

Abstract
Platelets are known contributors of hemostasis but have recently been shown to be important in inflammation and infectious diseases. Moreover, thrombocytopenia is often observed in patients with sepsis. We previously reported that platelets actively phagocytosed IgG-coated latex beads. In the current study, the capacity of human platelets to participate in host defense against bacterial infections was determined by assessing their ability to kill E. coli. Washed human platelets were incubated with unopsonized or IgG-opsonized E. coli and evaluated for binding and killing of E. coli. We found that although both unopsonized and IgG-opsonized E. coli associated with platelets, only IgG-opsonized E. coli were efficiently killed unless platelets were activated by a potent agonist. The bactericidal activity was dependent on FcγRIIA, was sensitive to cytochalasin D, but was not due to reactive oxygen metabolites. These data suggest that platelets may play an important role in protection against infection.
Keywords: FcγRIIA, IgG, E. coli, platelet
Introduction
Platelets are recognized for their critical role in maintaining hemostasis. More recently, additional activities are being attributed to platelets including participation in wound repair, angiogenesis, inflammation, and even host defense against infection (reviewed in (17)). For example, it is known that patients with sepsis often present with thrombocytopenia, which may be due to disseminated intravascular coagulation stimulated by bacterial products (6). Furthermore, platelets have been observed to interact with various bacterial species in vitro (reviewed in (4)). These interactions require various platelet surface proteins, including toll-like receptors, fibrinogen receptors and FcγRIIA(2, 5). FcγRIIA is the only IgG receptor present on human platelet surfaces and is responsible for binding and internalization of IgG-containing targets (1, 8, 23). Specifically, we have recently shown that platelets are capable of binding and internalizing IgG-coated latex beads, and the internalization is dependent on FcγRIIA and actin polymerization, similar to phagocytosis observed in leukocytes (1). Furthermore, others have observed platelets with internalized bacteria, viruses, liposomes, erythrocyte fragments and latex particles (4, 7, 12, 15, 26). Additional evidence for the bactericidal potential of platelets comes from studies examining platelet lysates which contain platelet microbicidal proteins (PMP) and β-defensin, both capable of killing Staphylococcus aureus (11, 24, 25). In light of the previous findings, the current study was designed to evaluate the ability of platelets to bind and kill bacterium, and the potential role of FcγRIIA in this process.
Materials and Methods
Platelet Preparation
Human blood from healthy volunteers was obtained via venipuncture consistent with the University of Toledo Biomedical Institutional Review Board policy. Blood was collected in Vacutainer® (BD, Franklin Lakes, NJ) tubes containing anticoagulant: 22.0 g/L trisodium citrate, 8.0 g/L citric acid, and 24.5 g/L dextrose. The tubes were immediately centrifuged for 10 minutes at 200 × G to obtain platelet-rich plasma (PRP). The PRP was washed two times with pH 6.5 tyrode buffer containing: 2.75 g/L trisodium citrate, 1.0 g/L citric acid, 3.125 g/L dextrose, and 8.5 g/L sodium chloride. The solution was centrifuged after each wash for 10 minutes at 440 × G. After the second wash, the platelets were resuspended in 500 μL of pH 7.4 tyrode buffer containing: 8.0 g/L sodium chloride, 0.2 g/L potassium chloride, 0.2 g/L magnesium chloride, 0.45 g/L sodium phosphate dibasic, and 0.9 g/L HEPES. Platelets were counted using a hemocytometer.
Bacteria Preparation
EGFP-expressing E. coli K12 (generously provided by Dr. Robert Blumenthal), were stored in glycerol stock at −80°C until use. Bacteria were grown overnight at 37°C in 10 mL of LB Miller’s broth (Difco, Detroit, MI). Bacteria was centrifuged at 4000 × G for 10 minutes then resuspended in 2 mL of Dulbecco’s Phosphate Buffered Saline (DPBS) buffer (Thermo Fisher Scientific, Waltham, MA) without calcium or magnesium. Some samples were then opsonized for 30 min at 37°C with goat anti-E. coli polyclonal IgG (abcam®, Cambridge, MA). Bacteria were washed twice with DPBS and counted using a hemocytometer.
Bacterial Killing Assay
Unstimulated platelets or platelets (1 × 107/100 μl) activated with 0.1 unit/ml thrombin (Chronolog,) and E. coli were mixed at a Multiplicity of Infection (MOI) of 1 then placed into a 12-well tissue culture plate (Corning), then centrifuged (300 × G for 60 sec) to induce contact. The plate was then floated on a 37°C water bath for 60 minutes before adding a 1% saponin solution to lyse platelets that had phagocytosed E. coli. Solutions were then serially diluted and plated on LB agar plates, incubated overnight at 37°C and CFU were determined for each plate. For inhibitor experiments, platelets were pre-treated with the inhibitor for 15 min at 37°C prior to adding bacteria. Cytochalasin D (Sigma Aldrich, St. Louis, MO) was used at a final concentration of 20 μM, anti-FcγRIIA mAb (clone IV.3, purified from hybridoma) was used at 20μg/ml, and diphenyleneiodonium chloride (DPI)(Sigma Aldrich) was used at 100nM. Each individual experiment was performed in triplicate wells and serial dilutions were plated in triplicate. Each experiment was performed at least three times.
Fluorescence Microscopy
Platelets and E. coli were mixed at an MOI of 1 and plated in 12-well tissue culture plates, as stated above. After incubation for 60 min at 37°C, the bacteria were stained using the Live-Dead BacLight bacterial viability kit (Invitrogen, Carlsbad, CA) and viewed using epifluorescence microscopy (Leica DMIRB, Leica Microsystems, Buffalo Grove, IL). Images were acquired by a QICAM Fast 1394 (Q-imaging, Surrey BC, Canada) and images were processed using Q-capture Pro software.
Statistical Analysis
Significance of bactericidal activity compared between two groups was assessed using two-tailed unpaired student’s t-test. Data shown are mean ± SE. A value of p<0.05 was considered significant.
Results
Previous reports from our group and others suggest that platelets can bind and internalize various targets of similar size to bacteria (1, 5, 7, 14, 19, 26). In order to determine if platelets bind E. coli we incubated freshly washed human platelets with bacteria and assessed viability using the Live Dead BacLight kit and epifluorescence illumination. We observed that platelets were bound to both unopsonized and IgG-opsonized E. coli (Fig. 1A). These data suggest that E. coli can be bound by platelets independent of plasma or serum proteins. Surprisingly, IgG-opsonization of E. coli did not appear to increase platelet binding as we observed similar numbers of bacteria per field of view (Figure 1B). Moreover, very few unopsonized E. coli were found to be stained with propidium iodide, suggesting that platelets failed to kill the bacteria even though they bound them. However, a slight (but statistically insignificant) decrease in bacterial viability was observed when E. coli were opsonized with IgG (Fig. 1B).
Figure 1. Association of E. coli with human platelets.
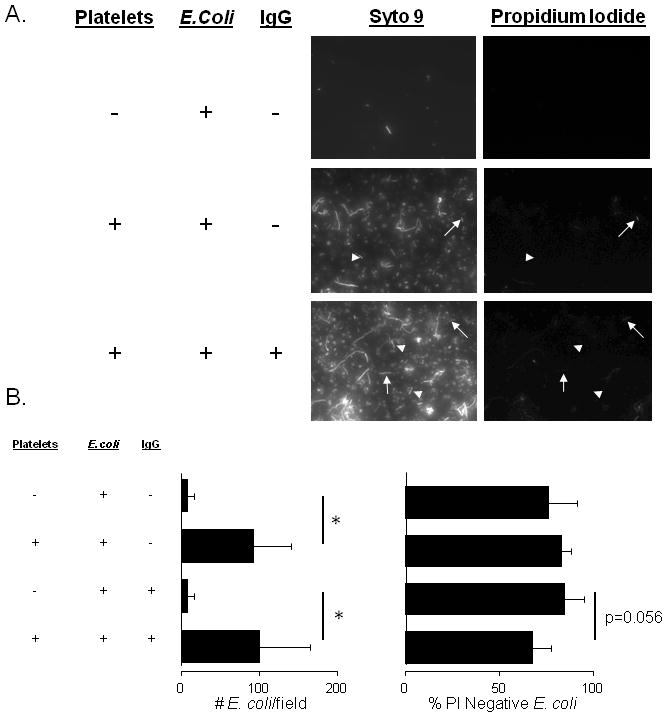
Washed human platelets were incubated with E. coli K12 strain at a MOI of 1 for 60 minutes. A: Plates were washed, stained with BacLight Live-Dead stain (Syto 9 and propidium iodide) and imaged using epifluorescence microscopy. Representative PI-positive E. coli are indicated by arrows and PI-negative E. coli by arrow-heads. B: Numbers of platelet-bound E. coli per field of view were counted and the % PI-negative E. coli was calculated. * p<0.05
As shown in Fig. 1B, experimental variability was considerable so we employed a more consistent assay to quantify bactericidal activity by measuring viable bacteria colony forming units (CFU) after exposure to platelets. After incubating platelets with unopsonized or IgG-opsonized E. coli, platelets were lysed and supernatents were serially diluted, plated on LB agar plates and the CFUs/ml were enumerated. Platelets incubated with unopsonized E. coli did not display any bactericidal activity (Fig. 2A). However, E. coli opsonized with IgG were efficiently killed (Fig. 2B). Because platelets only displayed bactericidal activity toward IgG-opsonized E. coli, we determined if this activity is attributed to the platelet IgG receptor, FcγRIIA. To determine if killing of IgG-opsonized E. coli is FcγRIIA dependent, platelets were pre-treated with a blocking anti-FcγRIIA mAb (clone IV.3) or an isotype control IgG. Killing of IgG-opsonized E. coli was inhibited in the presence of the FcγRIIA blocking antibody, but not isotype-control IgG (Fig. 2B). Bactericidal activity of FcγRIIA on traditional leukocyte populations involves phagocytosis, and we have shown that platelet FcγRIIA can mediate phagocytosis of IgG-coated beads that can be inhibited by cytochalasin D (1). Incubating platelets with cytochalasin D prior to adding IgG-opsonized bacteria produced a significant decrease in bactericidal activity, suggesting that phagocytosis is necessary for IgG-mediated killling. Since leukocyte bactericidal activity is also associated with the production of reactive oxygen metabolites (ROM), and platelets have been shown to produce significant amounts of ROM, we tested the effect of the ROM inhibitor, DPI, on bactericidal activity in platelets (13, 22). Surprisingly, DPI had no effect on bacterial killing. These data suggest that human platelets are capable of bactericidal activity that is dependent on FcγRIIA and actin, but does not require ROMs.
Figure 2.
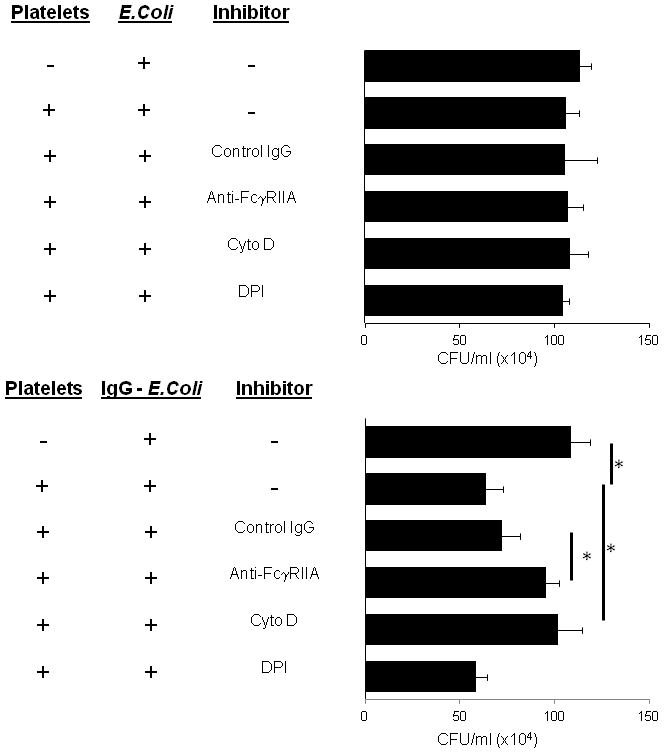
Human platelet killing of E. coli. A: Washed human platelets were incubated with E. coli K12 strain at a MOI=1 for 60 minutes at 37°C in triplicate wells of 12-well plates. Platelets were lysed and samples were serially diluted and plated on duplicate LB agar, incubated overnight at 37°C and colony forming units were counted and expressed as CFU/ml. B: Experiment performed exactly as in (A) except that E. coli were opsonized with IgG. Bars are mean ± SE (n=3). * p<0.05
FcγRIIA is known to induce platelet activation upon binding of IgG-coated latex beads (1). The activation leads to release of significant amounts of sCD40L and RANTES, expression of surface markers indicative of platelet activation (CD62P), and release of PMPs (1, 18). One possibility for the difference in killing between IgG-opsonized and non-opsonized E. coli could be the activation status of the platelets. To test this, the bactericidal activity of activated platelets was tested against IgG-opsonized and non-opsonized E. coli. Interestingly, activation of platelets with 0.1 unit/ml of thrombin (a potent platelet activating agent) resulted in the ability to kill non-opsonized E. coli while it had no significant additive affect on IgG-opsonized E. coli (Fig. 3). Of particular note, IgG-opsonized E. coli were killed more efficiently than non-opsonized E. coli in the presence of activated platelets. Therefore, we interpret this to mean that bactericidal activity is dependent on platelet activation which can be induced by either ligation of FcγRIIA or by thrombotic agents.
Figure 3.
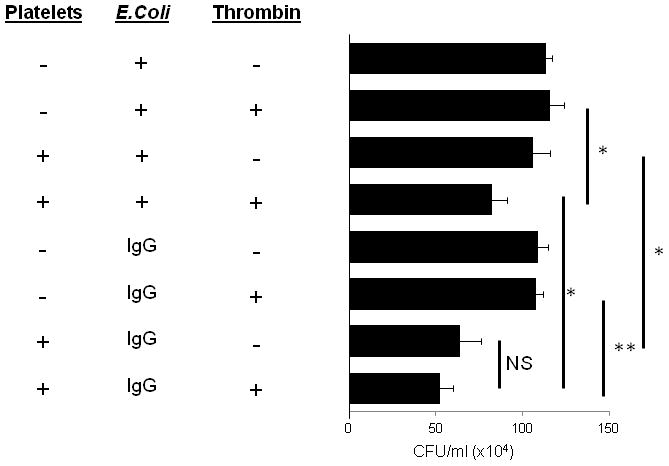
Killing of E. coli by activated platelets. IgG-opsonized or non-opsonized E. coli were incubated with 0.1 unit/ml thrombin in the presence or absence of platelets for 60 minutes at 37°C in triplicate wells of 12-well plates. Platelets were lysed and samples were serially diluted and plated on LB agar, incubated overnight at 37°C and colony forming units were counted and expressed as CFU/ml. Bars are mean ± SE (n=3). * p<0.05 NS=not significant
Discussion
Due to the strong evidence that platelets are capable of binding and internalizing particles similar in size to bacteria, and that platelets produce both ROM and PMP, we tested the hypothesis that platelets could kill bacteria, using a non-pathogenic laboratory strain of E. coli. We observed that platelets could bind E. coli in the absence and presence of opsonizing IgG, but that only IgG-opsonized E. coli were efficiently killed in a FcγRIIA-dependent manner. These observations imply that platelet-binding of non-opsonzed E. coli provides insufficient signals to elicit bactericidal activity by platelets. Most studies of platelet-bacterial interactions have found that plasma proteins (fibrinogen, collagen, von Willebrand factor) are required for binding with platelet surface proteins (αIIbβ3, GPVI, and GP1b, respectively) and these induce varying degrees of activation (4). In our studies, we observed E. coli binding in the absence of any plasma proteins. Although platelets express TLR4, which could mediate binding of Gram-negative bacteria, platelets do not express essential TLR4 binding partners CD14 or MD-2 (and our experiments were performed in PBS), so TLR4-mediated interaction is not likely. Platelets also express many other TLR family-members as well as scavenger receptors (CD36) which may participate in binding (3, 10, 16, 21, 27).
Our data also suggest that bactericidal activity is dependent on actin rearrangement, since the addition of cytochalasin D was able to block killing. In standard leukocyte assays, inhibition of actin rearrangement is interpreted as inhibiting phagocytosis. We have shown that treating platelets with cytochalasin D inhibits phagocytosis of IgG-opsonized latex beads (1). However, actin rearrangement is also involved in fusion of intracellular granules with the plasma membrane in platelets. Therefore, the ability of cytochalasin D to inhibit killing could also be due to blocking the secretion of granule contents, which may contain a number of microbicidal proteins (18). Of particular interest is the observation that bactericidal activity appears to be independent of ROM. Although platelets are known to produce ROM, the quantity of ROM produced may be insufficient to reach the concentration necessary to kill E. coli at an MOI of 1(22).
Previous reports suggest that platelets can kill S. aureus but have marginal bactericidal activity at a MOI=1, which is consistent with our observations (20). However, when the ratio of platelets to S. aureus is increased to 1000 or 10,000:1, platelets are able to kill nearly 100% of S. aureus and the killing is dependent on release of PMP after stimulation of adenine nucleotide receptors (P2X1 and P2Y12)(20). In addition to microbicidal proteins stored in granules, platelets also express β-defensin, which has recently been reported to be localized in the cytoplasm and is potent at killing S. aureus (11). Importantly, the β-defensin mediated killing is dependent on the production of S. aureusα-toxin which presumably permeabilizes platelets thus and causing β-defensin release (11). These data suggest that S. aureus killing most likely takes place independent of internalization and favors extracellular killing even though S. aureus has been reported to be phagocytosed by human platelets (26). Important to our studies, laboratory strains of E. coli (K12) (including the one we used) have not been reported to express any pore-forming molecules such as α-toxin (although some pathogenic strains express lytic molecules)(9).
Although platelets have been reported to internalize many different particles, the ability to kill bacteria is only now becoming appreciated. We report here that platelets can kill laboratory strains of E. coli. Interestingly, we did not observe significant differences in bactericidal activity using different MOIs or longer incubations times up to two hours (data not shown). However, killing appears to be dependent on activation induced by either ligating FcγRIIA or by thrombotic stimulation. We interpret these observations to mean than platelets can kill bacteria via internalization in a FcγRIIA-dependent manner but the majority of killing occurs by the release of PMPs from granules upon activation. However, further investigation is necessary to elucidate the bactericidal mechanism(s).
Conclusion
Human platelets can bind E. coli in the presence and absence of opsonizing IgG. Human platelets also have the potential to kill IgG-opsonized E. coli in a FcγRIIA-dependent and actin-dependent fashion.
Acknowledgments
The authors thank Dr. Robert Blumenthal for generously providing the EGFP-expressing E. coli and Robert Lee and Jixiao Liang for their support of these studies. This work was supported in part by an Arthritis Foundation Investigator Award (to R.G.W.), a Translational Research Stimulation Award by the University of Toledo College of Medicine (to R.G.W.), and by the National Institute of Allergy and Infectious Disease (1R01 AI073452) (to R.M.W). The authors have no financial disclosures to list.
References
- 1.Antczak AJ, Vieth JA, Singh N, Worth RG. Internalization of IgG-Coated Targets Results in Activation and Secretion of Soluble CD40 Ligand and RANTES by Human Platelets. Clin Vaccine Immunol. 2011;18:210–216. doi: 10.1128/CVI.00296-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, Hayashi C, Genco CA, Iafrati M, Freedman JE. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res. 2009;104:346–354. doi: 10.1161/CIRCRESAHA.108.185785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of Toll-like receptor molecules on human platelets. Immunol Cell Biol. 2005;83:196–198. doi: 10.1111/j.1440-1711.2005.01314.x. [DOI] [PubMed] [Google Scholar]
- 4.Fitzgerald JR, Foster TJ, Cox D. The interaction of bacterial pathogens with platelets. Nat Rev Microbiol. 2006;4:445–457. doi: 10.1038/nrmicro1425. [DOI] [PubMed] [Google Scholar]
- 5.Fitzgerald JR, Loughman A, Keane F, Brennan M, Knobel M, Higgins J, Visai L, Speziale P, Cox D, Foster TJ. Fibronectin-binding proteins of Staphylococcus aureus mediate activation of human platelets via fibrinogen and fibronectin bridges to integrin GPIIb/IIIa and IgG binding to the FcgammaRIIa receptor. Mol Microbiol. 2006;59:212–230. doi: 10.1111/j.1365-2958.2005.04922.x. [DOI] [PubMed] [Google Scholar]
- 6.Franchini M, Veneri D. Helicobacter pylori infection and immune thrombocytopenic purpura: an update. Helicobacter. 2004;9:342–346. doi: 10.1111/j.1083-4389.2004.00238.x. [DOI] [PubMed] [Google Scholar]
- 7.Jerushalmy Z, Kohn A, De Vries A. Interaction of myxoviruses with human blood platelets in vitro. Proc Soc Exp Biol Med. 1961;106:462–466. doi: 10.3181/00379727-106-26370. [DOI] [PubMed] [Google Scholar]
- 8.King M, McDermott P, Schreiber AD. Characterization of the Fc gamma receptor on human platelets. Cell Immunol. 1990;128:462–479. doi: 10.1016/0008-8749(90)90041-o. [DOI] [PubMed] [Google Scholar]
- 9.Koli P, Sudan S, Fitzgerald D, Adhya S, Kar S. Conversion of commensal Escherichia coli K-12 to an invasive form via expression of a mutant histone-like protein. MBio. 2011:2. doi: 10.1128/mBio.00182-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korporaal SJ, Van Eck M, Adelmeijer J, Ijsseldijk M, Out R, Lisman T, Lenting PJ, Van Berkel TJ, Akkerman JW. Platelet activation by oxidized low density lipoprotein is mediated by CD36 and scavenger receptor-A. Arterioscler Thromb Vasc Biol. 2007;27:2476–2483. doi: 10.1161/ATVBAHA.107.150698. [DOI] [PubMed] [Google Scholar]
- 11.Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, Kahr WH, Lindemann S, Seizer P, Yost CC, Zimmerman GA, Weyrich AS. Novel anti-bacterial activities of beta-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathog. 2011;7:e1002355. doi: 10.1371/journal.ppat.1002355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Male R, Vannier WE, Baldeschwieler JD. Phagocytosis of liposomes by human platelets. Proc Natl Acad Sci U S A. 1992;89:9191–9195. doi: 10.1073/pnas.89.19.9191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marcus AJ, Silk ST, Safier LB, Ullman HL. Superoxide production and reducing activity in human platelets. J Clin Invest. 1977;59:149–158. doi: 10.1172/JCI108613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Movat HZ, Weiser WJ, Glynn MF, Mustard JF. Platelet phagocytosis and aggregation. J Cell Biol. 1965;27:531–543. doi: 10.1083/jcb.27.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueller-Eckhardt C, Luscher EF. Immune reactions of human blood platelets. II. The effect of latex particles coated with gammaglobulin in relation of complement activation. Thromb Diath Haemorrh. 1968;20:168–179. [PubMed] [Google Scholar]
- 16.Shiraki R, Inoue N, Kawasaki S, Takei A, Kadotani M, Ohnishi Y, Ejiri J, Kobayashi S, Hirata K, Kawashima S, Yokoyama M. Expression of Toll-like receptors on human platelets. Thromb Res. 2004;113:379–385. doi: 10.1016/j.thromres.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 17.Smyth SS, McEver RP, Weyrich AS, Morrell CN, Hoffman MR, Arepally GM, French PA, Dauerman HL, Becker RC. Platelet functions beyond hemostasis. J Thromb Haemost. 2009;7:1759–1766. doi: 10.1111/j.1538-7836.2009.03586.x. [DOI] [PubMed] [Google Scholar]
- 18.Tang YQ, Yeaman MR, Selsted ME. Antimicrobial peptides from human platelets. Infect Immun. 2002;70:6524–6533. doi: 10.1128/IAI.70.12.6524-6533.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terada H, Baldini M, Ebbe S, Madoff MA. Interaction of influenza virus with blood platelets. Blood. 1966;28:213–228. [PubMed] [Google Scholar]
- 20.Trier DA, Gank KD, Kupferwasser D, Yount NY, French WJ, Michelson AD, Kupferwasser LI, Xiong YQ, Bayer AS, Yeaman MR. Platelet antistaphylococcal responses occur through P2X1 and P2Y12 receptor-induced activation and kinocidin release. Infect Immun. 2008;76:5706–5713. doi: 10.1128/IAI.00935-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valiyaveettil M, Podrez EA. Platelet hyperreactivity, scavenger receptors and atherothrombosis. J Thromb Haemost. 2009;7(Suppl 1):218–221. doi: 10.1111/j.1538-7836.2009.03422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wachowicz B, Olas B, Zbikowska HM, Buczynski A. Generation of reactive oxygen species in blood platelets. Platelets. 2002;13:175–182. doi: 10.1080/09533710022149395. [DOI] [PubMed] [Google Scholar]
- 23.Worth RG, Chien CD, Chien P, Reilly MP, McKenzie SE, Schreiber AD. Platelet FcgammaRIIA binds and internalizes IgG-containing complexes. Exp Hematol. 2006;34:1490–1495. doi: 10.1016/j.exphem.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 24.Yeaman MR, Norman DC, Bayer AS. Platelet microbicidal protein enhances antibiotic-induced killing of and postantibiotic effect in Staphylococcus aureus. Antimicrob Agents Chemother. 1992;36:1665–1670. doi: 10.1128/aac.36.8.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeaman MR, Puentes SM, Norman DC, Bayer AS. Partial characterization and staphylocidal activity of thrombin-induced platelet microbicidal protein. Infect Immun. 1992;60:1202–1209. doi: 10.1128/iai.60.3.1202-1209.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Youssefian T, Drouin A, Masse JM, Guichard J, Cramer EM. Host defense role of platelets: engulfment of HIV and Staphylococcus aureus occurs in a specific subcellular compartment and is enhanced by platelet activation. Blood. 2002;99:4021–4029. doi: 10.1182/blood-2001-12-0191. [DOI] [PubMed] [Google Scholar]
- 27.Zimman A, Podrez EA. Regulation of platelet function by class B scavenger receptors in hyperlipidemia. Arterioscler Thromb Vasc Biol. 2010;30:2350–2356. doi: 10.1161/ATVBAHA.110.207498. [DOI] [PMC free article] [PubMed] [Google Scholar]