

Abstract
NOTCH3 gene amplification plays an important role in the progression of many ovarian and breast cancers, but the targets of NOTCH3 signaling are unclear. Here we report the use of an integrated systems biology approach to identify direct target genes for NOTCH3. Transcriptome analysis showed that suppression of NOTCH signaling in ovarian and breast cancer cells led to downregulation of genes in pathways involved in cell cycle regulation and nucleotide metabolism. ChIP-on-chip analysis defined promoter target sequences, including a new CSL binding motif (N1) in addition to the canonical CSL binding motif, that were occupied by the NOTCH3/CSL transcription complex. Integration of transcriptome and ChIP-on-chip data demonstrated that the ChIP target genes overlapped significantly with the NOTCH-regulated transcriptome in ovarian cancer cells. From the set of genes identified we determined that the mitotic apparatus organizing protein DLGAP5 (HURP/DLG7) was a critical target. Both the N1 motif and the canonical CSL binding motif were essential to activate DLGAP5 transcription. DLGAP5 silencing in cancer cells suppressed tumorigenicity and inhibited cellular proliferation by arresting the cell cycle at the G2/M phase. In contrast, enforced expression of DLGAP5 partially counteracted the growth inhibitory effects of a pharmacological or RNAi-mediated inhibition in cancer cells. Our findings define direct target genes of NOTCH3 and highlight DLGAP5 in the tumor-promoting function of NOTCH3.
INTRODUCTION
NOTCH signaling has been shown to participate in cell fate determination and in progenitor cell maintenance during development. In mammals, there are four NOTCH receptors (NOTCH1-NOTCH4) which have distinct tissue expression patterns and are thought to function in specific cellular contexts. The NOTCH pathway is activated by receptor-ligand interactions on the cell membrane, which subsequently lead to a cascade of enzymatic cleavages of membrane NOTCH receptors by ADAM metalloprotease and γ-secretase complex. The cleaved product, intracellular fragment of NOTCH (NICD), translocates into the nucleus where it interacts with the nuclear DNA-binding factor, CSL (RBPJk), and recruits co-activators to activate transcription of target genes. In addition to its role in the developmental processes, aberrant activation of the NOTCH pathway has emerged as a mechanism in the pathogenesis of a variety of human neoplastic diseases (1). For example, a tumor-promoting role of NOTCH1 has been reported in human T-cell acute lymphoblastic leukemia (T-ALL) because activating point mutations of NOTCH1 involving the extracellular heterodimerization domain and/or the C-terminal PEST domain of NOTCH1 are present in more than half of T-ALLs (2, 3).
Amplification at the NOTCH3 genomic locus has been reported in ovarian high-grade serous carcinoma by us (4) and more recently by The Cancer Genome Atlas (5). Ovarian cancer cells with NOTCH3 gene amplification or overexpression are molecularly dependent on NOTCH signaling for cellular survival and growth (4), probably through a positive regulatory loop between NOTCH3 and its ligand, Jagged1 (6). In addition to ovarian cancer, NOTCH3 signaling aberrations have also been implicated in other types of cancers. Translocation of the NOTCH3 gene occurred in a subset of non-small-cell lung cancer (7) and constitutively expressed NOTCH3 induced neoplastic transformation in the breast, brain, and hematopoietic tissues (8-10). More recently, using an RNAi approach, NOTCH3 but not NOTCH1, was found to be critical in maintaining cellular proliferation of ErbB2-negative breast cancers (11).
To better understand the molecular mechanisms by which NOTCH pathway activation contributes to cancer development, investigators have identified and characterized several downstream target genes that are directly regulated by the NOTCH pathway (12). However, most of the studies have focused on NOTCH1; NOTCH3 regulated genes have remained largely unknown. In order to identify NOTCH3 direct target genes, we applied an integrated analysis of transcriptome and ChIP-on-chip in ovarian cancer cells with NOTCH3 amplification and over-expression to screen for genes whose mRNA levels are regulated by NOTCH, and whose promoters are bound by the NICD3/CSL transcription complex.
MATERIALS AND METHODS
Affymetrix GeneChip Analysis
Cell cultures were treated with 5 μM MRK003, and were harvested at 24 hr and 48 hr. As a control, DMSO was used in parallel under the same experimental conditions. Affymetrix GeneChip array, HG-U133 Plus 2.0, was used to analyze the transcriptome. The fold change of mRNA levels of each individual gene was calculated as the ratio of MRK003 treatment to control treatment at each time point. We used the logarithm of fold change as the data output (i.e., test statistic) and performed significance analysis to calculate p value, which is defined as the probability of obtaining a test statistic at least as extreme as the one that is actually observed under the null hypothesis. For null distribution we assumed that the test statistic followed a normal distribution where the mean and standard deviation were estimated from the control samples. We also implemented the Benjamini and Hochberg procedure (13) for multiple hypothesis testing and estimated the false discovery rate (FDR) for significantly expressed genes. Significantly up-regulated and down-regulated genes were finally determined by a predefined false discovery rate cutoff (FDR ≤ 0.1) and p value (p ≤ 0.003).
Chromatin Immunoprecipitation Analysis
OVCAR3 cells were first treated with 5 μM Dimethyl dithiobispropionimidate (Thermo Scientific) followed by crosslinking with formaldehyde. Cells were lysed in a buffer containing 1% SDS, 10 mM EDTA, and 50 mM Tris-HCl, pH 8.0, and sonicated. Lysates were incubated with Protein G Sepharose (Invitrogen) for 1 hr. Bead cleared lysates were incubated with anti-NOTCH3 antibody (sc-5593, Santa Cruz or #3446, Cell Signaling) at 4°C overnight. After washes, the complexes were eluted by incubating beads in buffer containing 1% SDS/0.1 M NaHCO3 and incubated at 37°C for 30 min. Samples were treated with RNase A and subsequently proteinase K. DNA was proceeded for ChIP-qPCR analysis as previously described (14). Threshold cycle numbers (Ct) were obtained using the iCycler Optical system interface software. Data were normalized using a standard curve, which was prepared from sonicated input (total) DNA. PCR primers are listed in Table S4.
ChIP-on-chip Analysis
The DNA sample from ChIP was amplified using a Sigma WGA2 kit and purified using a Qiagen QIAquick PCR purification kit. For input DNA control, 200 ng of purified DNA was used for WGA2 PCR. An aliquot of each amplified sample was diluted to 5 ng/μl and used for qPCR testing with positive and negative control primers. In cases in which 4 μg of DNA was not produced from the WGA2 PCR, 10 ng of the WGA2 reaction was reamplified using the WGA3 kit (Sigma) and retested with control primers. The NimbleGen 385K RefSeq promoter 1-plex array was used for sample hybridization. The NimbleGen program SignalMap was used to obtain signal peaks for each sample and to map these peaks to their corresponding genomic region. ChIP-on-chip data were analyzed by the Model-based analysis of two-color arrays (MA2C) method (Song, Johnson et al., 2007), and the signal intensity (raw) data on each chip were normalized at probe level. A window-based peak detection approach was implemented on the normalized data to detect peak regions. A MA2C score was assigned using the median of the probes in the window and p was calculated. A cutoff score of p = 0.005 was set for identifying significant peaks. Finally, all significant peak regions were mapped to the nearest genes.
De novo Motif Discovery and Enrichment Analysis
We performed de novo motif discovery on the NOTCH3 ChIP-on-chip binding peak regions. For pre-processing, we first used RepeatMasker to mask DNA sequences with interspersed repeats and low complexity. Then a Gibbs motif sampler algorithm in CisGenome was applied on the processed sequence data to extract top conserved motifs of 8 bp sequence length. To identify the most significant motifs, we performed enrichment analysis by comparing the occurrence frequency in ChIP-on-chip with occurrence in negative control regions which were extracted from the human genome with matched physical properties of ChIP-on-chip binding sequences. We defined the enrichment score as the ratio of these two occurrence rates, and the significance of enrichment was determined based on Fisher’s hypergeometric test.
Gene Set Enrichment Analysis
We performed gene set enrichment analysis for NOTCH3 ChIP target genes in transcriptome microarray data sets. Specifically, for each time point, we obtained the fold changes in gene expression and then calculated the test statistic, which was defined as the summation of the absolute value of logarithm of fold changes of overlapped NOTCH3 ChIP-chip target genes in the up/down-regulated transcriptome. To assess significance of NOTCH3 ChIP-chip targets enriched in the NOTCH3 up/down-regulated transcriptome, we performed a significance test and calculated the p-value. We repeated 10,000 times to randomly sample the same number of genes from the entre up/down-regulated gene population, and then calculated the test statistics to generate the null distribution. The empirical p-value was calculated from the frequency by which the test statistic in the null distribution was larger than in the observed distribution.
Electrophoretic Mobility Shift Assay
Electrophoretic mobility shift assay (EMSA) was performed using a LightShift Chemiluminescent EMSA kit (Pierce Biotechnology) according to the manufacturer’s protocol. Biotin-labeled oligonucleotide probes were manufactured by Sigma (St. Louis, MO), and complimentary probes were annealed. The biotin-labeled, double stranded probes were then incubated for 20 min at room temperature with nuclear lysates from HEK293T cells transfected with pcDNA-CSL-V5 or incubated with purified recombinant proteins (kind gifts from Dr. Stephen C. Blacklow, Harvard University). DNA/protein complexes were separated by electrophoresis, transferred to a nylon membrane, incubated with streptavidin-HRP, and visualized by chemiluminescence.
RESULTS
Genes and pathways that respond to NOTCH inactivation in ovarian and breast cancer cells
Based on gene expression microarray dataset, we selected OVCAR3 and MCF7 as the cell models to identify NOTCH3-regulated genes because both cell lines express the highest level of NOTCH3 mRNA among all four NOTCH receptors (Fig. 1A) (4). We chose to use a γ-secretase inhibitor, MRK003, to block NOTCH signals as MRK003 demonstrated specific blocking of NICD generation. Using an antibody that recognizes various secretase cleavage products of NOTCH3 in Western blot analysis, we observed a noticeable decrease of the post-γ-secretase cleavage product, NICD3, and an increase of the pre-γ-secretase cleavage product, NEXT (Notch Extracellular Truncation) after MRK003 treatment (Fig 1B). In contrast, MRK003 did not affect the levels either of full-length NOTCH3 or of the S1 cleavage product of NOTCH3, TM. The potency of MRK003 in suppressing γ-secretase was further confirmed in a Western blot analysis using an antibody specific to NICD1 (Fig 1B).
Fig 1. Differential expression of NOTCH3 in OVCAR3 and MCF7 cells, and transcriptome change in response to MRK003 NOTCH inhibitor.
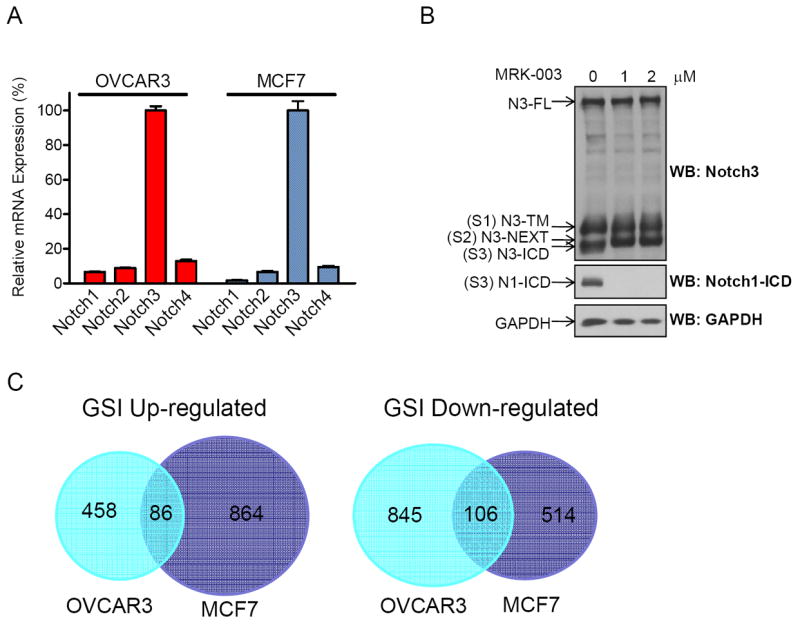
A. Transcript levels of four NOTCH receptors in OVCAR3 and MCF7 cells that were normalized to IOSE-80pc. B. Effects of MRK003 on OVCAR3 cells. Western blot analysis detects the cleaved NOTCH3 fragments and the amounts of NICD3 and NICD1 are significantly decreased by MRK003. N3-FL: full-length NOTCH3; S1 cleavage by a furin-like convertase generates N3-TM; S2 cleavage by metalloprotease generates N3-NEXT; S3 cleavage by γ-secretase produces NICD3. C. Venn diagrams representing the numbers of GSI-regulated genes that are unique or common in OVCAR3 and MCF7 cells.
Next, we performed Affymetrix gene expression array analysis in OVCAR3 and MCF7 cells 24 and 48 hrs after MRK003 treatment. Statistical analysis performed in the transcriptome dataset showed that in OVCAR3, 951 genes were significantly down-regulated and 544 genes up-regulated by MRK003 treatment in at least one of the two time points (p ≤ 0.003, FDR ≤ 0.1). In MCF7, 620 genes were down-regulated and 950 genes up-regulated (p ≤ 0.003, FDR ≤ 0.1). There was only a small fraction of genes shared by both cell lines, and the majority of NOTCH-regulated genes are specific to each cell line (Fig. 1C).
Ingenuity pathway analysis of the MRK003 downregulated genes performed in each cell line demonstrated that canonical pathways including purine and pyrimidine metabolism, cell cycle regulation, and glycolysis/gluconeogenesis were enriched in both OVCAR3 and MCF7. Analysis the MRK003 up-regulated genes, on the other hand, identified pathways involved in generating nitric oxide and reactive oxygen species are common in both cell lines (Table 1). The list of differentially expressed genes following MRK003 treatment and their fold changes at both 24 and 48 hrs have been deposited in https://jshare.johnshopkins.edu/twang16/OVCAR3_MCF7_MRK003 Genes
Table 1.
Canonical Pathways in Notch-Regulated Transriptome
| Ingenuity Canonical Pathways | MCF7 | OVCAR3 | |||
|---|---|---|---|---|---|
| P Value | Ratio | P Value | Ratio | ||
| MRK-003 Down-regulated | Cell Cycle Control of Chromosomal Replication | 0.00 | 0.47 | 0.04 | 0.17 |
| Pyrimidine Metabolism | 0.00 | 0.13 | 0.04 | 0.14 | |
| ATM Signaling | 0.00 | 0.19 | 0.00 | 0.15 | |
| Cyclins and Cell Cycle Regulation | 0.00 | 0.13 | 0.01 | 0.12 | |
| Breast Cancer Regulation by Stathmin1 | 0.00 | 0.09 | 0.03 | 0.08 | |
| Cell Cycle: G2/M DNA Damage Checkpoint Regulation | 0.00 | 0.17 | 0.00 | 0.21 | |
| Protein Ubiquitination Pathway | 0.00 | 0.07 | 0.02 | 0.08 | |
| Purine Metabolism | 0.00 | 0.07 | 0.01 | 0.09 | |
| p53 Signaling | 0.00 | 0.11 | 0.02 | 0.11 | |
| Germ Cell-Sertoli Cell Junction Signaling | 0.00 | 0.08 | 0.01 | 0.09 | |
| RAN Signaling | 0.01 | 0.17 | 0.01 | 0.22 | |
| Prostate Cancer Signaling | 0.01 | 0.21 | 0.04 | 0.19 | |
| Molecular Mechanisms of Cancer | 0.02 | 0.25 | 0.02 | 0.07 | |
| Glycolysis/Gluconeogenesis | 0.05 | 0.31 | 0.01 | 0.12 | |
| Antiproliferative Role of TOB in T Cell Signaling | 0.05 | 0.34 | 0.00 | 0.27 | |
| MRK-003 Up-regulated | Biosynthesis of Steroids | 0.00 | 0.29 | 0.00 | 0.33 |
| Glycine, Serine and Threonine Metabolism | 0.00 | 0.13 | 0.04 | 0.07 | |
| Production of Nitric Oxide and Reactive Oxygen Species in Macrophages | 0.00 | 0.09 | 0.03 | 0.06 | |
| TR/RXR Activation | 0.02 | 0.13 | 0.01 | 0.08 | |
| Metabolism of Xenobiotics by Cytochrome P450 | 0.02 | 0.14 | 0.04 | 0.07 | |
Identification of NOTCH3 direct target genes by ChIP-on-chip
In order to identify genes that were directly regulated by NOTCH3, we applied a ChIP-on-chip assay in OVCAR3 cells using the 385K RefSeq promoter array, which contains 18,028 promoter regions for 24,659 transcripts (NimbleGen). Our results showed that a total of 797 promoters were enriched by ChIP-on-chip using a NICD3 antibody (p < 0.005) (Table S1). To validate the ChIP-on-chip results, we performed quantitative PCR in 10 randomly selected gene promoters. The ChIP-qPCR showed that binding of NICD3 to 8 of 10 representative promoter regions could be confirmed, among which is a previously reported NOTCH1 target gene, PIN1 (Fig. S1)(15). To further validate the above result, we used a second anti-NOTCH3 antibody in the ChIP-qPCR analysis and obtained results consistent with the first anti-NOTCH3 antibody (Fig. S1).
De novo motif analysis of all 797 NICD3 ChIP-enriched promoter sequences revealed over-representation of the canonical CSL DNA binding motif, TTCCCA (p= 0.016). However, the most significantly enriched sequence motif was GTTGCCAT (designated as N1) which has not been previously described (p < 0.0001) (Fig. 2A). This new motif resembles the complementary sequence of the canonical CSL binding motif, albeit with several nucleotide substitutions (Fig. 2A). We assessed the distributions of ChIP-enriched DNA fragments including the canonical and the N1 motifs across the promoter regions (Fig. 2B). As compared to all ChIP-pull down DNAs (Fig. 2B, top), the binding sites of N1 and canonical motifs shared a very similar distribution- with more binding activity observed at, and immediately after, the transcription initiation site (Fig. 2B, middle and bottom). We further analyzed the promoter sequences bound by NOTCH3. Among 601 promoter sequences that were available in the BioMart database, the N1 motif was present in approximately 90% of the NOTCH3 ChIP target genes. Furthermore, the majority (86%) of the NOTCH3-bound promoters contained paired CSL and N1 motifs (Fig. S2A). The enrichment of the N1-CSL motif pair in the NOTCH3-bound promoters is significant as compared to their presence in the non-NOTCH3-bound promoters (p <0.01, Fisher’s exact test). The average distance between paired CSL and N1 motifs (defined as closest adjacent CSL and N1 motifs) was 238 base pairs; the distance distribution between paired CSL and N1 motifs is shown in Fig. S2B.
Fig 2. Identification of proteins that bind to promoters of potential NOTCH3-regulated genes.
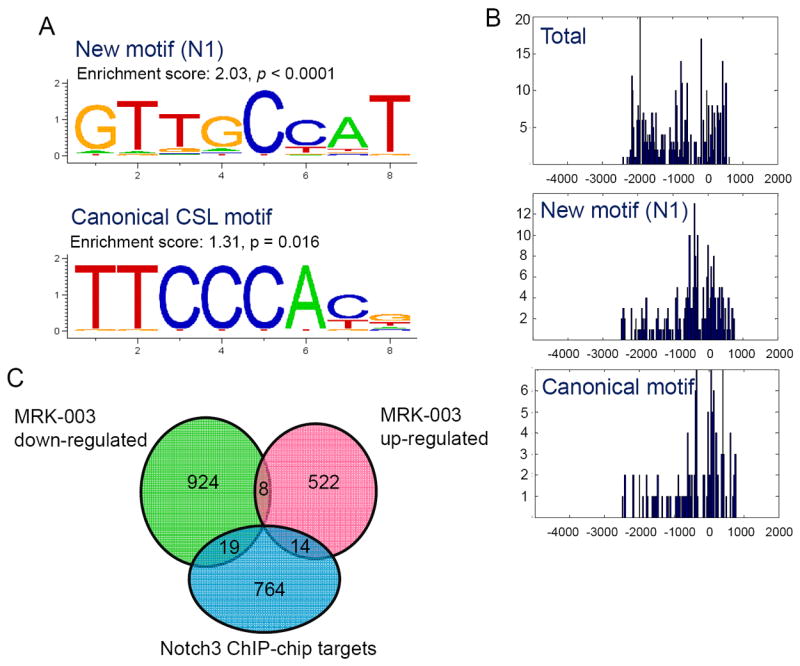
A. ChIP-on-chip analysis reveals enrichment of a previously un-described nucleotide sequence motif (N1). At each nucleotide position as shown, the height of a letter is proportional to the observed frequency of the corresponding nucleotide, and the height of each stack corresponds to the level of conservation, represented as information content (y-axis, bits). B. Distribution of chromatin immunoprecipitated DNA fragments at gene promoters and at both N1 and canonical CSL binding sites. The “0” on the x-axis refers to the transcription start site. Top: total DNA fragments; middle: new binding motifs; bottom: canonical CSL binding motifs. C. Venn diagram showing co-occurrence of positive hits from ChIP-on-chip targets and MRK003-regulated genes in OVCAR3.
We also compared the current NOTCH3 ChIP-on-chip data with a previous NOTCH1 ChIP-on-chip study which was performed using a RefSeq promoter array (16). There was minimal overlap of the ChIP target genes between these two studies, with the exception of one gene, LGMN. However, we found that in addition to the canonical CSL binding motif, N1 motif was also significantly enriched in the NOTCH1-ChIP target promoters (16) (Fig. S3). The above results indicate that both NOTCH3/CSL and NOTCH1/CSL transcriptional complexes directly bound to the N1 motif or to the region in close proximity to the N1 motif.
To test if these 797 NOTCH3-ChIP target genes were enriched in the NOTCH3-regulated transcriptome based on the MRK003 study, we performed Gene Set Enrichment Analysis in both OVCAR3 and MCF7 transcriptome datasets. Our analysis demonstrated that in the OVCAR3 cells, ChIP targets were significantly enriched in the NOTCH3 up-regulated transcriptome 24 hours after MRK003 treatment and in down-regulated transcriptome 48 hours after treatment (p < 0.05). In contrast, ChIP targets were not enriched in NOTCH-regulated transcriptome of MCF7 cells at either of the time points (Table S2).
By integrating NOTCH3-ChIP targets with the NOTCH3-regulated transcriptome in OVCAR3 cells, we identified a total of 33 genes that overlapped in both datasets (Fig 2C and Table S3). To test if these genes were expressed in ovarian carcinoma tissues, we analyzed a previously published transcriptome dataset using Oncomine™. In ovarian cancer, we found that NOTCH3 and five of its regulated genes, including EHF, DLGAP5, DNAJC19, NCOA5, and NRAS, were over-expressed in ovarian carcinoma tissue as compared to normal ovarian surface epitheliums (p < 0.05)(Fig. S4). We selected DLGAP5 (Disks large-associated protein 5, also known as HURP, hepatoma up-regulated protein) for further characterization because its binding intensity in the ChIP assay was the highest among all ChIP target genes (Table S3).
Binding of the NICD3/CSL transcription complex to the new N1 motif
The promoter region of DLGAP5 contains three canonical CSL binding motifs, which are located at -2354 bp (C1), -1862 (C2), and +496 (C3). The ATG translation initiation codon is located in exon 2; thus, it is of note that C3 is located in the intron between exon 1 and exon 2. The novel NICD3 binding motif (N1) as discovered in this study is located at +508 bp, adjacent to the 3rd canonical CSL motif, C3 (Fig. 3A & 3B, top panels). To determine promoter sequences specifically involved in the direct binding of the NICD3/CSL transcription complex, we designed PCR primers flanking each of the three canonical CSL binding motifs and performed ChIP-qPCR using a NICD3 antibody. The distance between C3 and N1 was only 6 bp, thus the third PCR primer pair was designed to amplify the C3/N1 motif region. The results demonstrated that the C3/N1 region was significantly enriched by ChIP (Fig. 3A, bottom). To further confirm direct binding of the NICD3/CSL protein complex to the unique C3/N1 pair, we performed EMSA assays. Wildtype biotinylated DNA probes encompassing the C3/N1 region were mixed with nuclear extract prepared from HEK293T cells, which contains abundant endogenous CSL and NOTCH3 proteins. A specific band shift was observed, a phenomenon that could be competitively blocked by unlabeled wildtype probes in a dose-dependent manner. This band shift was not blocked if the competing probe contained either C3 or N1 mutated sequences (Fig. 3B). These results indicate that both canonical and N1 motif sequences are required for the formation and/or stabilization of the NICD3/CSL/DNA complex. Next, we determined whether the NICD3/CSL protein complex interacted with the N1 motif alone. Using oligonucleotide probes that encompassed only the N1 motif, we detected a specific band shift in EMSA when the nuclear extract was prepared from HEK293T cells. This band shift was competitively abolished by unlabeled wildtype N1 probe but not by the mutant N1 probe (Fig. 3C).
Fig 3. Binding of NOTCH3 transcription complex with the N1 motif.
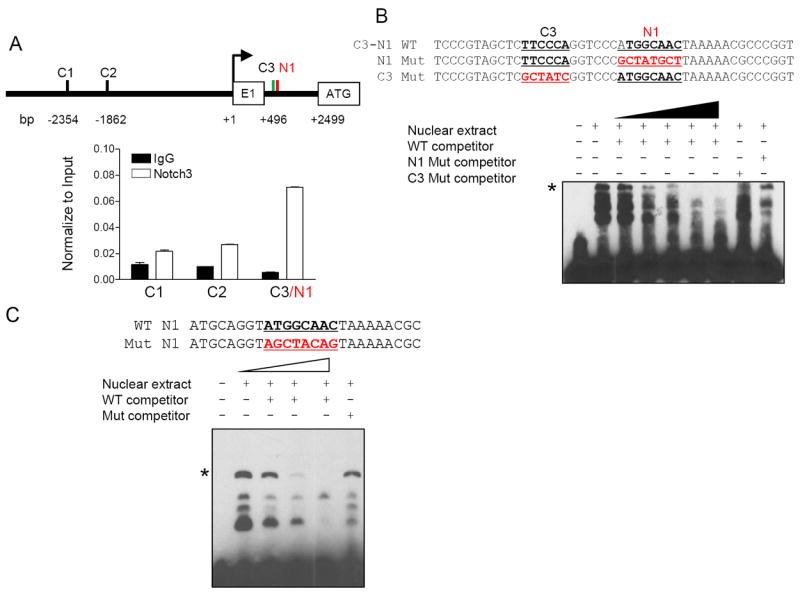
A. Distribution of the three canonical CSL binding sites (C1-C3) and the new binding motif (N1) in the DLGAP5 promoter region (top panel). Chromatin immunoprecipitation was performed followed by qPCR using primers flanking C1, C2, and C3/N1 promoter regions (bottom panel). IgG was used as the normalization control. B. The probes for the EMSA assay were generated from the C3/N1, mutant N1 (with wildtype C3) and mutant C3 sequences (with wildtype N1) (upper panel). Nuclear extracts from HEK293T cells were mixed with biotin-labeled probes (bottom panel). An asterisk indicates the specific shifted band that can be competed away by increasing concentrations of unlabeled wildtype probes but not by either mutant N1 or mutant C3 probes. The mutated sequences are highlighted. C. Nucleotide sequences containing only the N1 region were used for EMSA assay (top panel). A specific band shift (asterisk) can be competed away by increasing concentrations of unlabeled wildtype probes, but not by unlabeled N1 mutant probes.
To confirm that CSL protein directly bound to the N1 motif, we performed EMSA assay by mixing affinity-purified CSL recombinant protein with biotinylated N1 probe. A band shift was identified, indicating that CSL protein interacted with the N1 probe (lane 2, Fig. 4). The binding could be competitively reduced by excessive unlabeled wildtype N1 probe but not by the mutated N1 probe (lane 3 and 5, Fig. 4). Given that the N1 motif contains three consecutive G or C nucleotides which are also present in the canonical CSL motif, it is possible that the N1 site represents a variant of the CSL binding motif. To test this possibility, we generated a new mutant N1 sequence by replacing GGC from the N1 motif with CCC from the canonical CSL binding motif and used it in the EMSA competition assay (Fig. 4, top). The results demonstrated that this new probe (N1 backbone but with CCC rather than GGC in the middle) could efficiently compete with the parental wildtype N1 probe (lane 4, Fig. 4), suggesting functional similarity between these two motifs. To determine if the NICD3 protein could complex with the N1 DNA probe and CSL protein, we performed additional EMSA experiments, and demonstrated a supershift when both NICD3 and CSL proteins were present as compared to the control groups with individual proteins (lane7, Fig. 4). Next, we determined if the N1 site could be bound by NOTCH1/CSL complex. As shown in lane 12 of Fig. 4, a similar “supershift” was detected in the presence of both NICD1 and CSL proteins. This band shift could also be competitively blocked by unlabeled wildtype N1 probe and unlabeled CSL probe, but not by the mutant N1 probe.
Fig 4. Notch-CSL protein complex interacts with the N1 motif.
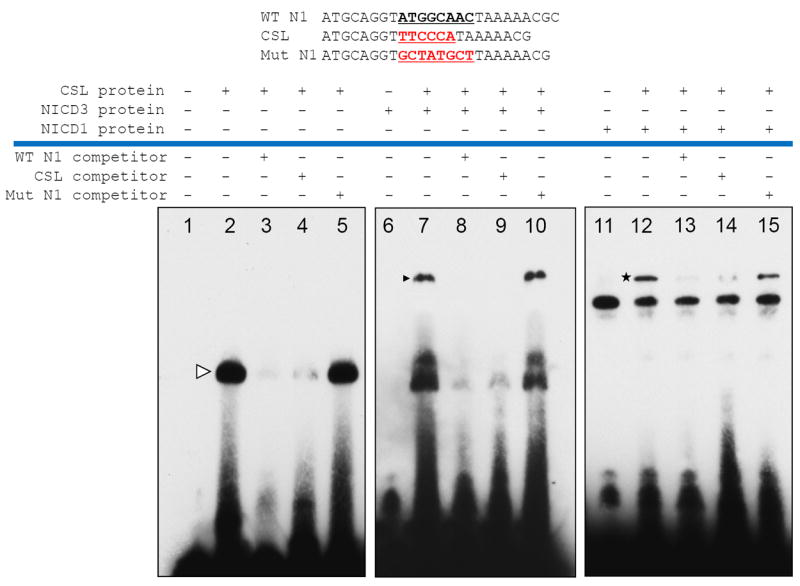
Top: nucleotide sequences of EMSA probes encompassing the N1 region. A band shift is detected when CSL protein was incubated with the biotin-labeled N1 probes (lane 2), and this band can be competitively reduced by either unlabeled wildtype N1 probe or CSL probe (lane 3-4) but not by mutant N1 probe (lane 5). When CSL protein was mixed with NICD3 or NICD1 protein, a higher molecular weight band shift is detected (lane 7 and lane 12). This shifted band was competitively abolished in the presence of unlabeled wildtype N1 probe or CSL probe (lane 8-9 and lane 13-14) but not by the mutant N1 probe.
N1 binding motif is essential for transcription of NOTCH3-regulated genes
To determine if the C3/N1 promoter region was involved in transcriptional regulation of DLGAP5, we generated a luciferase DLGAP5 promoter reporter containing the C3/N1 paired motif sequences. A deletion mutant lacking both C3 and N1 binding motifs was used as a control (Fig. 5A). In addition, to determine the individual contributions of C3, N1, and their adjacent sequences to DLGAP5 transcription, we created a series of DLGAP5 promoter reporter constructs with different structural modifications including microdeletion at the C3 or N1 motif, and various truncations in the promoter region (Fig. 5A). The results demonstrated that the wildtype reporter yielded a higher level of luciferase activity in cells ectopically expressing NICD3 than those without. In contrast, microdeletion in either the C3 or N1 sequence reduced promoter activity in the presence of NICD3 to the level obtained without NICD3. Interestingly, microdeletion in both N1 and C3 sites did not lead to a further reduction in the reporter activity, suggesting that both C3 and N1 sequences are required for DLGAP5 promoter activity, and may play an equivalent role in transcriptional regulation of DLGAP5. The four truncating deletion reporters all exhibited very low basal levels of reporter activity as compared to reporters with wildtype sequences or with C3/N1 microdeletions. Of note, Truc-2 showed that the presence of C3/N1 site alone was not sufficient to support the basal transcriptional activity of DLGAP5. The data from truncation mutants suggest that the C3/N1 sequences in the DLGAP5 promoter are required but not sufficient to confer full transcriptional activity of DLGAP5 (Fig. 5A).
Fig 5. N1 motif is required for NICD3-dependent transcription.
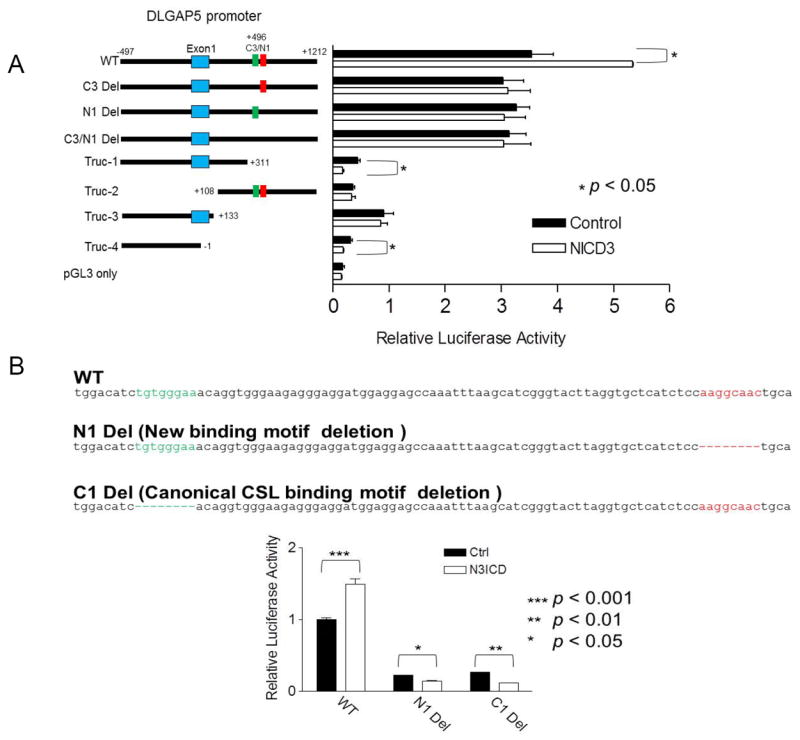
A. Left panel: Schematic presentation of luciferase reporter constructs containing the wildtype, microdeleted, and truncated DLGAP5 promoters. Green bar: C3 motif, red bar: N1 motif, Blue box: exon1. Right panel: Luciferase reporter activity of different DLGAP5 promoter constructs. Ectopic expression of NICD3 leads to increased luciferase activity using the wildtype promoter construct, while microdeletion of N1, C3, or both, loses NICD3-regulated transcriptional activity. For each comparison group, only p values with significance are indicated. B. Top panel: nucleotide sequences of the PIN1 promoter and the deletion mutants. Canonical CSL motif: green; N1 motif: red. Bottom panel: Luciferase reporter activity of the wildtype PIN promoter and mutant reporter with either N1 or C1 microdeletion.
The above ChIP-on-chip study also identified PIN1 (peptidyl-prolyl cis/trans isomerase-1) as a potential NOTCH3-regulated gene. PIN1 has been reported as a NOTCH1 direct target gene (15), but its transcriptional regulation by NOTCH3 has not been established. Analysis of the PIN1 promoter sequence demonstrated that the N1 motif was present at 65 bp proximal to the previously reported canonical CSL binding site (Fig. 5B). Deletion mutants of either the CSL motif or the N1 motif in the PIN1 promoter were subsequently created, and the results demonstrated that microdeletion of either the canonical CSL binding site or the N1 site led to a similar effect in reducing PIN1 transcriptional activity (Fig. 5B).
Next, we determined if NOTCH3 protein was involved in regulating the transcriptional activity of DLGAP5. OVCAR3 cells were transfected with a DLGAP5 wildtype promoter reporter and were treated with either MRK003 or NOTCH3 shRNAs to reduce the NICD3 protein level. We found that either NOTCH3 shRNA or MRK003 treatment significantly suppressed DLGAP5 reporter activity (Fig. S5A) and the protein levels of NOTCH3 and DLGAP5 were selectively decreased by NOTCH3 shRNAs (Fig. S5B).
DLGAP5 is a potential mediator of NOTCH3 signaling in cancer cells
We next asked if constitutive DLGAP5 expression could rescue the growth suppressive effect caused by inactivation of the NOTCH3 pathway, a phenotype that has been previously reported in other cancer types (4, 11). OVCAR3 and MCF7 cancer cells were transfected with a CMV-driven expression vector to ectopically express DLGAP5 (Fig. 6A) and the cells were then treated with either MRK003 or NOTCH3 shRNAs to suppress NOTCH3 signaling. As compared to transfection with the vector only control, transfection with the DLGAP5 cDNA expressing construct resulted in significant protection from the anti-proliferative effect of MRK003 or NOTCH3 shRNA (Fig. 6B). The above result suggests that DLGAP5 expression could reverse, at least in part, the anti-proliferative effect of NOTCH3 pathway inactivation.
Fig. 6. Ectopic expression of DLGAP5 partially reverses the anti-proliferative effect by MRK003 and NOTCH3 shRNAs in cancer cells.
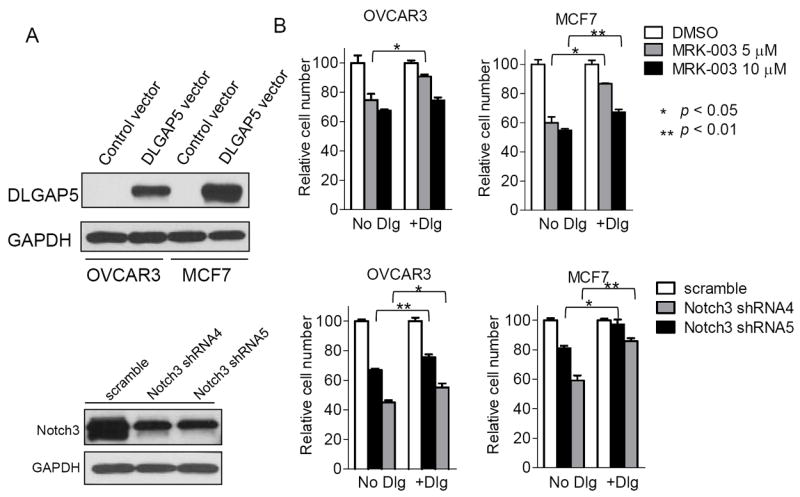
A. Western blot shows robust expression of DLGAP5 (~95 kD) in OVCAR3 and MCF7 cells after transfection with a DLGAP5 expression vector but not in cells transfected with a control vector. B. The effect of MRK003 or NOTCH3 shRNAs on relative cell numbers in DLGAP5-transfected OVCAR3 and MCF7 cells. In both MRK003 and NOTCH3 shRNA-treated groups, the number of DLGAP5-expressing cells significantly increased as compared to cells without ectopic DLGAP5 expression. For each comparison group, only p values with significance are indicated.
Biological effects of DLGAP5 expression in cancer cells
Next, we determined if DLGAP5 expression was essential for cellular survival and growth of ovarian cancer cells. We have previously shown that ovarian cancer frequently harbors NOTCH3 amplification and overexpression, and more importantly, ovarian cancer cells are molecularly dependent on the NOTCH3 pathway activation for cellular proliferation and survival (4). OVCAR3, OVCAR5, and A2780 ovarian cancer cell lines were selected for this functional analysis because they express high levels of DLGAP5 mRNA (Fig. S6). The efficiency of two DLGAP5 shRNAs in reducing DLGAP5 transcript levels was demonstrated by qRT-PCR (Fig. S7). After gene knockdown using either DLGAP5 shRNA1 or shRNA3, the percentage of cells in the G2/M phase was significantly increased as compared to control shRNA-treated cells (Fig. S8A). Cellular proliferation was significantly reduced in DLGAP5-shRNA treated groups in all three ovarian cancer cell lines (Fig. S8B). However, no evidence of increased apoptotic activity was observed in cells treated with DLGAP5 shRNAs (data not shown). In addition, DLGAP5 shRNA-treated OVCAR5 and A2780 cells failed to form subcutaneous tumors in nude mice, whereas control cells treated with scrambled shRNA were highly tumorigenic (Fig. S9).
DISCUSSION
In this study, we found that pathways involving nucleotide biosynthesis and cell cycle checkpoint regulation are enriched in the NOTCH transcriptome of both OVCAR3 and MCF7 cells; interestingly, these pathways were also affected by NOTCH signaling in NOTCH1-predominant T-ALLs (16, 17). However, further analysis shows that there is only small overlap in the NOTCH-regulated genes among OVCAR3, MCF7, and T-ALLs (16). This finding indicates that although cell lines from different lineages can share common NOTCH-regulated pathways, the genes that are directly involved could be quite distinct. The fact that the NOTCH3 ChIP targets performed in OVCAR3 were only over-representative in the NOTCH transcriptome of OVCAR3 cells but not in the transcriptome obtained from MCF7 further supports the view that direct target genes of NOTCH are highly context- and cell type- specific. It is plausible that unique co-factors present in distinct cell type play a significant role in dictating the DNA binding specificity and/or affinity of the NOTCH transcription complex.
A new NICD3/CSL binding motif (N1) was identified in this study and promoter reporter analysis in DLAGP5 and PIN demonstrated that the N1 motif likely plays an equally important role as the canonical CSL binding motif in transcriptional regulation. Since the majority of NOTCH3 target genes contain paired N1-CSL motifs at the promoter, it is plausible that N1 and canonical CSL motif sequences collaborate to facilitate the formation and/or stabilization of the NICD3/CSL transcription complex. In fact, we think the N1 motif may represent a “degenerative” version of the CSL binding site, as the N1 motif contains a core sequence of GCC which is very similar to the CCC that is found in the core of the canonical CSL binding motif. In addition, the sequences flanking the G-C core are either A or T bases, implying conservative nucleotide usage at these positions. Therefore, it is not surprising that the N1 motif is present in target sequences pulled down by both Notch3- and Notch1- ChIP. Our EMSA data further demonstrated binding of the N1 motif sequence to NICD3/CSL and NICD1/CSL complexes, providing a plausible role of N1 motif in NOTCH1 and NOTCH3 transcriptional regulation.
Since the occurrence of the N1 motif is more frequent in the Notch3-ChIP target sequences than in the Notch1-ChIP target sequences (Fig. S3), it is likely that the NICD3 transcription complex has a higher binding affinity to the N1 motif than does the NICD1 complex. However, this interpretation is tentative, because the differences may merely reflect the use of different anti-Notch antibodies. On the other hand, we have recently performed ChIP-chip using antibodies reacting to other transcriptional factors or chromatin remodeling proteins including PBX1, RNA Polymerase II, RSF-1, and SNF2H but did not identify the enrichment of N1 and canonical CSL motif sequences, indicating that NOTCH complexes specifically interact with both motifs. Further genome-wide ChIP-chip or ChIP-seq study is required to address whether the upstream regulatory regions including enhancers of which the sequences were not covered by the arrays could also interact with NOTCH3.
We also have established direct transcriptional regulation of DLGAP5 by NOTCH3 in the current study. DLGAP5 has been shown to be an essential component of the mitotic apparatus (18, 19) and its depletion could lead to prolonged pro-metaphase and aberrant chromatin segregation (20). In fact, DLGAP5 was implicated as an oncogenic target of Aurora A, a major mitotic serine/threonine kinase (21), and phosphorylation of DLGAP5 by Aurora A is prerequisite in regulating spindle assembly and function during mitosis (19). The current study provides new evidence that DLGAP5 is required for the G2/M transition, and its expression is required for tumor growth in a mouse xenograft model. Therefore, the demonstration that DLGAP5 is a direct downstream target of NOTCH3 establishes a new molecular wiring which may, at least partially, explain how NOTCH3 activation contributes to ovarian cancer pathogenesis from the perspective of aberration of mitotic apparatus.
In conclusion, the integrated transcriptome and promoter occupation analysis has allowed us to discover a new NICD/CSL binding motif and to identify NOTCH3 direct target genes in ovarian cancer cells. These results could serve as a molecular foundation for future studies aimed at understanding the mechanisms of NOTCH3 signaling in tumorigenesis and may facilitate the design of NOTCH3-based cancer therapy.
Supplementary Material
Acknowledgments
We thank Dr. Stephen C. Blacklow for critical reading of this manuscript and for gifts including purified CSL and NOTCH proteins, Dr. Giannino Del Sal for the Pin1 promoter reporter plasmid, and Dr. Masayuki Futagami for help in constructing the Pin1 promoter deletion mutants. This work was supported by the Ovarian Cancer Research Fund (TLW), American Cancer Society (TLW), and NIH/NCI grants, RO1CA148826 (TLW), RO1CA129080 (IMS), and RO1CA103937 (IMS).
References
- 1.Roy M, Pear WS, Aster JC. The multifaceted role of Notch in cancer. Curr Opin Genet Dev. 2007;17:52–9. doi: 10.1016/j.gde.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Weng AP, Ferrando AA, Lee W, Morris JPt, Silverman LB, Sanchez-Irizarry C, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–71. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 3.Grabher C, von Boehmer H, Look AT. Notch 1 activation in the molecular pathogenesis of T-cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2006;6:347–59. doi: 10.1038/nrc1880. [DOI] [PubMed] [Google Scholar]
- 4.Park JT, Li M, Nakayama N, Davidson B, Eberhart CG, Kurman RJ, et al. Notch-3 gene amplification in ovarian cancer. Cancer Res. 2006;66:6312–8. doi: 10.1158/0008-5472.CAN-05-3610. [DOI] [PubMed] [Google Scholar]
- 5.TCGA network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen X, Stoeck A, Lee SJ, Shih IM, Wang MM, Wang TL. Jagged1 expression regulated by Notch3 and Wnt/beta-catenin signaling pathways in ovarian cancer. Oncotarget. 2010;1:210–8. doi: 10.18632/oncotarget.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dang TP, Gazdar AF, Virmani AK, Sepetavec T, Hande KR, Minna JD, et al. Chromosome 19 translocation, overexpression of Notch3, and human lung cancer. J Natl Cancer Inst. 2000;92:1355–7. doi: 10.1093/jnci/92.16.1355. [DOI] [PubMed] [Google Scholar]
- 8.Bellavia D, Campese AF, Alesse E, Vacca A, Felli MP, Balestri A, et al. Constitutive activation of NF-kappaB and T-cell leukemia/lymphoma in Notch3 transgenic mice. Embo J. 2000;19:3337–48. doi: 10.1093/emboj/19.13.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu C, Dievart A, Lupien M, Calvo E, Tremblay G, Jolicoeur P. Overexpression of activated murine notch1 and notch3 in transgenic mice blocks mammary gland development and induces mammary tumors. Am J Pathol. 2006;168:973–90. doi: 10.2353/ajpath.2006.050416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dang L, Fan X, Chaudhry A, Wang M, Gaiano N, Eberhart CG. Notch3 signaling initiates choroid plexus tumor formation. Oncogene. 2006;25:487–91. doi: 10.1038/sj.onc.1209074. [DOI] [PubMed] [Google Scholar]
- 11.Yamaguchi N, Oyama T, Ito E, Satoh H, Azuma S, Hayashi M, et al. NOTCH3 signaling pathway plays crucial roles in the proliferation of ErbB2-negative human breast cancer cells. Cancer Res. 2008;68:1881–8. doi: 10.1158/0008-5472.CAN-07-1597. [DOI] [PubMed] [Google Scholar]
- 12.Ong CT, Cheng HT, Chang LW, Ohtsuka T, Kageyama R, Stormo GD, et al. Target selectivity of vertebrate notch proteins. Collaboration between discrete domains and CSL-binding site architecture determines activation probability. J Biol Chem. 2006;281:5106–19. doi: 10.1074/jbc.M506108200. [DOI] [PubMed] [Google Scholar]
- 13.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Methodological. 1995;57:289–300. [Google Scholar]
- 14.Buckhaults P, Zhang Z, Chen YC, Wang TL, St Croix B, Saha S, et al. Identifying tumor origin using a gene expression-based classification map. Cancer Res. 2003;63:4144–9. [PubMed] [Google Scholar]
- 15.Rustighi A, Tiberi L, Soldano A, Napoli M, Nuciforo P, Rosato A, et al. The prolyl-isomerase Pin1 is a Notch1 target that enhances Notch1 activation in cancer. Nat Cell Biol. 2009;11:133–42. doi: 10.1038/ncb1822. [DOI] [PubMed] [Google Scholar]
- 16.Palomero T, Lim WK, Odom DT, Sulis ML, Real PJ, Margolin A, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci U S A. 2006;103:18261–6. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rao SS, O’Neil J, Liberator CD, Hardwick JS, Dai X, Zhang T, et al. Inhibition of NOTCH signaling by gamma secretase inhibitor engages the RB pathway and elicits cell cycle exit in T-cell acute lymphoblastic leukemia cells. Cancer Res. 2009;69:3060–8. doi: 10.1158/0008-5472.CAN-08-4295. [DOI] [PubMed] [Google Scholar]
- 18.Koffa MD, Casanova CM, Santarella R, Kocher T, Wilm M, Mattaj IW. HURP is part of a Ran-dependent complex involved in spindle formation. Curr Biol. 2006;16:743–54. doi: 10.1016/j.cub.2006.03.056. [DOI] [PubMed] [Google Scholar]
- 19.Wong J, Lerrigo R, Jang CY, Fang G. Aurora A regulates the activity of HURP by controlling the accessibility of its microtubule-binding domain. Mol Biol Cell. 2008;19:2083–91. doi: 10.1091/mbc.E07-10-1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong J, Fang G. HURP controls spindle dynamics to promote proper interkinetochore tension and efficient kinetochore capture. J Cell Biol. 2006;173:879–91. doi: 10.1083/jcb.200511132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu CT, Hsu JM, Lee YC, Tsou AP, Chou CK, Huang CY. Phosphorylation and stabilization of HURP by Aurora-A: implication of HURP as a transforming target of Aurora-A. Mol Cell Biol. 2005;25:5789–800. doi: 10.1128/MCB.25.14.5789-5800.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.