reacts quantitatively with 0.5 equiv of 2,5-ditbutyl-p-benzoquinone (tBu2Q) to give 1 and 2,5-ditbutyl-p-hydroquinone (tBu2H2Q) (eq 2 and Figure 1). Similar reactivity is also observed with duraquinone and ubiquinone. Hydroubiquinone is the biological reactant for the Rieske cluster in the mitochondrial bc1 complex Q-cycle. Because of the difference in BDFEs, the biological reaction runs in the opposite direction, the Rieske cluster oxidizing the hydroquinone to the semiquinone radical.4b,7 Mechanistic studies of quinone reactions are in progress. These are mechanistically complex as they are 2e–/2H+ reductions that proceed via the anionic and/or neutral semiquinone, SQ•—/HSQ•. Preliminary results indicate that 4 reacts with [tBu2SQ•—]Na(15-crown-5) to give mixtures of 1 (45 %) and 2 (55 %). The semiquinone anion has been observed spectroscopically in the bc1 complex (albeit under conditions that favor superoxide formation),8b and thus it may be a relevant species in the H-atom transfer to the Rieske cluster.
 |
(2) |
In sum, we have prepared and characterized all four congeners of a bis-benzimidazolate ligated [2Fe-2S] cluster system which serves as a Rieske cluster model. The ability to prepare the protonated congeners 3 and 4 allows for the complete thermochemical properties of the system to be established, including pKa values for the diferric and ferric/ferrous [Fe-S] clusters. Though the BDFE of 4 is ca. 14 kcal mol-1 less than that observed at biological clusters, a similar degree of thermodynamic coupling is observed. Mixed-valence and protonated 4 undergoes concerted proton-electron transfer with TEMPO to give oxidized and deprotonated 1, the first example of CPET involving a ferrous/ferric [Fe-S] cluster. Cluster 4 also reacts with quinones to give 1. The latter is the microscopic reverse of the proposed first step in the mitochondrial Q-cycle.
Supplementary Material
ACKNOWLEDGMENTS
We thank Prof. Stefan Stoll for help with EPR experiments.
Funding Sources
We gratefully acknowledge financial support from the U.S. National Institute of Health (R01GM50422 to J.M.M. and 1F32GM099316 to C.T.S.)
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental procedures and characterization data are available (.pdf), as well as crystallographic details for 1 and 2 (.cif). This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.a Lippard SJ, Berg JM. Principles of Bioinorganic Chemistry. University Science Books; Mill Valley, CA: 1994. [Google Scholar]; b Beinert H, Holm RH, Münck E. Science. 1997;277:653. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 2.a Zu Y, Fee JA, Hirst J. J. Am. Chem. Soc. 2001;123:9906. doi: 10.1021/ja016532c. [DOI] [PubMed] [Google Scholar]; b Bak DW, Zuris JA, Paddock ML, Jennings PA, Elliott SJ. Biochemistry. 2009;48:10193. doi: 10.1021/bi9016445. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Camba R, Jung Y-S, Hunsicker-Wang LM, Burgess BK, Stout CD, Hirst J, Armstrong FA. Biochemistry. 2003;42:10589. doi: 10.1021/bi035021v. [DOI] [PubMed] [Google Scholar]; d Lanzilotta WN, Christiansen J, Dean DR, Seefeldt LC. Biochemistry. 1998;37:11376. doi: 10.1021/bi980048d. [DOI] [PubMed] [Google Scholar]
- 3.a Franz JA, Lee S-J, Bowden TA, Alnajjar MS, Appel AM, Birnbaum JC, Bitterwolf TE, Dupuis M. J. Am. Chem. Soc. 2009;131:15212. doi: 10.1021/ja904602p. [DOI] [PubMed] [Google Scholar]; b Olsen MT, Rauchfuss TB, Wilson SR. J. Am. Chem. Soc. 2010;132:17733. doi: 10.1021/ja103998v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a Zu Y, Couture MMJ, Kolling DRJ, Crofts AR, Eltis LD, Fee JA, Hirst J. Biochemistry. 2003;42:12400. doi: 10.1021/bi0350957. [DOI] [PubMed] [Google Scholar]; b Hsueh K-L, Westler WM, Markley JL. J. Am. Chem. Soc. 2010;132:7908. doi: 10.1021/ja1026387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferraro DJ, Gakhar L, Ramaswamy S. Biochem. Biophys. Res. Commun. 2005;338:175. doi: 10.1016/j.bbrc.2005.08.222. [DOI] [PubMed] [Google Scholar]
- 6.a Osyczka A, Moser CC, Dutton PL. Trends Biochem. Sci. 2005;30:176. doi: 10.1016/j.tibs.2005.02.001. [DOI] [PubMed] [Google Scholar]; b Berry EA, Guergova-Kuras M, Huang L-S, Crofts AR. Annu. Rev. Biochem. 2000;69:1005. doi: 10.1146/annurev.biochem.69.1.1005. [DOI] [PubMed] [Google Scholar]
- 7.a Lhee S, Kolling DRJ, Nair SK, Dikanov SA, Crofts AR. J. Biol. Chem. 2010;285:9233. doi: 10.1074/jbc.M109.043505. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cape JL, Bowman MK, Kramer DM. J. Am. Chem. Soc. 2005;127:4208. doi: 10.1021/ja043955g. [DOI] [PubMed] [Google Scholar]
- 8.a Cape JL, Aidasani D, Kramer DM, Bowman MK. Biochemistry. 2009;48:10716. doi: 10.1021/bi901205w. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Cape JL, Bowman MK, Kramer DM. Proc. Natl. Acad. Sci. U.S.A. 2007;104:7887. doi: 10.1073/pnas.0702621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a Tanaka K, Moriya M, Tanaka T. Inorg. Chem. 1986;25:835. [Google Scholar]; b Henderson RA. Coord. Chem. Rev. 2005;249:1841. [Google Scholar]
- 10.a Beardwood P, Gibson JF. J. Chem. Soc., Chem. Commun. 1986:490. [Google Scholar]; b Beardwood P, Gibson JF. J. Chem. Soc., Dalton Trans. 1992:2457. [Google Scholar]; c Ding XQ, Bill E, Trautwein AX, Winkler H, Kostikas A, Papaefthymiou V, Simopoulos A, Beardwood P, Gibson JF. J. Chem. Phys. 1993;99:6421. [Google Scholar]
- 11.Albers A, Demeshko S, Dechert S, Bill E, Bothe E, Meyer F. Angew. Chem. Int. Ed. 2011;50:9191. doi: 10.1002/anie.201100727. [DOI] [PubMed] [Google Scholar]
- 12.Rao PV, Holm RH. Chem. Rev. 2004;104:527. doi: 10.1021/cr020615+. [DOI] [PubMed] [Google Scholar]
- 13.Bertini I, Luchinat C, Parigi G. Solution NMR of Paramagnetic Molecules Applications to Metallobiomolecules and Models. Vol. 2. Elsevier; Amsterdam: 2001. [Google Scholar]
- 14.Beardwood P, Gibson JF, Bertrand P, Gayda J-P. Biochimica et Biophysica Acta (BBA) Protein Structure and Molecular Enzymology. 1983;742:426. doi: 10.1016/0167-4838(83)90330-8. [DOI] [PubMed] [Google Scholar]
- 15.Izutsu K. Acid-Base Dissociation Constants in Dipolar AproticSolvents. Blackwell Scientific Publications; Oxford: 1990. [Google Scholar]
- 16.Bates K, Garrett B, Henderson RA. Inorg. Chem. 2007;46:11145. doi: 10.1021/ic7015484. [DOI] [PubMed] [Google Scholar]
- 17.Wong GB, Bobrik MA, Holm RH. Inorg. Chem. 1978;17:578. [Google Scholar]
- 18.Kaljurand I, Kütt A, Sooväli L, Rodima T, Mäemets V, Leito I, Koppel IA. J. Org. Chem. 2005;70:1019. doi: 10.1021/jo048252w. [DOI] [PubMed] [Google Scholar]
- 19.Warren JJ, Tronic TA, Mayer JM. Chem. Rev. 2010;110:6961. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mader EA, Davidson ER, Mayer JM. J. Am. Chem. Soc. 2007;129:5153. doi: 10.1021/ja0686918. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.