

Abstract
Both cell-autonomous and non-cell-autonomous factors contribute to tumor growth and metastasis of melanoma. The function of Caveolin-1 (Cav1), a multifunctional scaffold protein known to modulate several biological processes in both normal tissue and cancer, has been recently investigated in melanoma cancer cells, but its role in the melanoma microenvironment remains largely unexplored. Here, we show that orthotopic implantation of B16F10 melanoma cells in the skin of Cav1KO mice increases tumor growth, and co-injection of Cav1-deficient dermal fibroblasts with melanoma cells is sufficient to recapitulate the tumor phenotype observed in Cav1KO mice. Using indirect co-culture experiments with fibroblasts and melanoma cells combined with cytokine analysis, we found that Cav1-deficient fibroblasts promoted the growth of melanoma cells via enhanced paracrine cytokine signaling. Specifically, Cav1-deficient fibroblasts displayed increased ShhN expression, which heterotypically enhanced the Shh signaling pathway in melanoma cells. In contrast to primary tumor growth, the ability of B16F10 melanoma cells to form lung metastases was significantly reduced in Cav1KO mice. This phenotype was associated mechanistically with the inability of melanoma cells to adhere to and to transmigrate through a monolayer of endothelial cells lacking Cav1. Together, our findings demonstrate that Cav1 may regulate different mechanisms during primary melanoma tumor growth and metastatic dissemination.
INTRODUCTION
Tumors are heterogeneous microenvironments that consist of both neoplastic and non-neoplastic cells (tumor-stroma). Tumor growth and the consequent metastatic dissemination of tumor cells result from continuous reciprocal interactions between cancer cells and their surrounding stroma (1, 2). Cutaneous melanoma remains the most aggressive type of skin cancer and both cell-autonomous and non cell-autonomous mechanisms are necessary for melanoma growth and metastasis (3). Recent research, in fact, has demonstrated that stromal cells (fibroblasts and endothelial cells) support the growth and dissemination of melanoma cells by modulating angiogenesis, secreting growth factors and cytokines, and contributing to extracellular matrix (ECM) deposition and degradation (4). Thus, identifying novel mechanisms critically regulating tumor-stroma interactions may be therapeutically relevant in this type of cancer.
Caveolae are specialized microdomains of the plasma membrane enriched in the scaffold protein Caveolin-1 (Cav1) (5, 6). Due to the multitude of interacting proteins described, Cav1 has been implicated in the modulation of many biological processes in both normal tissue and cancer (8, 35). Although much research has primarily focused on determining the function of Cav1 in cancer cells, recent studies have started to investigate the function of Cav1 protein in the tumor microenvironment (9, 10). Indeed, Cav1 is highly expressed in endothelial cells and fibroblasts, two of the cell types that are normally involved in stromal remodeling during melanoma progression (3). In addition, the angiogenesis defects (11) and impaired skin wound-healing (12) displayed by Cav1KO mice suggest that loss of Cav1 in the stromal compartment may functionally affect tumor-stromal interactions in melanomagenesis.
To examine this issue we used Cav1KO mice to determine whether stromal Cav1 may affect the growth and metastatic ability of B16F10 melanoma cells. We show that absence of Cav1 promotes the growth of intradermally implanted B16F10 melanoma cells in mice. Indirect co-culture experiments and co-injections of fibroblasts and melanoma cells demonstrate that lack of Cav1 in dermal fibroblasts promotes the growth of melanoma cells in vitro and in vivo via paracrine cytokine signaling. In contrast, the ability of B16F10 cells to form lung metastases in Cav1KO mice was significantly impaired. These results were consistent with the inability of B16F10 cells to transmigrate through a monolayer of HUVEC cells lacking Cav1. Collectively, our data suggest functionally distinct roles for stromal Cav1 in melanoma primary tumor growth and metastasis.
METHODS
Materials
Antibodies and their sources were as follows: Cav1 (N-20), PECAM-1 (M20), eGFP (sc-8334) and Shh(N-19) were from Santa Cruz. Keratin-14 (K14) was from Covance. Rat anti-mouse VCAM1 and ICAM1 were from R&D. Gli-1 was from Cell Signaling. β-Tubulin, was from Sigma and glyceraldehyde-3-phosphate-dehydrogenase (GAPDH) was from Fitzgerald.
Animal Studies
3-4-month-old Cav1WT (WT), Cav1KO (13), and Cav2KO (14) C57Bl/6 female mice were used for orthotopic and i.v. injections of B16F10 cells. For co-injection experiments, 3-4-month-old athymic female mice (NCr-Nu; Taconic) were used (15, 16). All in vivo studies were approved by the Institutional Animal Care and Use Committee (IACUC) of Thomas Jefferson University.
Cell lines
B16F10 and A-375 were from ATCC, while human immortalized dermal foreskin fibroblasts (hTBJ1) were originally from Clontech. Human Umbilical Vein Endothelial Cells (HUVEC) were purchased from ALLCELLS. Only early passages of these cell lines were used in experiments.
Neonatal dermal fibroblasts
Dermal fibroblasts (FB) and primary mouse keratinocytes (MK) were isolated from the skin of newborn mice (1-3-day-old) as described in details by others (18). FB were suspended in medium with 10% FBS-DMEM (Invitrogen) and initially plated at a density of 5,000 cells/cm2.
Co-injection experiments
WT, Cav1KO and Cav2KO neonatal FB (see above) were intradermally co-injected with 105 B16F10 cells in nude mice at 5:1 ratios. Similarly, hTBJ1 fibroblasts were co-injected with human A-375 melanoma cells (19, 20).
Lentiviral vectors
For lentivirus mediated silencing of the Cav1 gene, pre-designed control shRNAmiR (shCtrl-miR) and shRNAmiRs (shCAV1-miRs) targeting the human CAV1 mRNA (NM_001753.3) were purchased from Invitrogen and subcloned into the pRRLsin.cPPT.hCMV.eGFP.WPRE (LV-eGFP) lentiviral vector (21). The resulting constructs (LV-shCtrl-miR-eGFP and LV-shCAV1-miRs-eGFP) were packaged according to standard protocols (21). Effective CAV1 knockdown in target cells was determined by Western Blot analysis of FACS sorted eGFP positive cells. A lentiviral vector (Lv105-Puro) encoding mCherry cDNA was from Genecopoeia whereas ready to use shCtrl-Puro and shGli-1-Puro (gene ID: 14632) lentiviral particles were from Santa Cruz.
Co-cultures of Fibroblasts and Melanoma cells
Direct cocultures (direct cell-cell contact) were established by seeding 104/cm2 mCherry-labeled melanoma cells on eGFP-labeled (shCtrl/shCAV1 cells) or on unlabeled FB (1:5 ratios) that had previously been serum-activated as described below for the conditioned medium experiments. 48h or 72h after the initial plating (22) co-cultures were harvested and their relative growth determined by Flow Cytometry analysis (FACS). Transwell cocultures of fibroblasts and melanoma cells (no cell-cell contact) were established as described above by using Transwell inserts with 0.4μm-pore-size membrane inserts (23).
Conditioned medium experiments
Conditioned medium (CM) was collected from dermal FB that were maintained in serum free medium for 48h (SFM). FB were then serum activated by maintaining them for 12h in 10%FBS-DMEM and 24h in 1%FBS-DMEM (24). CM from these cells (SA-CM) was then incubated with melanoma cells for 48h. For Shh-pathway inhibition studies, Cyclopamine (Selleck) or DMSO were added to CM for 48h. Recombinant Shh protein (Peprotech) was used for hedgehog signaling activation in melanoma cells.
Cytokine Array and ELISA
Cytokine array and ELISA for ShhN were performed on FB-CM using commercially available kits from Raybiotech and R&D.
Immunohistochemistry
S100 (RAb;Dako) immunohistochemical staining of 5μm paraffin embedded tumor sections was performed as previously described by us (15).
Tumor angiogenesis
Microvessel density (MVD) was determined by CD31 (1:500; sc-1506) immunohistochemical staining of 5μm paraffin embedded tumor sections (25). CD31-positive vessels were counted in 5-6 specimens per group (five fields/sample) using a 20x objective and an ocular grid (0.25 mm2 per field). CD31 immunoblotting of whole-tumor lysates was also performed (26, 27).
[3H]Thymidine Incorporation Assay and Growth Curves
DNA synthesis was determined by incubating asynchronously growing cells with 0.5 μCi/ml of [3H]Thymidine (Perkin-Elmer) for 18 hours (28). Growth of melanoma cells was assessed by MTT assay at 0, 24, 48, 72, and 96 hours after plating (29).
Western blotting
Homogenized tissue samples and cells were sonicated and lysed in a modified radioimmunoprecipitation assay buffer and processed for immunoblot analysis as previously described (27).
Tumor-Cell Transendothelial Migration Assay
HUVEC (5×104) were grown to confluence (72h) on top of an 8-μm-pore size gelatin-coated membrane (Transwells; BD Biosciences). 1×105 [3H]Thymidine labeled or mCherry labeled B16F10 cells in 500μl of 0.1%BSA-DMEM were added to the transwell inserts. Serum-free NIH3T3 conditioned medium (48h) was used as a chemoattractant. After 6h, transwells were washed with PBS and wiped with cotton swabs. Membranes were removed and the amount of radioactivity determined by liquid scintillation counting (LSC) (30). Alternatively, inserts were fixed with 4% PFA, wiped with cotton swabs, and mounted onto glass slides. Migrated mCherry-B16F10 cells were imaged by confocal microscopy (LSM510.META.Confocal; Zeiss).
Tumor-Endothelial Cell Adhesion Assay
[3H]-Thymidine labeled B16F10 cells (1×105) were incubated on top of a monolayer of HUVEC cells in 0.1%BSA-DMEM at 37 °C for 30 min. After being washed with PBS, cells were solubilized in 0.5N NaOH/0.1% SDS and the amount of radioactivity determined by LSC (30). For neutralizing antibodies experiments, HUVEC monolayers were pre-treated for 30min with VCAM-1 (sc-20070), ICAM-1 (sc-59787), or IgG isotype control (10μg/ml each) (31).
TNFα induced ICAM1 and VCAM1 expression in mice
3-4-month-old WT and Cav1KO female mice were IP injected with 25 μg/kg of mouse recombinant TNFα (R&D). After 5 h, mice were sacrificed and lungs were cleared of blood by infusing cold PBS through the right ventricle (32). The lung left lobe was then collected and processed for western blot analysis (27).
Statistical Analysis
Results are represented as means ± SEM. Statistical analysis was performed using GraphPad Software.
RESULTS
Cav1 ablation in mice promotes the growth of B16F10 melanoma cells independently of Cav2
In order to determine whether absence of Cav1 in the skin affects B16F10 cell growth, we examined the growth of B16F10 cells orthotopically (intradermally) implanted in the skin of WT and Cav1KO C57Bl/6 female mice. After 18 days, analysis of tumor size revealed that their growth was enhanced (~2 fold) in Cav1KO mice (Fig. 1A). Caveolin-2 (Cav2) is normally co-expressed and heteroligomerizes with Cav1. Previous studies have reported reduced Cav2 levels in Cav1KO mice (13). Thus, the Cav1KO tumor phenotype may be confounded by Cav2 loss or reduced function. Relative to WT and Cav1KO animals, B16F10 cells implanted in the skin of Cav2KO mice grew more slowly (~1.5 fold), indicating that the Cav1KO tumor phenotype was Cav2 independent (Fig.1B). Similar reductions in tumor growth were observed in subcutaneously injected Cav2KO mice (Fig.S1), indicating a growth promoting role for Cav2 in melanoma. CD31 immunohistochemical staining of Cav1KO tumor sections revealed increased MVD relative to WT and Cav2KO tumors, respectively. These results were corroborated by CD31 immunoblots of whole tumors lysates (Fig.1C). Taken together, these findings suggest that growth of B16F10 in Cav1KO mice correlates with their MVD and this effect is independent of Cav2.
Figure 1. Cav1 ablation in mice promotes the growth of B16F10 melanoma cells independently of Cav2.
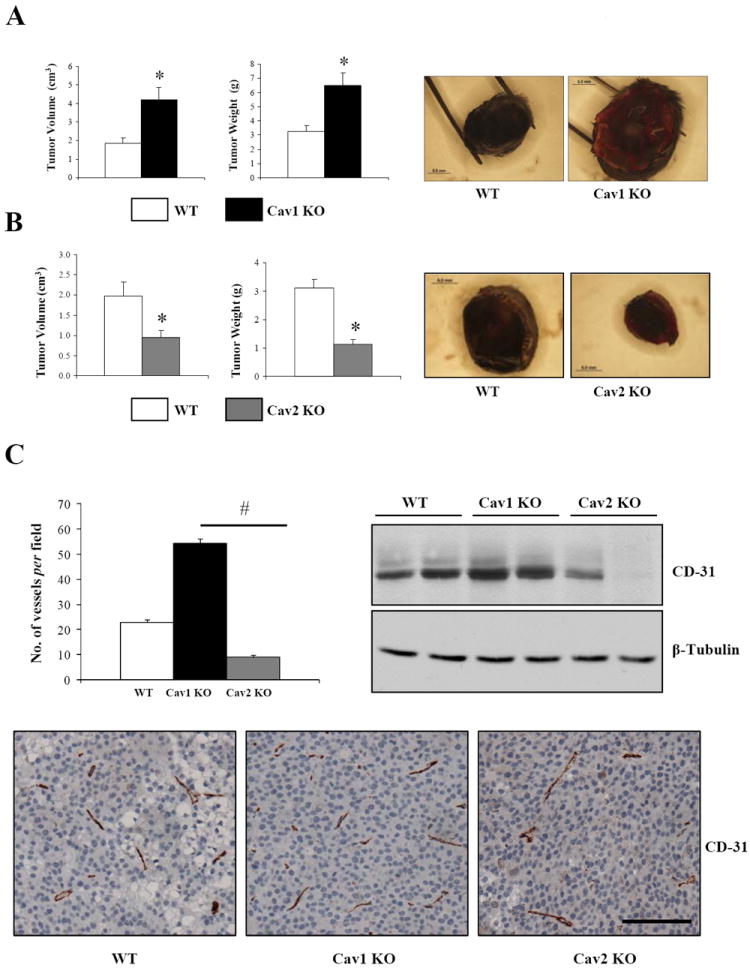
106 B16F10 melanoma cells were orthotopically (intradermally) implanted in the skin of 3-4-month-old WT, Cav-1 KO (A) and Cav2KO (B) C57Bl/6 female mice (n≥8, per group). After 18 days, tumors were excised and their size determined. Representative images of tumors are displayed on the right. C, CD31 immunohistochemistry of tumor sections showing that microvascular density correlates with tumor size in Cav1KO and Cav2KO mice (n=5, per group). CD31 immunoblotting of whole-tumor lysates is shown. Results are means ± SEM shown (* #, P < 0.05, by two tailed Mann-Whitney and by Dunnett’s Multiple Comparisons Test; Scale Bar, 100μm).
Absence of Cav1 but not Cav2 in dermal fibroblasts enhances the growth of melanoma cells in co-injection experiments
Dermal fibroblasts are the main cell components of the skin important in maintaining the normal physiological functions of this organ. To determine whether loss of Cav1 in dermal fibroblasts was sufficient to recapitulate the tumor phenotype observed in Cav1KO mice, dermal xenografts were established in nude mice by coinjecting WT and Cav1KO neonatal dermal fibroblasts with B16F10 melanoma cells at 5:1 ratios. After 14 days, analysis of tumor size showed that Cav1KO fibroblasts promoted the growth of B16F10 cells. S100b immunohistochemical staining (melanoma cell marker) and trichrome staining of tumor sections showed that collagen deposition and/or stromal cell proliferation was unchanged in B16F10/WT and B16F10/Cav1KO tumors (Fig.2A). In contrast, the growth of tumors resulting from coinjecting B16F10/Cav2KO fibroblasts was comparable to the growth of their controls. Interestingly, immunoblot analysis revealed that Cav1 expression was maintained in Cav2KO fibroblasts whereas Cav2 levels were slightly diminished in Cav1KO fibroblasts (Fig.2B). The tumor promoting role of Cav1 deficient fibroblasts was further demonstrated by coinjecting human A-375 melanoma cells with hTBJ1-shCtrl or hTBJ1-shCAV1 fibroblasts in nude mice (Fig.2C). Together, these results demonstrate that Cav1 deficient fibroblasts, but not Cav2KO cells, are sufficient to recapitulate the tumor phenotype of Cav1KO mice.
Figure 2. Absence of Cav1 but not Cav2 in dermal fibroblasts enhances the growth of melanoma cells in co-injection experiments.
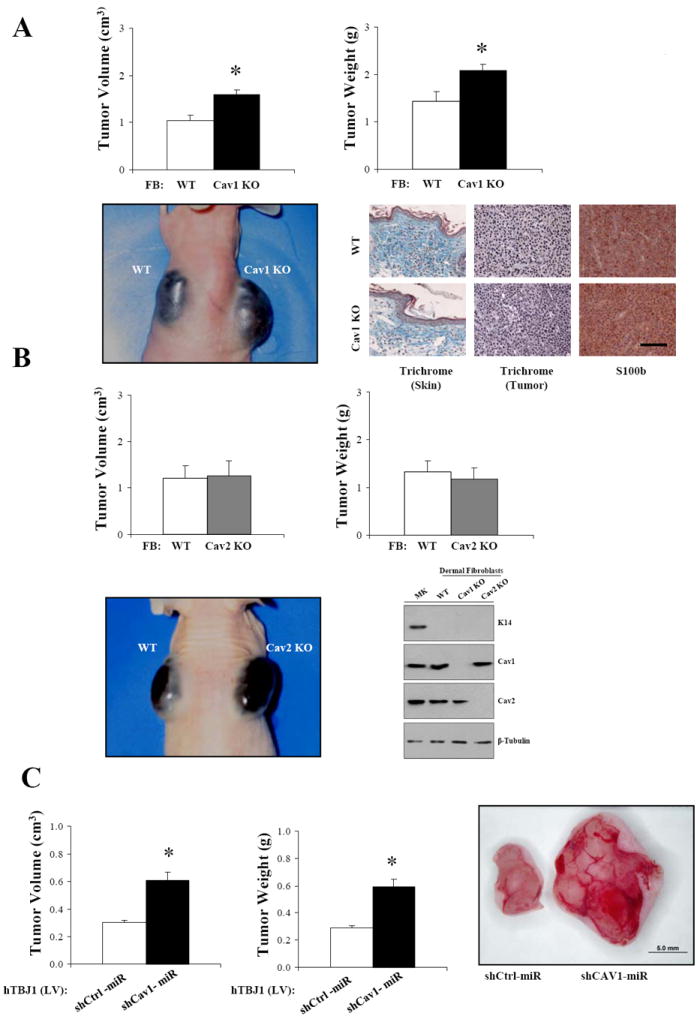
A, B16F10 melanoma cells (1×105) were intradermally co-injected with Cav1KO neonatal dermal fibroblasts at 1:5 ratios in 3-4-month-old nude female mice. After 14 days, tumors were dissected and their size determined. Masson’s trichrome staining and S100b (melanoma cell marker) immunohistochemistry of tumor sections revealed similar intratumoral collagen deposition between WT-FB/B16F10 and Cav1KO FB/B16F10 xenografts (n=5, per group). B, tumor size of B16F10 cells coinjected with WT and Cav2KO dermal fibroblasts as in (A). K14 (keratinocyte marker), Cav1 and Cav2 immunoblots of freshly isolated dermal fibroblast and keratinocytes (MK) are also shown. β-Tubulin immunoblot is shown as loading control. C, lentivirus mediated CAV1 silencing (Lv-shCAV1miR) in hTBJ cells promotes the growth of A-375 cells as determined by co-injection experiments. Results are means ± SEM (n≥5, per group; *, P < 0.05, by two tailed Mann-Whitney Test; Scale Bar, 100μm).
Fibroblasts lacking Cav1 promote the growth of melanoma cells in non-contact cocultures but not in direct-contact cocultures
To investigate possible mechanisms whereby Cav1 deficient fibroblasts may promote B16F10 tumor growth, we cocultured (under cell-cell contact conditions) mCherry-B16F10 cells with serum-activated WT or Cav1KO dermal fibroblasts or mCherry-A375 cells with serum-activated eGFP-labeled hTBJ1-shCtrl or hTBJ1-shCAV1 at 1:5 ratios in low-serum medium (1% FBS). After 48h and 72h, FACS analysis of mCherry-B16F10 or mCherry-A375 cells revealed that Cav1 deficient fibroblasts were unable to promote the growth of melanoma cells under these conditions. Immunoblot analysis of primary Cav1KO FBs or shCAV1 FBs confirms absence/knockdown of Cav1 protein relative to WT and shCtrl FBs, respectively. Furthermore, primary cultures were negative for the keratinocyte cell marker K14, confirming the purity of these cell populations (Fig.3A and B). In contrast, a [3H]Thymidine incorporation assay demonstrates that proliferation of melanoma cells was significantly increased when cocultured (72h) with fibroblasts lacking Cav1 under non contact conditions (Fig.3C). These findings suggest that soluble secreted factors may be mediating the pro-proliferative effects of Cav1 deficient fibroblasts on melanoma cells.
Figure 3. Fibroblasts lacking Cav1 promote the growth of melanoma cells in non-contact cocultures but not in direct-contact cocultures.
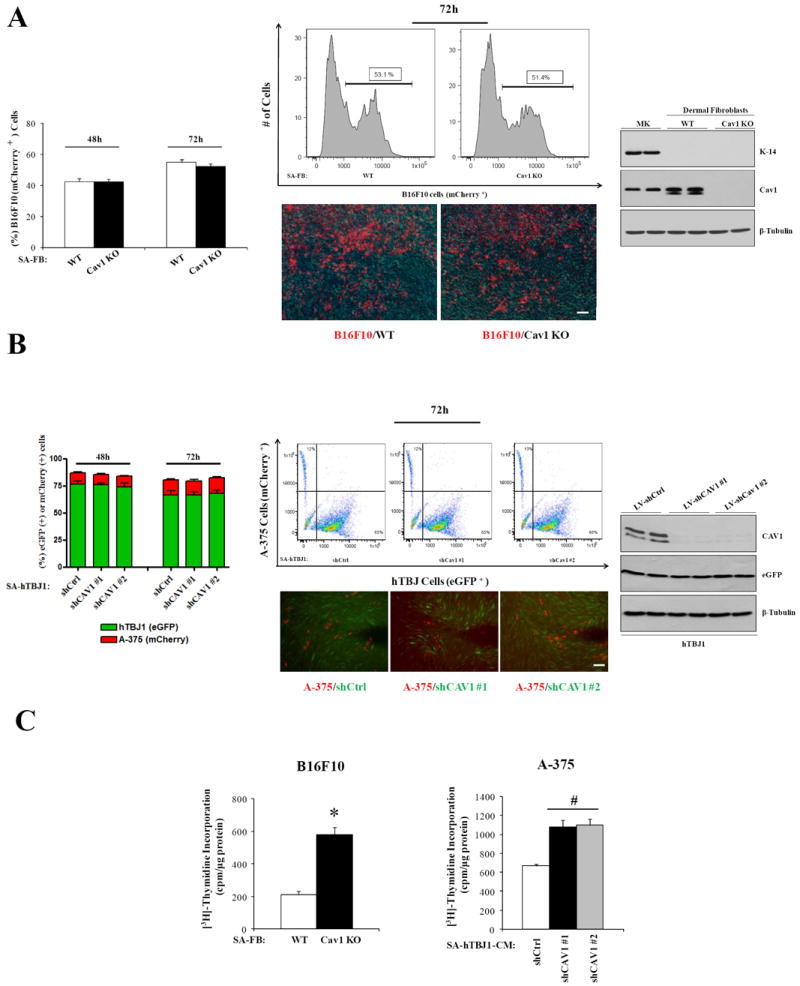
A, representative flow cytometry plots, photomicrographs and quantification of B16F10-mCherry cells cocultured for 48-72h with serum-activated (SA) WT and SA-Cav1KO neonatal dermal fibroblasts in 1%FBS-DMEM (n= 5 per group). Cav1 and K14 (keratinocyte marker) immunoblots of freshly isolated dermal fibroblasts and keratinocytes are shown (right). B, representative flow cytometry plots, photomicrographs and quantification of A-375-mCherry cells growth, cocultured for 48-72h with SA-eGFP-labeled hTBJ1-shCtrlmiR or SA-hTBJ1-shCAV1miR dermal fibroblasts in 1%FBS-DMEM (n= 5 per group). Cav1 and eGFP immunoblots of SA-hTBJ1-shCtrlmiR or SA-hTBJ1-shCAV1miR are shown (right). β-Tubulin immunoblots are shown as loading controls. C, increased proliferation ([3H]-Thymidine incorporation assay) of B16F10 and A-375 cells cocultured in absence of cell-cell contact (tranwell; 48h) with SA-FBs lacking Cav1 (n = 5 per group). Results are means ± SEM (* #, P < 0.05, by two tailed Student’s t test and by Dunnett’s Multiple Comparisons Test; Scale Bar, 100μm).
Serum-activated Cav1KO dermal fibroblasts display increased amounts of protumorigenic cytokines
In order to determine whether Cav1 expression may regulate secreted soluble factors in fibroblasts, a cytokine array was performed on conditioned medium (CM) from serum activated WT and Cav1KO dermal fibroblasts. CM from serum activated Cav1KO fibroblasts displays increased expression of ShhN, bFGF and MMP2/3, cytokines known to promote proliferation, invasion and angiogenesis during melanomagenesis. Decoy receptors (HGFR, VEGFR2) and an inhibitor of MMPs (TIMP1) were reduced in Cav1KO CM (Fig.4A). Increased expression of ShhN was also confirmed by ELISA assay and by immunoblot analysis of CM and cell lysates from Cav1 deficient fibroblasts (Fig.4A). In addition, B16F10 cells incubated with CM from serum-activated Cav1KO fibroblasts (48h) displays increased cell proliferation/growth and hyperactivation of the Shh signaling pathway as evidenced by increased Cyclin D1/A and Gli-1 expression (a Shh target gene) and by increased [3H]-Thymidine incorporation and MTT assay (Fig.4B). Similar outcomes were obtained when A-375 cells were incubated with CM from serum-activated hTBJ1-sh-CAV1 cells (Fig.4A and C). Thus, our results indicate that fibroblasts lacking Cav1 secrete factors that promote proliferation, invasion and angiogenesis.
Figure 4. Serum-activated Cav1KO dermal fibroblasts display increased amounts of protumorigenic cytokines.
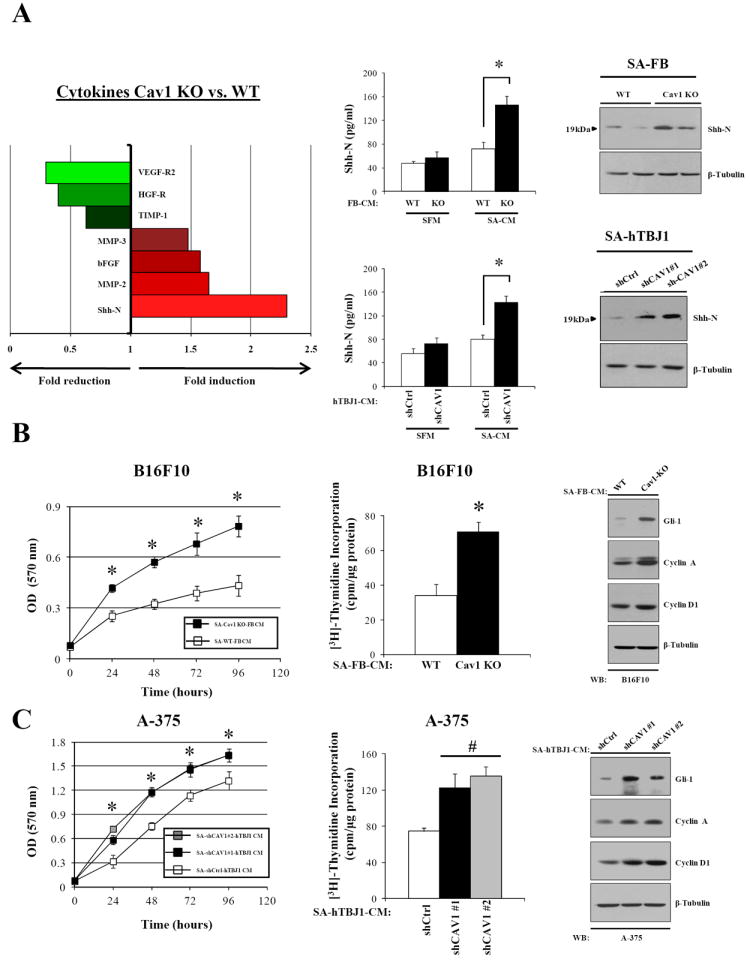
A, left, cytokines differentially regulated in conditioned medium from SA-WT and SA-Cav1KO neonatal dermal fibroblasts. Center, ELISA showing increased ShhN levels in the conditioned medium of serum-activated dermal fibroblasts (SA-CM) lacking Cav1 (n= 4 per group). ShhN immunoblots of serum-activated dermal fibroblasts are displayed (right). B, MTT-assay and [3H]-Thymidine incorporation assay (48h) of B16F10 melanoma cells treated with SA-CM from WT and Cav1KO fibroblasts. Immunoblot analysis showing increased expression of Gli-1, Cyclin D1 and Cyclin A in B16F10 melanoma cells incubated (48h) with SA-CM from Cav1KO dermal fibroblasts (n= 8 per group). Similar results are shown (C) for human A-375 melanoma cells treated with SA-CM from hTBJ1-shCtrl and hTBJ1-shCAV1 cells. Results are means ± SEM (n ≥ 6 per group; *, #, P < 0.05, by two tailed Student’s t test and by Dunnett’s multiple comparisons test).
Inhibition of Shh signaling pathway in melanoma cells reverses the proproliferative/protumorigenic effects of fibroblasts lacking Cav1
Recent studies have shown aberrant activation of Shh signaling in several cancer types including melanoma (33). To determine whether pharmacological inhibition of the Shh pathway in B16F10 cells can reverse the pro-proliferative effect of Cav1KO-CM, we performed a [3H]-thymidine incorporation assay on melanoma cells incubated for 48h with Cav1KO-CM containing Cyclopamine, a specific inhibitor of the Shh pathway. Our results show that low concentrations of Cyclopamine (5-10 μM) were effective in blocking the pro-proliferative effects of Cav1KO-CM on B16F10 melanoma cells. Interestingly, the proliferation of B16F10 cells incubated with WT-CM containing Cyclopamine remains unchanged (Fig.5A). Similarly, Cyclopamine prevented the pro-proliferative effects of CM from serum-activated hTBJ1-shCAV1 cells on A-375 human melanoma cells (Fig.5B). To examine whether inhibition of the Shh signaling pathway in B16F10 melanoma cells abolishes the protumorigenic properties of Cav1 deficient fibroblasts in vivo, we stably silenced the Gli-1 gene by lentiviral shRNA technology. Complete Gli-1 knockdown and reduced Gli-1 expression levels were achieved in absence and presence of Shh, respectively. Similar Gli-1 expression levels were also observed in A-375 cells treated with Shh, suggesting a fully functional Shh signaling pathway in both melanoma cell types (Fig.5C, left). Coinjection experiments performed as in Fig.2A demonstrated that Gli-1 knockdown in B16F10 cells was sufficient to reverse the tumor promoting effects of Cav1 deficient fibroblasts (Fig.5C, right). In summary, these data show that Shh heterotypic signaling is critical for B16F10 melanoma cell proliferation and melanoma tumor growth when Cav1 is absent in dermal fibroblasts.
Figure 5. Inhibition of Shh signaling pathway in melanoma cells reverses the pro-proliferative/protumorigenic effects of fibroblasts lacking Cav1.
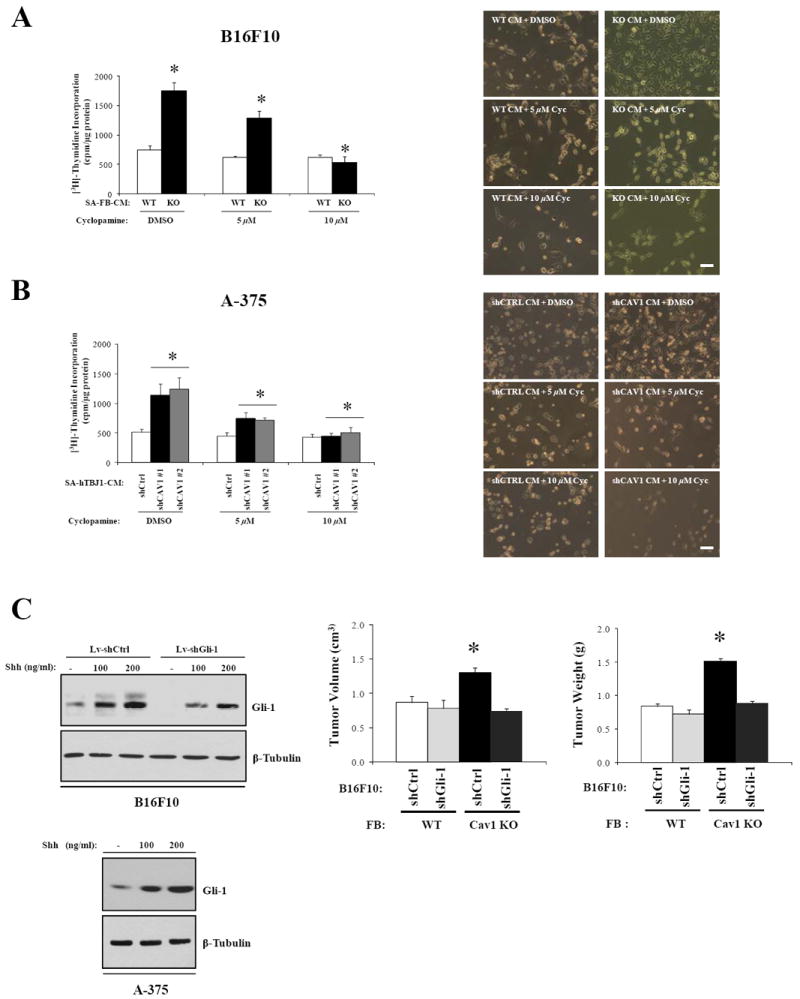
A, [3H]Thymidine incorporation assay of B16F10 melanoma cells treated with DMSO or cyclopamine following incubation with SA-CM from WT and Cav1KO dermal fibroblasts. Representative phase-contrast images of B16F10 cells treated with SA-CM from WT and Cav1KO fibroblasts with cyclopamine are shown on the right. Similar experiments (B) were performed with A-375 cells incubated with SA-CM from hTBJ1-shCtrl and hTBJ1-shCAV1 cells. C, Gli-1 and β-Tubulin immunoblots of B16F10-shCtrl, B16F10-shGli-1 and A-375 cells before and after being treated with Shh (left). Gli-1 knock-down in B16F10 cells reverses the tumor promoting effects of Cav1KO FBs as (right) determined by coinjection experiments. Results are means ± SEM (n ≥ 4 per group; *, P < 0.05, by Tukey’s Multiple Comparisons Test; Scale Bar, 200μm).
Cav1 deficiency inhibits lung colonization and transendothelial migration of B16F10 melanoma cells
Given the absence of spontaneous metastasis formation in B16F10 orthotopic tumor bearing WT and Cav1KO mice (data not shown), we i.v. injected 105 B16F10 cells to determine their ability to colonize the lungs of WT and Cav1KO mice (experimental metastasis assay). Interestingly, the ability of B16F10 cells to colonize the lungs of Cav1KO mice was significantly impaired (Fig.6A). To identify possible mechanisms accounting for these findings, we determined the ability of mCherry labeled or [3H]thymidine labeled B16F10 cells to transmigrate through a monolayer of lentivirally transduced shCtrl and shCAV1-HUVEC cells. Consistent with our in vivo data, the ability of B16F10 cells to adhere to and to transmigrate through a HUVEC monolayer was significantly reduced in CAV1 knockdown cells (Fig.6B and C). Interestingly, incubation of HUVEC with ICAM-1 and VCAM-1 antibodies reduced the adhesion of B16F10 cells to levels similar to those observed with the HUVEC-shCAV1, suggesting a critical role for Cav1 in regulating the process of metastatic extravasation (Fig.6C, left). TNFα induced VCAM-1 and ICAM-1 expression has been described as being critical in cancer cells-endothelium interactions (34). Interestingly, our in vitro results were corroborated by reduced VCAM-1 and ICAM-1 expression levels in lungs of 5h-TNFα-treated Cav1KO mice (Fig.6C, right). Collectively, these results demonstrate that CAV1 has a key role in the endothelium and regulates processes such as adhesion and transmigration that are ultimately relevant for the establishment of lung metastases in vivo.
Figure 6. Cav1 deficiency inhibits lung colonization and transendothelial migration of B16F10 melanoma cells.
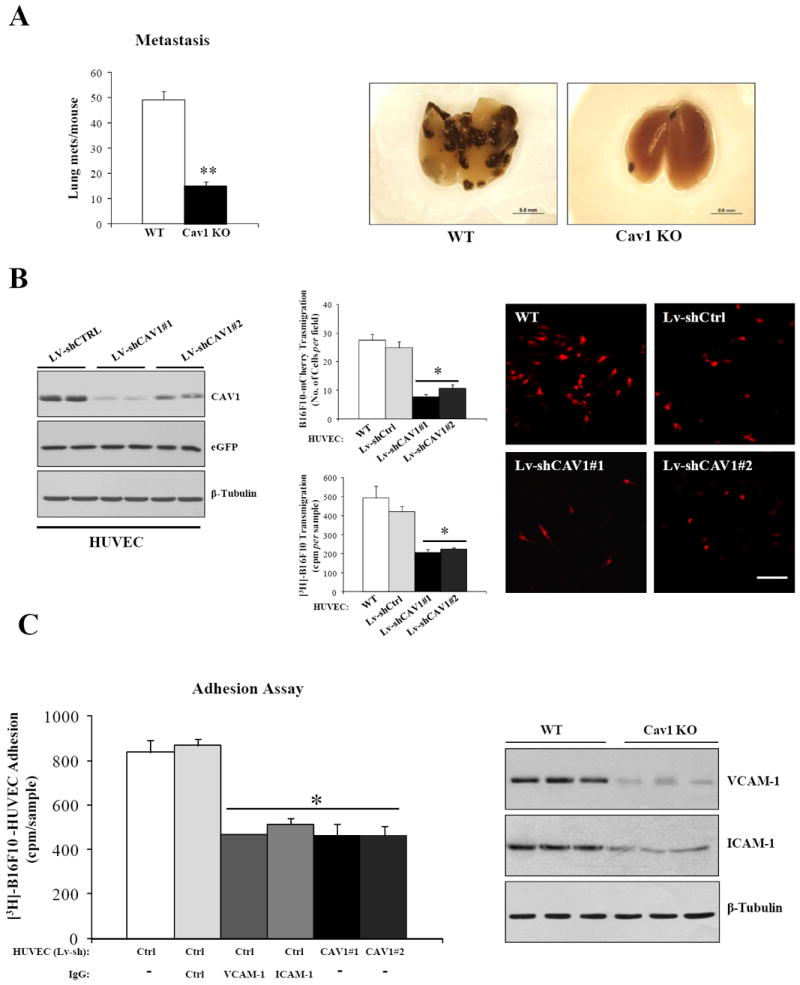
A, left, experimental lung metastasis assay demonstrating that the ability of B16F10 cells to colonize the lungs of Cav1KO mice is markedly reduced in (n = 9 per group). Representative images of WT and Cav1KO lungs dissected 18 days after i.v. injections of 105 B16F10 cells are shown (right). B, transmigration (6h) of mCherry and/or [3H]thymidine labeled B16F10 cells across a confluent monolayer of HUVEC-WT, HUVEC-Lv-shCtrl and HUVEC-Lv-shCAV1-#1/#2 (n=4 per group). Representative images of transmigrated B16F10-mCherry cells are shown (right). C, adhesion assay of [3H]thymidine labeled B16F10 cells on confluent monolayers of HUVEC-shCtrl and HUVEC-shCAV1 untreated or treated with IgG1-isotype, ICAM-1 and VCAM-1 antibodies (n=4 per group). VCAM-1, ICAM-1 immunoblots of lungs from TNFα treated WT and Cav1KO mice (right). Results are means ± SEM (**,*, P < 0.05, by two tailed Mann-Whitney Test and by Dunnett’s Multiple Comparisons Test; Scale Bar, 100μm).
DISCUSSION
In the current study, we show that Cav1 gene disruption promotes the growth of B16F10 melanoma cells in the skin of mice, whereas it inhibits the formation of lung metastases. Our data indicate that lack of Cav1 in dermal fibroblasts contributes to primary melanoma tumor growth by increased paracrine cytokine signaling, whereas the inability of B16F10 cells to form lung metastases is attributed to defects in VCAM1 and ICAM1 mediated adhesion to endothelial cells. Although the function of Cav1 has been recently examined in melanoma cancer cells (15, 35), the role of stromal Cav1 in melanoma tumor growth and metastasis remains less well studied. Here, we demonstrate that the difference in tumor growth seen in Cav1KO and Cav2KO mice seems to correlate well with differences in their microvascular density. However, in contrast to our results, previous studies have shown reduced tumor growth and reduced MVD in Cav1KO mice subcutaneously injected with B16F10 cells (36, 17), indicating that the injection site (intradermal vs. subcutaneous) and consequently the different tumor microenvironments may significantly affect melanoma tumor growth (7, 44). Although our findings are consistent with other studies showing a direct positive relationship between lack of Cav1 and higher microvascular density in vivo (11, 50), we cannot exclude the possibility that other stromal factors other than the endothelial cells are responsible for the Cav1KO and Cav2KO tumor phenotypes. Dermal fibroblasts, in fact, are abundant cellular components of the skin and they exert important biological functions to maintain normal skin homeostasis (37). Our coinjection experiments show that absence of Cav1 in dermal fibroblasts is sufficient to recapitulate the tumor phenotype of Cav1KO mice. Interestingly, Cav2 deficient fibroblasts that express Cav1 (14), fail to replicate the tumor phenotype of Cav2KO (and Cav1KO) mice, indicating that the tumor promoting effects of Cav1 deficient fibroblasts were Cav2 independent.
Based on these findings, we postulated that Cav1 deficient fibroblasts promote the growth of melanoma cells by either direct cell-cell contact or paracrine signaling. To test this hypothesis we performed direct and indirect cocultures of fibroblasts and melanoma cells. Interestingly, our results from coculture experiments suggest that the growth promoting features of Cav1 deficient fibroblasts may be attributed to enhanced paracrine signaling that does not require direct cell-cell contact. However, the inability of Cav1 deficient fibroblasts to promote the growth of melanoma cells in direct cell-cell contact cocultures can most likely be attributed to direct cell-cell contact inhibitory mechanisms exerted by normal fibroblasts (primary and/or immortalized cells) that are able to overcome the proproliferative effects of secreted soluble factors. To identify possible secreted factors we performed a cytokine array on conditioned medium from serum activated dermal fibroblasts. The increased secretion of cytokines such as ShhN, MMP2/3 and bFGF and the reduced expression of VEGFR2, HGF-R (decoy receptors (38)) and TIMP1 (MMPs inhibitor) observed in Cav1KO dermal fibroblasts further confirms their protumorigenic phenotype, and this cytokine signature correlates well with the tumor phenotype of Cav1KO mice. In addition, these results are in agreement with many published studies that identified similar factors associated with the stromal remodeling of tumors (3, 39). A key finding of our study is the increased amount of the soluble form of the Shh protein (ShhN) observed in the CM of serum activated Cav1KO dermal fibroblasts. Aside from having an essential role in embryonic development, Shh modulates many aspects of skin biology including wound-healing (40), proliferation, and transformation (41). Furthermore, although Shh has been described to mainly function in an autocrine manner in melanomagenesis (33), it is now becoming increasingly evident that Shh may contribute to tumor growth in a paracrine manner (42). Our results, showing increased DNA synthesis and increased Gli-1 expression in melanoma cells incubated with CM from Cav1 deficient FBs, provide evidence that absence of Cav1 enhances Shh heterotypic signaling. Consequently, the pro-proliferative and protumorigenic effects of Cav1 deficient fibroblasts are reversed by inhibiting the Shh pathway with cyclopamine and by silencing Gli-1 in B16F10 cells. Another important key finding of our study is the inability of B16F10 cells to form lung metastases in Cav1KO mice. The dissemination of cancer cells to metastatic sites is a stepwise process that begins with the invasion of the dermis surrounding the primary tumor and ends with metastatic extravasation and colonization of ectopic sites (43, 7). Metastatic extravasation from the bloodstream is a critical last step of the metastatic cascade that similarly to leukocyte transmigration requires the firm binding of cancer cells to the endothelial adhesion molecules VCAM-1 and ICAM-1. Blockade of VCAM-1 and ICAM-1 mediated interactions has been shown to effectively prevent the development of metastasis in a preclinical setting (31, 45, 46). Given these considerations, our adhesion assay results and our data showing reduced ICAM-1 and VCAM-1 expression in the lungs of TNFα treated Cav1KO mice, suggest that the inability of B16F10 cells to form metastases and to extravasate may be attributed to defects in VCAM1 and ICAM1 mediated adhesion to endothelial cells. Given our primary tumor results, the metastasis phenotype of Cav1KO mice seems quite paradoxical. However, recent work reveals that Cav1KO mice display several non cancer related phenotypes that support our observations. For instance, previous studies have shown that the resistance of Cav1KO mice to atherosclerosis development may be attributed to impaired endothelial VCAM-1 and ICAM-1 functions that ultimately result in reduced inflammation and impaired macrophage migration throughout the endothelium (47-49). Thus, it appears that similar endothelial defects may cause resistance to atherosclerosis and reduce melanoma metastasis in Cav1KO mice.
In summary, we show that loss of Cav1 promotes the growth of B16F10 tumors in the skin, while it suppresses B16F10 lung metastasis. Mechanistically, this phenotype is associated with enhanced paracrine cytokine signaling in Cav1KO dermal fibroblasts and with defects in endothelial cell mediated transmigration of melanoma cells (Fig.7). Thus, these findings support the notion that effective anticancer therapies will have to take into account the complex interactions between cancer cells and their microenvironment in both primary tumors and metastases.
Figure 7.
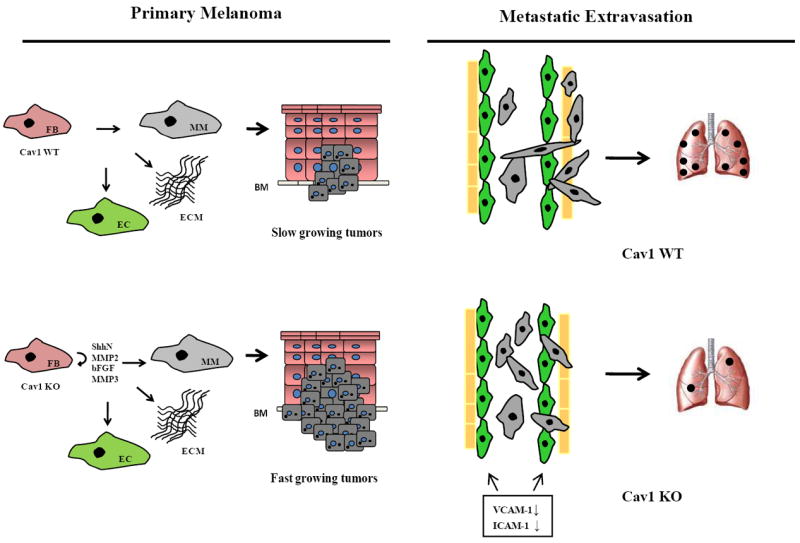
Schematic representation of Cav1 mediated mechanisms regulating primary melanoma tumor growth and metastasis in mice. In primary melanoma, absence of Cav1 in dermal fibroblasts promotes the growth of B16F10 cells by enhanced expression of protumorigenic cytokines (left). In contrast, absence of Cav1 results in reduced VCAM1/ICAM1 expression levels and in inhibition of transendothelial migration and lung colonization of B16F10 cells (right). FB, dermal fibroblasts; EC, endothelial cells; MM, malignant melanoma; BM, basement membrane.
Supplementary Material
Acknowledgments
FC was supported by a grant from the American Heart Association (BGIA). MPL was supported by NIH/NCI grants (R01CA120876; R01CA098779), the Susan G. Komen Breast Cancer Foundation, the Margaret Q. Landenberger Research Foundation and in part by the Pennsylvania Department of Health. CT was supported by NIH Graduate Training Program Grant T32-CA09678. We thank Dr. Philippe Frank for kindly providing WT/Cav2KO C57Bl/6 mice, Matthew Farabaugh (TJU) for excellent technical assistance with FACS analysis, Dr. Kyung-Min Noh (Rockefeller University) and Dr. Gino Cingolani (TJU) for the critical reading of the manuscript and insightful discussions.
Abbreviations
- BSA
Bovine Serum Albumin
- HH
HEDGEHOG
- Shh
Sonic HH
- FBS
Fetal Bovine Serum
- ELISA
Enzyme-Linked Immunosorbent Assay
- FB
Fibroblasts
- VCAM-1
Vascular-Cell-Adhesion-Molecule-1
- ICAM-1
Intercellular-Cell-Adhesion-Molecule-1
- MTT
(3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliu-bromide
- CM
conditioned-medium
- SFM
Serum-Free-Medium
Footnotes
Author contributions:
FC designed research and supervised experiments. CT, DWM, RCC, SK performed experiments. FC, AF made the LV constructs and stable cell lines. FC, CT isolated primary FB and performed coinjections in mice. GL, MC, FS, RGP, MPL shared reagents. FC wrote the manuscript.
References
- 1.Kopfstein L, Christofori G. Metastasis: cell-autonomous mechanisms versus contributions by the tumor microenvironment. Cell Mol Life Sci. 2006;63:449–68. doi: 10.1007/s00018-005-5296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mcallister SS, Weinberg RA. Tumor-Host Interactions: A Far-Reaching Relationship. J Clin Oncol. 28:4022–4028. doi: 10.1200/JCO.2010.28.4257. [DOI] [PubMed] [Google Scholar]
- 3.Ruiter D, Bogenrieder T, Elder D, Herlyn M. Melanoma-stroma interactions: structural and functional aspects. Lancet Oncol. 2002;3:35–43. doi: 10.1016/s1470-2045(01)00620-9. [DOI] [PubMed] [Google Scholar]
- 4.Villanueva J, Herlyn M. Melanoma and the tumor microenvironment. Curr Oncol Rep. 2008;10:439–46. doi: 10.1007/s11912-008-0067-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glenney JR, Soppet D. Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in RSV-transformed fibroblasts. Proc Natl Acad Sci USA. 1992;89:10517–21. doi: 10.1073/pnas.89.21.10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palade GE, Bruns RR. Structural modification of plasmalemma vesicles. J Cell Biol. 1968;37:633–49. doi: 10.1083/jcb.37.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fidler IJ. Critical factors in the biology of human cancer metastasis:twenty-eighth G.H.A. Clowes memorial award lecture. Cancer Res. 1990;50:6130–8. [PubMed] [Google Scholar]
- 8.Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54:431–67. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 9.Goetz JG, Minguet S, Navarro-Lerida I, Lazcano JJ, Samaniego R, Calvo E, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 146:148–63. doi: 10.1016/j.cell.2011.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol. 2009;174:2023–34. doi: 10.2353/ajpath.2009.080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeWever J, Frerart F, Bouzin C, Baudelet C, Ansiaux R, Sonveaux P, et al. Caveolin-1 is critical for the maturation of tumor blood vessels through the regulation of both endothelial tube formation and mural cell recruitment. Am J Pathol. 2007;171:1619–28. doi: 10.2353/ajpath.2007.060968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lizarbe TR, Garcia-Rama C, Tarin C, Saura M, Calvo E, Lopez JA, et al. Nitric oxide elicits functional MMP-13 protein-tyrosine nitration during wound repair. Faseb J. 2008;22:3207–15. doi: 10.1096/fj.07-103804. [DOI] [PubMed] [Google Scholar]
- 13.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- 14.Razani B, Wang XB, Engelman JA, Battista M, Lagaud G, Zhang XL, et al. Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without disruption of caveolae. Mol Cell Biol. 2002;22:2329–44. doi: 10.1128/MCB.22.7.2329-2344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trimmer C, Whitaker-Menezes D, Bonuccelli G, Milliman JN, Daumer KM, Aplin AE, et al. CAV1 inhibits metastatic potential in melanomas through suppression of the integrin/Src/FAK signaling pathway. Cancer Res. 2010;70:7489–99. doi: 10.1158/0008-5472.CAN-10-0900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valente P, Fassina G, Melchiori A, Masiello L, Cilli M, Vacca A, et al. TIMP-2 overexpression reduces invasion and angiogenesis and protects B16F10 melanoma cells from apoptosis. Int J Cancer. 1998;75:246–53. doi: 10.1002/(sici)1097-0215(19980119)75:2<246::aid-ijc13>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 17.Chang SH, Feng D, Nagy JA, Sciuto TE, Dvorak AM, Dvorak HF. Vascular permeability and pathological angiogenesis in caveolin-1-null mice. Am J Pathol. 2009;175:1768–76. doi: 10.2353/ajpath.2009.090171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lichti U, Anders J, Yuspa SH. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat Protoc. 2008;3:799–810. doi: 10.1038/nprot.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–48. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 20.Zhang W, Matrisian LM, Holmbeck K, Vick CC, Rosenthal EL. Fibroblast-derived MT1-MMP promotes tumor progression in vitro and in vivo. BMC Cancer. 2006;6:52. doi: 10.1186/1471-2407-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Follenzi A, Battaglia M, Lombardo A, Annoni A, Roncarolo MG, Naldini L. Targeting lentiviral vector expression to hepatocytes limits transgene-specific immune response and establishes long-term expression of human antihemophilic factor IX in mice. Blood. 2004;103:3700–9. doi: 10.1182/blood-2003-09-3217. [DOI] [PubMed] [Google Scholar]
- 22.Berking C, Takemoto R, Schaider H, Showe L, Satyamoorthy K, Robbins P, et al. Transforming growth factor-beta1 increases survival of human melanoma through stroma remodeling. Cancer Res. 2001;61:8306–16. [PubMed] [Google Scholar]
- 23.Zujovic V, Taupin V. Use of cocultured cell systems to elucidate chemokine-dependent neuronal/microglial interactions: control of microglial activation. Methods. 2003;29:345–50. doi: 10.1016/s1046-2023(02)00358-4. [DOI] [PubMed] [Google Scholar]
- 24.Iyer VR, Eisen MB, Ross DT, Schuler G, Moore T, Lee JC, et al. The transcriptional program in the response of human fibroblasts to serum. Science. 1999;283:83–7. doi: 10.1126/science.283.5398.83. [DOI] [PubMed] [Google Scholar]
- 25.Capozza F, Williams TM, Schubert W, McClain S, Bouzahzah B, Sotgia F, et al. Absence of caveolin-1 sensitizes mouse skin to carcinogen-induced epidermal hyperplasia and tumor formation. Am J Pathol. 2003;162:2029–39. doi: 10.1016/S0002-9440(10)64335-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bolontrade MF, Stern MC, Binder RL, Zenklusen JC, Gimenez-Conti IB, Conti CJ. Angiogenesis is an early event in the development of chemically induced skin tumors. Carcinogenesis. 1998;19:2107–13. doi: 10.1093/carcin/19.12.2107. [DOI] [PubMed] [Google Scholar]
- 27.Capozza F, Combs TP, Cohen AW, Cho YR, Park SY, Schubert W, et al. Caveolin-3 knockout mice show increased adiposity and whole body insulin resistance, with ligand-induced insulin receptor instability in skeletal muscle. Am J Physiol Cell Physiol. 2005;288:C1317–C1331. doi: 10.1152/ajpcell.00489.2004. [DOI] [PubMed] [Google Scholar]
- 28.Pyun BJ, Choi S, Lee Y, Kim TW, Min JK, Kim Y, et al. Capsiate, a nonpungent capsaicin-like compound, inhibits angiogenesis and vascular permeability via a direct inhibition of Src kinase activity. Cancer Res. 2008;68:227–35. doi: 10.1158/0008-5472.CAN-07-2799. [DOI] [PubMed] [Google Scholar]
- 29.Romijn JC, Verkoelen CF, Schroeder FH. Application of the MTT assay to human prostate cancer cell lines in vitro: establishment of test conditions and assessment of hormone-stimulated growth and drug-induced cytostatic and cytotoxic effects. Prostate. 1988;12:99–110. doi: 10.1002/pros.2990120112. [DOI] [PubMed] [Google Scholar]
- 30.DeLisser H, Liu Y, Desprez PY, Thor A, Briasouli P, Handumrongkul C, et al. Vascular endothelial platelet endothelial cell adhesion molecule 1 (PECAM-1) regulates advanced metastatic progression. Proc Natl Acad Sci U S A. 107:18616–21. doi: 10.1073/pnas.1004654107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ronald JA, Ionescu CV, Rogers KA, Sandig M. Differential regulation of transendothelial migration of THP-1 cells by ICAM-1/LFA-1 and VCAM-1/VLA-4. J Leukoc Biol. 2001;70:601–9. [PubMed] [Google Scholar]
- 32.Henninger DD, Panes J, Eppihimer M, Russell J, Gerritsen M, Anderson DC, et al. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs of the mouse. J Immunol. 1997;158:1825–32. [PubMed] [Google Scholar]
- 33.Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, et al. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci U S A. 2007;104:5895–900. doi: 10.1073/pnas.0700776104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miles FL, Pruitt FL, van Golen KL, Cooper CR. Stepping out of the flow: capillary extravasation in cancer metastasis. Clin Exp Metastasis. 2008;25:305–24. doi: 10.1007/s10585-007-9098-2. [DOI] [PubMed] [Google Scholar]
- 35.Williams TM, Lisanti MP. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am J Physiol Cell Physiol. 2005;288:C494–C506. doi: 10.1152/ajpcell.00458.2004. [DOI] [PubMed] [Google Scholar]
- 36.Woodman SE, Ashton AW, Schubert W, Lee H, Williams TM, Medina FA, Wyckoff JB, Combs TP, Lisanti MP. Caveolin-1 knockout mice show an impaired angiogenic response to exogenous stimuli. Am J Pathol. 2003;162:2059–68. doi: 10.1016/S0002-9440(10)64337-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. 2002;99:12877–82. doi: 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonecchi R, Savino B, Borroni EM, Mantovani A, Locati M. Chemokine decoy receptors: structure-function and biological properties. Curr Top Microbiol Immunol. 341:15–36. doi: 10.1007/82_2010_19. [DOI] [PubMed] [Google Scholar]
- 39.Pavlakovic H, Becker J, Albuquerque R, Wilting J, Ambati J. Soluble VEGFR-2: an antilymphangiogenic variant of VEGF receptors. Ann N Y Acad Sci. 1207(Suppl 1):E7–15. doi: 10.1111/j.1749-6632.2010.05714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le H, Kleinerman R, Lerman OZ, Brown D, Galiano R, Gurtner GC, et al. Hedgehog signaling is essential for normal wound healing. Wound Repair Regen. 2008;16:768–73. doi: 10.1111/j.1524-475X.2008.00430.x. [DOI] [PubMed] [Google Scholar]
- 41.McMahon AP, Ingham PW, Tabin CJ. Developmental roles and clinical significance of hedgehog signaling. Curr Top Dev Biol. 2003;53:1–114. doi: 10.1016/s0070-2153(03)53002-2. [DOI] [PubMed] [Google Scholar]
- 42.Yauch RL, Gould SE, Scales SJ, Tang T, Tian H, Ahn CP, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406–10. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
- 43.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 331:1559–64. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 44.Loi M, Di Paolo D, Becherini P, Zorzoli A, Perri P, Carosio R, Cilli M, Ribatti D, Brignole C, Pagnan G, Ponzoni M, Pastorino F. The use of the orthotopic model to validate antivascular therapies for cancer. Int J Dev Biol. 2011;55:547–55. doi: 10.1387/ijdb.103230ml. [DOI] [PubMed] [Google Scholar]
- 45.Nobumoto A, Nagahara K, Oomizu S, Katoh S, Nishi N, Takeshita K, et al. Galectin-9 suppresses tumor metastasis by blocking adhesion to endothelium and extracellular matrices. Glycobiology. 2008;18:735–44. doi: 10.1093/glycob/cwn062. [DOI] [PubMed] [Google Scholar]
- 46.Wang HH, Qiu H, Qi K, Orr FW. Current views concerning the influences of murine hepatic endothelial adhesive and cytotoxic properties on interactions between metastatic tumor cells and the liver. Comp Hepatol. 2005;4:8. doi: 10.1186/1476-5926-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernandez-Hernando C, Yu J, Davalos A, Prendergast J, Sessa WC. Endothelial-specific overexpression of caveolin-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2010;177:998–1003. doi: 10.2353/ajpath.2010.091287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernandez-Hernando C, Yu J, Suarez Y, Rahner C, Davalos A, Lasuncion MA, et al. Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell Metab. 2009;10:48–54. doi: 10.1016/j.cmet.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Frank PG, Lee H, Park DS, Tandon NN, Scherer PE, Lisanti MP. Genetic ablation of caveolin-1 confers protection against atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:98–105. doi: 10.1161/01.ATV.0000101182.89118.E5. [DOI] [PubMed] [Google Scholar]
- 50.Lin MI, Yu J, Murata T, Sessa WC. Caveolin-1-deficient mice have increased tumor microvascular permeability, angiogenesis, and growth. Cancer Res. 2007;67:2849–56. doi: 10.1158/0008-5472.CAN-06-4082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.