

Abstract
The dynamic kinetic resolution of β-aryl α-keto esters has been accomplished using a newly designed (arene)RuCl(monosulfonamide) transfer hydrogenation catalyst. This dynamic process generates three contiguous stereocenters with remarkable diastereoselectivity through a reduction/lactonization sequence. The resulting enantioenriched, densely functionalized γ-butyrolactones are of high synthetic utility as highlighted by several secondary derivatizations.
The conversion of racemic compounds into enantiomerically enriched products via a dynamic kinetic transformation is a powerful approach to asymmetric synthesis.1 The advantages of this technique are fully realized when the racemic starting materials are trivial to access and a racemization pathway is simple to implement. As such, the asymmetric hydrogenation of configurationally labile α-substituted β-keto esters (Figure 1a) remains the archetypal dynamic kinetic resolution (DKR);2 this process is used to create greater than 100 tons of enantioenriched material each year.3 The success of the reaction relies upon the acidity of the carbon acid that facilitates interconversion of the two enantiomers through the achiral enol. In contrast, the analogous resolution of less acidic α-keto esters has remained essentially undeveloped,4 despite its enormous potential synthetic utility and complementary nature of the products (Figure 1b). Herein we describe the first highly enantioselective dynamic kinetic asymmetric transfer hydrogenation (DKR-ATH) of β-aryl α-keto esters, a transformation that (1) allows direct access to trisubstituted γ-butyrolactones; (2) establishes three contiguous stereocenters with complete diastereocontrol; (3) employs a newly designed ruthenium catalyst; (4) exhibits high catalyst efficiency, and (5) utilizes readily available starting materials.
Figure 1.

Dynamic Kinetic Resolution of Keto Esters
At the reaction design stage, we applied the self-imposed constraint to not only establish a convenient and scalable route to the α-keto ester precursors, but also to strategically incorporate structural elements that would allow us to productively merge a dynamic process with downstream complexity-building events. Specifically, we envisioned a diastereoselective dynamic reduction of α-keto ester 1 to create the α- and β-stereocenters; the incorporation of a diester at the γ-position was projected to facilitate substrate synthesis and also allow a third stereocenter to be established by concomitant lactonization of the nascent hydroxyl group (Figure 1c). The functionality presented in α-keto ester 1 collectively constitutes a retron for a carbene-catalyzed Stetter addition between ethyl glyoxylate and readily available benzylidene malonate derivatives.5 The implementation of the strategy outlined in Figure 1c would provide direct access to synthetically useful enantioenriched γ-butyrolactones, which notably would arise from four commercial reagents (aldehyde, dimethyl malonate, ethyl glyoxylate, and formic acid) and three catalytic steps (Knoevenagel, Stetter, DKR/lactonization). As revealed by Table 1, the new glyoxylate Stetter catalyzed by the Rovis triazolium carbene5c is efficient for a number of substrates and can be performed on a multigram scale. A major challenge associated with stereoselective carbonyl umpolung catalysis, viz., the configurational vulnerability of the stereocenter next to the ketone, is rendered moot here since the racemate is the desired product.
Table 1.
Glyoxylate Stetter Substrate Scopea
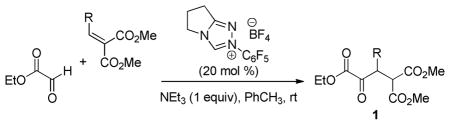 | |||
|---|---|---|---|
| Entry | R | Product | % yieldb |
| 1 | C6H5 | 1a | 96 |
| 2 | 4Cl-C6H5 | 1b | 95 |
| 3 | 4Me-C6H5 | 1c | 92 |
| 4 | 4MeO-C6H5 | 1d | 93 |
| 5 | 4NC-C6H5 | 1e | 90 |
| 6 | 2Me-C6H5 | 1f | 78 |
| 7c | piperonyl | 1g | 91 |
| 8 | 2-furyl | 1h | 87 |
| 9 | 3-N-TsIndole | 1i | 89 |
| 10 | 3-N-BocIndole | 1j | 84 |
| 11 | CH2CHPh | 1k | 93 |
Conditions: Unless otherwise noted, all reactions were performed on a 2.0 mmol scale in PhCH3 (4 mL) at ambient temperature for 16 h.
Isolated yield.
Reaction performed on a 10 mmol scale.
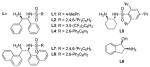
Our point of departure for this study with respect to catalyst development was Noyori’s (arene)RuCl(monosulfonamide-DPEN),6 a catalyst that has tremendous efficacy in both the asymmetric reduction of simple ketones7 and the dynamic reduction of α-substituted β-keto esters and amides.8 We elected to employ formic acid as the organic reductant and triethylamine as the base using 1a as the test substrate. Initial studies identified [RuCl2(p-cymene)]2 as the optimal precatalyst 9 and a number of chiral 1,2-diaminoethane monosulfonamide ligands were screened for selectivity (Table 2). Subjecting 1a to 2 mol % of the ruthenium dimer and Noyori’s ligand L1 (Ru atom: L mole ratio 1:4) in DMF at 70 °C provided the desired γ-butyrolactone in high yield (90%) and diastereoselectivity (>20:1 dr), but with low levels of enantiocontrol (57:43 er, entry 1). A significant increase in the enantioselectivity was observed by modification of the sulfonamide (L2–L4), but desirable levels of selectivity were not obtained (entries 2–4). Employing 1,2-diaminocyclohexane and 1,2-aminoindanol to serve as the chiral backbone (L5 and L6) yielded comparable results (entries 5–6).
Table 2.
Evaluation of Chiral Diamine Ligands
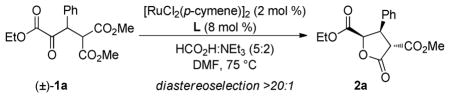 | |||
|---|---|---|---|
| entry | ligand | yield (%) | er |
| 1 | L1 | 90 | 57:43 |
| 2 | L2 | 87 | 70:30 |
| 3 | L3 | 84 | 70:30 |
| 4 | L4 | 84 | 72:28 |
| 5 | L5 | 73 | 73:27 |
| 6 | L6 | 88 | 60:40 |
| 7 | L7 | 82 | 88:12 |
| 8 | L8 | 92 | 94:6 |
 | |||
Conditions: 2a (1.0 equiv), [RuCl2(p-cymene)]2 (0.02 equiv), L (0.08 equiv), [2a]0 = 0.1 M in DMF, 70 °C, 16 h.
Based on these observations, it was clear that a new chiral ligand would need to be identified to achieve high levels of enantiocontrol. The development of the “mother diamine”/diaza-Cope approach to the synthesis of C2-symmetric 1,2-diamines10 allowed facile screening of a number of chiral diamine backbones. We found that the α-naphthyl/triisopropylbenzenesulfonamide ligand L7 considerably increased the selectivity (88:12 er, entry 7). To further optimize the ligand structure, perturbations of the sulfonamide were examined due to its apparent ability to directly impact the chiral environment (Table 2, entry 1 vs. entry 2). A number of diverse sulfonyl chlorides were accessed through a one-pot double alkylation/sulfonylation of 1,3-dichlorobenzene.11 The simplest m-terphenyl sulfonamide variant L8, distinguished itself as being uniquely effective for providing high levels of enantioselectivity for the title reaction (94:6 er, entry 8). The α-naphthyl backbone and m-terphenylsulfonamide operate synergistically; no improvement in enantiocontrol with the DPEN/m-terphenylsulfonamide ligand L4 was observed. The α-naphthyl ethylenediamine backbone has been used sporadically in asymmetric synthesis and the use of the m- terphenylsulfonamide for enantioselective catalysis is rarer still.12
As outlined in Table 3, we have found this DKR-ATH to be broad in scope for a number of β-aryl α-keto esters.13 High yields and enantioselectivities (up to 94% yield and 96.5:3.5 er) were obtained for substrates incorporating electron-rich, electron-poor, and heteroaryl substituents at the β-position. The product lactones exhibit high incidence of crystallinity. The syn, anti-relationship of the trisubstituted γ-butyrolactone products was determined by NOESY experiments and the absolute configuration was established by X-ray crystallographic analysis of 2b.14
Table 3.
DKR-ATH Substrate Scopea
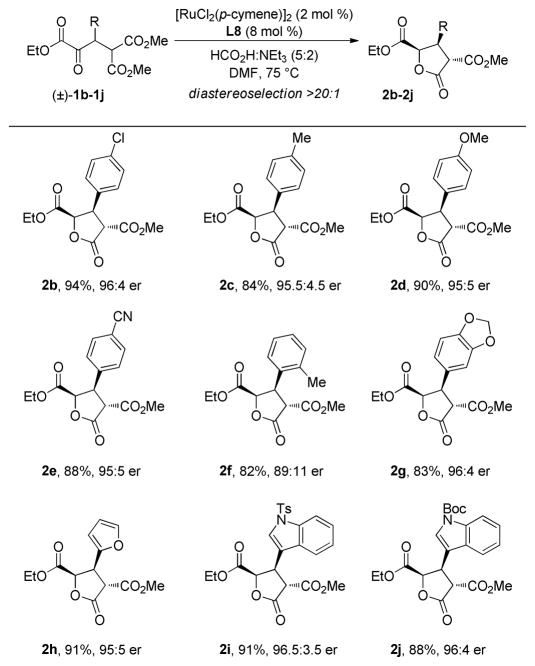 |
Conditions: As described for Table 2.
To demonstrate the synthetic utility and catalytic efficiency of this method, the reaction was performed on multigram scale (Scheme 1). Additional experimentation revealed that the catalyst loading could be lowered to 1 mol % with no loss in reaction efficiency. The reduction was trivially performed on a 10 g scale with 1g yielding lactone 2g (er 95:5); recrystallization yielded enantiomerically pure lactone in 72% yield.
Scheme 1.

Catalyst Efficiency and Scalability
The reduction products present functionality immediately amenable to further manipulation (Scheme 2). Lactone 2a undergoes facile diastereoselective alkylation upon treatment with allyl bromide and DBU, yielding tetrasubstituted lactone 3a bearing an all-carbon quaternary carbon center (94% yield). Krapcho decarboxylation15 afforded α-unsubstituted lactone 3b (86% yield), which is primed to undergo further enolate-based bond constructions. Of particular importance, alkylation with dibromomethane followed by dehalodecarboxylation16 afforded α-alkylidene γ-butryolactone 3c. This substructure is featured in 3% of all known natural products and members of this subclass exhibit a wide range of biological activity.17 Due to the versatility of the product classes obtainable, we expect this method to be of significant value in the areas of both natural product and medicinal agent synthesis.
Scheme 2.

Synthetic Utility of Lactone Products
Conditions: (a) allyl bromide (2 equiv), DBU (2 equiv), THF, rt, 2h, 94%. (b) LiCl, DMSO, 140 °C, 87%. (c) i) K2CO3, dibromomethane, 84%. ii) LiCl, DMSO, 140 °C, 87%.
In summary, we have designed a new asymmetric transfer hydrogenation catalyst that has led to the successful development of the first dynamic kinetic resolution of β-aryl α-keto esters. Under the reaction conditions, spontaneous lactonization of the α-hydroxyl group onto the pendant ester occurs to provide trisubstituted γ-butyrolactones with complete diastereocontrol. With respect to the rubric of green chemistry, it is instructive to inventory the three steps that lead to the lactones 2: (i) the Knoevenagel condensation is amine-catalyzed and generates water as the byproduct; (ii) the glyoxylate Stetter addition is carbene-catalyzed and is 100%-atom economical, generating no stoichiometric by-products; (iii) the DKR-ATH reaction uses a chiral ruthenium catalyst, formic acid as the reductant and generates only CO2 and CH3OH as byproducts. Further studies to understand the origin of selectivity, extension of this method to other β-stereogenic α-keto esters, and identification of other green, dynamic transformations with α-keto esters are ongoing in our laboratory.
Supplementary Material
Acknowledgments
The project described was supported by Award No. R01 GM084927 from the National Institute of General Medical Sciences, Novartis, and Amgen. K.M.S. acknowledges an NIH NRSA Fellowship to Promote Diversity in Health-Related Research.
Footnotes
Supporting Information. Experimental procedures, spectral and HPLC data and crystallographic data (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Noyori R, Tokunaga M, Kitamura M. Bull Chem Soc Jpn. 1995;68:36.Genet JP. Acc Chem Res. 2003;36:908. doi: 10.1021/ar020152u.For recent reviews on dynamic kinetic resolutions see: Pellissier H. Tetrahedron. 2008;64:1563.Pellissier H. Tetrahedron. 2011;67:3769.
- 2.Noyori R, Ikeda T, Ohkuma M, Widhalm M, Kitamura H, Takaya S, Akutagawa S, Sayo N, Saito T. J Am Chem Soc. 1989;111:9134. [Google Scholar]
- 3.Shimizu H, Nagasaki I, Matsumura K, Sayo N, Saito T. Acc Chem Res. 2007;40:1385. doi: 10.1021/ar700101x. [DOI] [PubMed] [Google Scholar]
- 4.For example: Patel RN, Banerjee A, Howell JM, McNamee DB, Mirfakhrae D, Nanduri V, Thottathil JK, Szarka LJ. Tetrahedron: Asymmetry. 1993;4:2069.Steward KM, Johnson JS. Org Lett. 2010;12:2864. doi: 10.1021/ol100996w.
- 5.For examples of the Stetter reaction with glyoxamides see: Liu Q, Perreault S, Rovis T. J Am Chem Soc. 2008;130:14066. doi: 10.1021/ja805680z.Liu Q, Rovis T. Org Lett. 2009;11:2856. doi: 10.1021/ol901081a.Vora HU, Lathrop HP, Reynolds NT, Kerr MS, Read de Alaniz J, Rovis T. Org Synth. 2010;87:350.
- 6.Hashiguchi S, Fujii A, Ikariya T, Noyori R. J Am Chem Soc. 1995;117:7562. [Google Scholar]
- 7.Noyori R, Hashiguchi S. Acc Chem Res. 1997;30:97. [Google Scholar]
- 8.(a) Ros A, Magriz A, Dietrich H, Lassaletta JM, Fernández R. Tetrahedron. 2007;63:6755. [Google Scholar]; (b) Limato J, Krska SW, Dorner BT, Vazquez E, Yoshikawa N, Tan L. Org Lett. 2010;12:512. doi: 10.1021/ol902715d. [DOI] [PubMed] [Google Scholar]; (c) Seashore-Ludlow B, Villo P, Häcker C, Somfai P. Org Lett. 2010;12:5274. doi: 10.1021/ol102323k. [DOI] [PubMed] [Google Scholar]
- 9.See the Supporting Information for details.
- 10.Kim H, Nguyen Y, Yen CPH, Chagal L, Lough AJ, Kim BM, Chin J. J Am Chem Soc. 2008;130:12184. doi: 10.1021/ja803951u. [DOI] [PubMed] [Google Scholar]
- 11.(a) Saednya A, Hart H. Synthesis. 1996:1455. [Google Scholar]; (b) Luning U, Baumgartner H, Manthey C, Meynhardt B. J Org Chem. 1996;61:7922. doi: 10.1021/jo961094r. [DOI] [PubMed] [Google Scholar]
- 12.α-Naphthyl ethylenediamine: Denmark SE, Su X, Nishigaichi Y, Coe DM, Wong KT, Winter SBD, Choi JY. J Org Chem. 1999;64:1958. doi: 10.1021/jo9820723.Denmark SE, Pham SM, Stavenger RA, Su X, Wong KT, Nishigaichi Y. J Org Chem. 2006;71:3904. doi: 10.1021/jo060243v.Basak AK, Shimada N, Bow WF, Vicic DA, Tius MA. J Am Chem Soc. 2010;132:8266. doi: 10.1021/ja103028r.m-Terphenylsulfonamide: Kosugi Y, Akakura M, Ishihara K. Tetrahedron. 2007;63:6191.
- 13.Subjecting ketone 1k to the reaction conditions in Table 3 afforded the corresponding γ-butyrolactone in 67:33 er. Studies are ongoing to optimize the DKR-ATH to include β-alkyl α-keto ester substrates.
- 14.CCDC. 871630 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Centre via. www.ccdc.cam.ac.uk/data_request/cif.
- 15.Kraphcho AP. Synthesis. 1982:893. [Google Scholar]
- 16.Bukowsa M, Kolodziejek W, Prejzner J. Pol J Chem. 1990;64:573. [Google Scholar]
- 17.Kitson RRA, Millemaggi A, Taylor RJK. Angew Chem, Int Ed. 2009;48:9426. doi: 10.1002/anie.200903108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.