

Abstract
This paper demonstrates that the secondary hydroxyl can be functionalized in preference to the primary hydroxyl of a 1,2-diol. The site selectivity is achieved by using an enantioselective organic catalyst that is able to reversibly and covalently bond to the diol. The reaction was parlayed into a divergent kinetic resolution on a racemic mixture, providing access to highly enantioenriched secondary protected 1,2-diols in a single synthetic step.
Selective manipulation of multiple similar functional groups within a molecular ensemble is a significant challenge to chemical synthesis. This problem is generally addressed by employing reaction sequences wherein the more reactive groups are functionalized first.1 The ability to functionalize a less reactive position in the presence of a more reactive group may enable new synthesis strategies. The challenge of such a transformation is that most synthetic catalysts cannot change the energetics of the reaction coordinate sufficiently to overcome inherent biases in substrate reactivity.
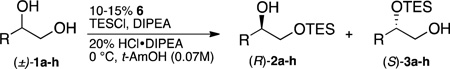
Notable exceptions are enzymes that routinely functionalize less reactive sites within a complex molecule.2 Enzymes are able to achieve these dramatic changes in selectivity by binding substrates in a specific orientation that places the target functional group near a catalytic residue. The entropic price for this exquisite control is paid for through multiple enthalpically favored interactions between the substrate and the amino acids that line the substrate-binding pocket. In analogy to enzymes synthetic catalysts are able to functionalize less reactive sites when they use non-covalent substrate binding as a critical component in determining the overall selectivity.3 For example, both Miller3f and Kawabata3c have reported catalysts that functionalize glucose at the C4 position. In both cases it is postulated that non-covalent interactions are used to bind to the more accessible primary hydroxyl, which directs the reaction to the more hindered site. The size of these organocatalysts is relatively large (although significantly smaller than enzymes), reflecting the need to have a well-defined array of non-covalent interactions to overturn the inherent reactivity. As a complimentary method, using reversible covalent bonding4,5 instead of non-covalent interactions would allow for efficient substrate-catalyst association through a single point of contact, minimizing the catalyst architecture devoted to substrate binding, while retaining the proximity effects necessary to achieve site selectivity. Recent work by Taylor and co-workers has shown that covalent interactions in boron-based catalysts can be used to selectively functionalize sugars.6 We report a chiral organic catalyst that uses reversible covalent bonding between the catalyst and substrate to functionalize a secondary alcohol in preference to an adjacent primary alcohol. Because the catalyst is chiral, the enantiomers of a racemic mixture react at different rates such that a regiodivergent silylation of a racemic mixture ensues (eq 1). This delivers the protected secondary alcohol product in excellent enantioselectivity in a single synthetic step (up to >98% ee, Table 2).7,8
 |
(1) |
Table 2.
Substrate scope for regiodivergent RRM
 | |||||
|---|---|---|---|---|---|
| entry | R | yield 2a (%) |
ee 2b (%) |
yield 3a (%) |
ee 3b (%) |
| 1c | -Cy | 52 | 81 | 41 | 97 |
| 2d | -(CH2)3CH3 | 54 | 79 | 40 | 98 |
| 3c | -CH2CH(CH3)2 | 53 | 82 | 40 | 98 |
| 4e | -CH3 | 48 | 70 | 36 | 92 |
| 5f | -CH2Ph | 46 | 80 | 40 | 96 |
| 6d | -CH2OBn | 56 | 74 | 40 | 99 |
| 7g | -CH2OPh | 44 | 78 | 32 | 96 |
| 8e | -CH=CH2 | 53 | 57 | 37 | 91 |
| 9c | -CH2Cl | 52 | 90 | 45 | 97 |
| 10c | -CH2Br | 50 | 91 | 41 | 98 |
yields and ee’s are an average of two runs; note in all cases 2 and 3 are separable by SiO2 column chromatography such that the isolated yields are of the pure constitutional isomers.
ee determined by GC or HPLC analysis.
Run with 15% 6b, 1.3 eq TESCl and DIPEA for 45 min.
Run with 10% 6a, 1.2 eq TESCl and DIPEA for 1.5 h.
Run with 15% 6b, 1.2 eq TESCl and DIPEA for 25 min.
Run with 10% 6b, 1.2 eq TESCl and DIPEA for 45 min.
Run with 15% 6a, 1.4 eq TESCl and DIPEA for 45 min.
We thought that a scaffolding catalyst previously developed for the desymmetrization of 1,2-diols9 could be used to perform the site-selective functionalization of a secondary hydroxyl over a primary hydroxyl. As shown in Scheme 1, each enantiomer of diol will have an independent site selectivity based on both the affinity of the 1° vs. 2° hydroxyls for the catalyst (Keq and K’eq), and relative rates of functionalization (k1 vs. k2 and k3 vs. k4).10 We hypothesized that one of the enantiomers of the diol would allow us to effectively combine the binding selectivity and stereoselectivity to overturn the large inherent substrate bias for primary functionalization. In this report we show that the scaffolding catalyst gives dramatically different site selectivities for the enantiomers of the diol, such that an effective divergent kinetic resolution occurs.
Scheme 1.

Curtin-Hammett kinetics as a basis for site and enantioselectivity
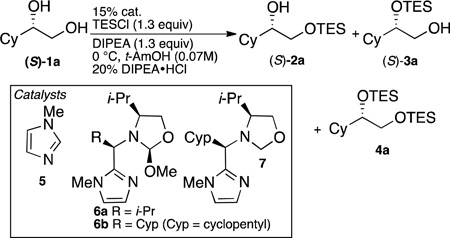
To simplify this complex problem, the site-selective silylation of a single enantiomer of terminal 1,2-diol (S)-1a (Table 1) was investigated. As expected, a control reaction examining silylation with triethylsilyl chloride and catalysis with N-methylimidazole resulted in silylation of the primary alcohol (2a:3a = 98:2, Table 1 entry 1). Preliminary studies showed that, under acetal exchange conditions, primary alcohols have ~8 fold higher affinity for 6a than secondary alcohols (see SI), suggesting that the binding selectivity alone would not be sufficient to overturn the inherent substrate bias (98:2). Using 6a in the silylation of (S)-1a results in a dramatic change in selectivity such that the secondary protected product is favored (2a:3a = 18:82, Table 1, entry 2). Furthermore, switching to scaffold 6b, increases the selectivity to 12:88 (2a:3a, Table 1, entry 3) with an isolated yield of 74% of (S)-3a. The net site selectivity change is between 2 and 3 orders of magnitude compared to N-methyl imidazole. A second control reaction using 7 as the catalyst, which does not have a substrate-binding site, results in selective formation of the primary protected product (2a:3a = 91:9; Table 1, entry 4), demonstrating the necessity of covalent bonding between the substrate and catalyst. This reaction was also the only reaction that suffered from low conversion (33% conv), consistent with the fact that covalent bonding to the catalyst is also necessary for rate acceleration. Application of 6b to (R)-1a results in a complete reversal in site selectivity so that silylation of the primary alcohol occurs almost exclusively (Table 1, entry 5).
Table 1.
Catalyst screening for 2° alcohol protection
 | ||||
|---|---|---|---|---|
| entry | catalyst | yield 4a (%)a | 2a:3aa | yield 3a (%)a |
| 1 | 5 | 9 | 98:2 | <2 |
| 2 | 6a | 5 | 18:82 | 58 |
| 3 | 6b | 5 | 12:88 | 76 (74)b |
| 4 | 7 | 8 | 91:9 | <1 |
| 5c | 6b | 7 | >98:2 | <2 |
GC yields and selectivities based on internal standard (trimethoxybenzene).
isolated yield of 3a.
(R)-1a used as substrate
These foregoing results suggested that kinetic resolution could be achieved on the racemic substrate. When the racemic substrate is silylated under the standard conditions both the primary and secondary silylated products are enantioenriched, with 3a forming in 97% ee in favor of the (S)-configuration and 2a in 81% ee favoring the (R)-configuration (Table 2, entry 1). Such a kinetic resolution falls under the category of a regiodivergent reaction on a racemic mixture (regiodivergent RRM).11,12,13
Investigation of the substrate scope for the resolution revealed that the reaction is permissive of both sterically demanding groups14 such as cyclohexyl and sec-butyl (Table 2, entry 3), and smaller alkyl groups such butyl (Table 2, entry 2). In most cases the enantioselectivity of the secondary protected product was found to be ≥96% ee with the exception of Me and vinyl substituents (92% and 91% ee, Table 2, entries 4 and 8). The methyl and vinyl substrates highlight the importance of stereoselectivity as a means of controlling site selectivity, because as the smallest substituents they are the most challenging to differentiate. Both vinyl and OPh groups also pose the additional challenge that the secondary alcohols are electronically deactivated towards silylation. Consequently, background silylation (including bis silylation) becomes more competitive with 2° alcohol functionalization lowering the overall yield of 3. When R is benzyl and OBn, the reaction proceeds in good yield (40%) and high enantioselectivity (96% and 99%, Table 2, entries 5 and 6). Additionally, both – Cl and –Br substituents are tolerated under the reaction conditions, and provide excellent enantioselectivities for 3 (Table 2, entries 9 and 10).
The divergent resolution allows for access to (S)-3a–h in high ee, but the formation of (R)-2a–h occurs with more modest enantioselectivities. The lower enantioselectivity for (R)-2a–h directly results from the site selectivity observed for the S-diol (Table 1, entry 3; also see Scheme 1 boxes). The formation of (R)-2a–h is faster than (S)-3a–h, suggesting that silylation of the primary alcohol is also catalyzed by 6.15 Therefore, analogous to a standard kinetic resolution, the enantioselectivity for (R)-2 can be increased by lowering the overall conversion. However, unlike a traditional kinetic resolution, which generally is effective at providing starting material in high ee,16,17 the divergent resolution makes it possible to isolate the products in high ee and yield. For example, by using 0.6 equiv of TESCl, (R)-2a (R = Cy) is isolated in 47% yield and in 92% ee (eq 2) vs. 56% yield and 81% ee with 1.3 equiv TESCl (Table 2, entry 1). From a practical perspective these results mean that either the 1° or 2° protected products can be accessed in high enantioselectivity simply by adjusting the amount of TESCl employed.
 |
(2) |
The synthesis of enantioenriched diols is a process that has been achieved with several noteworthy enantioselective reactions including asymmetric dihydroxylation of olefins,18 hydrolytic kinetic resolution of terminal epoxides,19 and asymmetric diboration/oxidation of olefins and alkynes.20 However, to access compounds where a secondary alcohol is protected in the presence of a primary alcohol would require an additional 2–3 synthetic steps from the enantiopure diol. Using the regiodivergent RRM not only resolves the enantiomers of starting material but also chemically differentiates the primary and secondary alcohols. This procedure can also be used to selectively functionalize the secondary over the primary hydroxyl of enantioenriched 1,2-diols in a single step. The ability to overturn the large substrate bias was made possible by implementing a catalyst that is highly stereoselective and has a preference for binding less hindered hydroxyls covalently. We believe that exploiting binding selectivity and proximity will be a productive means of functionalizing inherently less reactive sites on complex molecules.
Supplementary Material
ACKNOWLEDGMENT
We dedicate this paper to Robert G. Bergman on the occasion of his 70th birthday. We thank Omar DePaolis for experimental support. We thank the Alfred P. Sloan foundation (KLT), LaMattina Fellowship (XS and ADW), NSF (CHE-1150393) and NIGMS (RO1GM087581) for funding of this project. Mass spectrometry instrumentation at Boston College is supported by funding from the NSF (DBI-0619576).
ABBREVIATIONS
- DIPEA
diisopropylethyl amine
- Cy
cyclohexyl
- Cyp
cyclopentyl
- Ph
phenyl
- Bn
benzyl
- t-AmOH
tert-amyl alcohol
- t-BuOH
tert-butanol
- TESCl
triethylsilyl chloride
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental details, compound characterization, equilibrium data, and estimations of site selectivities for (R) and (S) diols from table 2. This information is available free of charge via the Internet at http://pubs.acs.org/.
REFERENCES
- 1.For reviews on synthetic strategy see: Gaich T, Baran PS. J. Org. Chem. 2010;75:4657. doi: 10.1021/jo1006812. Shenvi RA, O'Malley DP, Baran PS. Acc. Chem. Res. 2009;42:530. doi: 10.1021/ar800182r. Tatsuta K, Hosokawa S. Chem. Rev. 2005;105:4707. doi: 10.1021/cr040630+. Harmata M. Strategies and Tactics in Organic Synthesis. Vol. 5. London: Elsevier; 2004. Nicolaou KC, Snyder A. Classics in Total Synthesis II: Targets, Strategies, Methods. Chichester: Wiley; 2003. Corey EJ, Cheng X–M. Logic of Chemical Synthesis. New York: John Wiley & Sons, Inc; 1995.
- 2.For examples of site selective enzymatic reactions see: Thibodeaux CJ, Melancon CE, Liu H. Angew. Chem. Int. Ed. 2008;47:9814. doi: 10.1002/anie.200801204. Thibodeaux CJ, Melancon CE, Liu H. Nature. 2007;446:1008. doi: 10.1038/nature05814. Koeller KM, Wong C-H. Chem. Rev. 2000;100:4465. doi: 10.1021/cr990297n.
- 3.(a) Jordan PA, Miller SJ. Angew. Chem. Int. Ed. 2012;51:2907. doi: 10.1002/anie.201109033. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yoshida K, Furuta T, Kawabata T. Angew. Chem. Int. Ed. 2011;50:4888. doi: 10.1002/anie.201100700. [DOI] [PubMed] [Google Scholar]; (c) Kawabata T, Furuta T. Chem. Lett. 2009;38:640. [Google Scholar]; (d) Ohshima T, Iwasaki T, Maegawa Y, Yoshiyama A, Mashima K. J. Am. Chem. Soc. 2008;130:2944. doi: 10.1021/ja711349r. [DOI] [PubMed] [Google Scholar]; (e) Lewis CA, Miller SJ. Angew. Chem. Int. Ed. 2006;45:5616. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]; (f) Griswold KS, Miller SJ. Tetrahedron. 2003;59:8869. [Google Scholar]
- 4.For reviews on using reversible covalent bonding in catalysis see: Rousseau G, Breit B. Angew. Chem. Int. Ed. 2011;50:2450. doi: 10.1002/anie.201006139. Tan KL. ACS Cat. 2011;1:877.
- 5.For a recent example of a catalyst that uses temporary intramolecularity in enantioselective catalysis see, MacDonald MJ, Schipper DJ, Ng PJ, Moran J, Beauchemin AM. J. Am. Chem. Soc. 2011;133:20100. doi: 10.1021/ja208867g.
- 6.(a) Gouliaras C, Lee D, Chan L, Taylor MS. J. Am. Chem. Soc. 2011;133:13926. doi: 10.1021/ja2062715. [DOI] [PubMed] [Google Scholar]; (b) Lee D, Taylor MS. J. Am. Chem. Soc. 2011;133:3724. doi: 10.1021/ja110332r. [DOI] [PubMed] [Google Scholar]; (c) Chan L, Taylor MS. Org. Lett. 2011;13:3090. doi: 10.1021/ol200990e. [DOI] [PubMed] [Google Scholar]
- 7.For examples of catalyzed silyl transfer see: Rodrigo JM, Zhao Y, Hoveyda AH, Snapper ML. Org. Lett. 2011;13:3778. doi: 10.1021/ol2010819. Grajewksa A, Oestreich M. Synlett. 2010;16:2482. Yan H, Jang HB, Lee JW, Kim HK, Lee SW, Yang JW, Song CE. Angew. Chem. Int. Ed. 2010;49:8915. doi: 10.1002/anie.201004777. You Z, Hoveyda AH, Snapper ML. Angew. Chem. Int. Ed. 2009;48:547. doi: 10.1002/anie.200805338. Yu Z, Mitra AW, Hoveyda AH, Snapper ML. Angew. Chem. Int. Ed. 2007;46:8471. doi: 10.1002/anie.200703650. Zhao Y, Rodrigo J, Hoveyda AH, Snapper ML. Nature. 2006;443:67. doi: 10.1038/nature05102. Isobe T, Fukuda K, Araki Y, Ishikawa T. Chem. Commun. 2001;3:243.
- 8.For a review on Si-O coupling reaction including metal-catalyzed reactions see: Weickgenannt A, Mewald M, Oestreich M. Org. Biomol. Chem. 2010;8:1497–1504. doi: 10.1039/b925722e.
- 9.Sun X, Worthy AD, Tan KL. Angew. Chem. Int. Ed. 2011;50:8167. doi: 10.1002/anie.201103470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.For a review on Curtin Hammett Winstein-Holness kinetics see: Seeman JI. Chem. Rev. 1983;83:83.
- 11.For reviews on divergent kinetic resolutions see: Miller LC, Sarpong R. Chem. Soc. Rev. 2011;40:4550. doi: 10.1039/c1cs15069c. Kumar RR, Kagan HB. Adv. Synth. Catal. 2010;352:231. Vedejs E, Jure M. Angew. Chem. Int. Ed. 2005;44:3974. doi: 10.1002/anie.200460842.
- 12.For an example of a regiodivergent RRM using silyl transfer see ref 7a.
- 13.In this case the formation of constitutional isomers is derived from site selectivity, so a more appropriate term maybe site divergent RRM. Regioselectivity generally refers to differentiating positions within the same functional group; in this case we are differentiating the same type of functional group within the same molecule or siteselectivity.
- 14.Attempts at silylating 3,3-dimethyl-1,2-butanediol (R=t-Bu) results in no protection of the secondary hydroxyl. These results suggest the current catalyst system is unable to overcome the large substrate bias for primary alcohol protection for this substrate.
- 15.Reacting (R)-1a with control catalyst 7 results in modest conversion (60% conv) and low yield (16%) of (R)-2a, suggesting that covalent bonding between 6b and (R)-1a is necessary for efficient catalysis.
- 16.For a review of kinetic resolutions see: Kagan HB, Fiaud JC. Topics in Stereochemistry. Vol. 18. New York: Wiley; 1988. Kinetic Resolution; pp. 249–330.
- 17.For an example of kinetic resolution of 1,2-diols via silylation see ref 7e.
- 18.Kolb HC, Van Nieuwenhze MS, Sharpless KB. Chem. Rev. 1994;94:2483. [Google Scholar]
- 19.(a) White DE, Jacobsen EN. Tetrahedron: Asymmetry. 2003;14:3633. [Google Scholar]; (b) Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Gould AE, Furrow ME, Jacobsen EN. J. Am. Chem. Soc. 2002;124:1307. doi: 10.1021/ja016737l. [DOI] [PubMed] [Google Scholar]; (c) Tokunaga M, Larrow JF, Kakiuchi F, Jacobsen EN. Science. 1997;277:936. doi: 10.1126/science.277.5328.936. [DOI] [PubMed] [Google Scholar]
- 20.(a) Lee Y, Jang H, Hoveyda AH. J. Am. Chem. Soc. 2009;131:18234. doi: 10.1021/ja9089928. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kliman LT, Mlynarski SN, Morken JP. J. Am. Chem. Soc. 2009;131:13210. doi: 10.1021/ja9047762. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Morgan JB, Miller SP, Morken JP. J. Am. Chem. Soc. 2003;125:8702. doi: 10.1021/ja035851w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.