

Graphical abstract

Abbreviations: ABC, ATP-binding cassette; BCRP, breast cancer resistance protein; BSA, bovine serum albumin; CDNB, 1-chloro-2,4-dinitrobenzene; CSA, cyclosporin A; DMF, dimethylformamide; DMSO, dimethyl sulfoxide; ESI-MS, electrospray ionization mass spectrometry; GSH, glutathione; GST, glutathione-S-transferase; KP1089, [bis(2-acetylpyridine 4,4-dimethylthiosemicarbazonato-N,N,S)gallium(III)] tetrachloridogallate; KP1550, 2-acetylpyridine 4,4-dimethylthiosemicarbazone; KP1657, [bis(2-acetylpyridine thiosemicarbazonato-N,N,S)gallium(III)] nitrate; KP1719, [bis(3-aminopyridine-2-carboxaldehyde 4,4-dimethylthiosemicarbazonato-N,N,S)gallium(III)] hexafluorophosphate; KP1740, [bis(2-formylpyridine 4,4-dimethylthiosemicarbazonato-N,N,S)gallium(III)] hexafluorophosphate; LRP, lung resistance protein; MDR, multidrug resistance; MRP, multidrug resistance-related protein; PBS, phosphate-buffered saline; P-gp, P-glycoprotein; TD, terminal dimethylation; Triapine, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone
Keywords: Resistance, Thiosemicarbazones, Multidrug resistance, Triapine, ABC transporter, Glutathione
Abstract
Triapine is an α-N-heterocyclic thiosemicarbazone with promising anticancer activity against hematologic malignancies but widely ineffective against solid tumor types in clinical trials. The anticancer activity of thiosemicarbazones can be dramatically increased by terminal dimethylation. KP1089 is a gallium compound containing two terminal dimethylated thiosemicarbazone ligands. To gain insights on the vulnerability of this highly active terminal dimethylated thiosemicarbazone to drug resistance mechanisms, a new cell model with acquired resistance against the lead compound KP1089 was established. Subsequent genomic analyses (arrayCGH and FISH) revealed amplification of the ABCC1 gene on double minute chromosomal DNA in KP1089-resistant cells as well as overexpression of ABCC1 and ABCG2 on the protein level. KP1089 was further confirmed as a substrate of ABCC1 and ABCG2 but not of ABCB1 using a panel of ABC transporter-overexpressing cell models as well as ABC transporter inhibitors. Moreover, glutathione depletion strongly enhanced KP1089 activity, although no glutathione conjugate formation by glutathione-S-transferase was observed. Thus, a co-transport of KP1089 together with glutathione is suggested. Finally, a panel of thiosemicarbazone derivatives was tested on the new KP1089-resistant cell line. Notably, KP1089-resistant cells were not cross-resistant against thiosemicarbazones lacking terminal dimethylation (e.g. Triapine) which are less active than KP1089. This suggests that terminal dimethylation of thiosemicarbazones – linked with distinctly enhanced anticancer activity – leads to altered resistance profiles compared to classical thiosemicarbazones making this compound class of interest for further (pre)clinical evaluation.
1. Introduction
The promising anticancer activity of thiosemicarbazone-based drugs is well known and resulted in the clinical development of the ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (Triapine, Fig. 1). Indeed, several studies already proofed the activity of Triapine against advanced leukemia in clinical phase I trials [1,2]. However, several clinical phase II studies revealed that Triapine is ineffective against a variety of solid tumors including advanced adenocarcinoma of the pancreas [3], non-small-cell lung cancer [4], and renal cell carcinoma [5]. Although an impact of multidrug-resistance proteins (especially of ABCB1) was already suggested [6–8], the reasons for the general inefficacy of Triapine against solid tumors are widely speculative. In general, the mechanisms of drug resistance are numerous and, frequently, resistance is based not only on one mechanism but is found to be multifaceted and complex. Thus, resistance at the cellular level might be, on the one hand, directly associated with the mode of action of the respective anticancer agent (such as changed target expression or target mutation). On the other hand, tumors frequently develop cross-resistance to several, often structurally and mechanistically unrelated drugs, a phenomenon called multidrug resistance (MDR) [9]. One of the most prominent and best investigated mechanisms underlying MDR is the (over)expression of ATP-driven membrane-located export proteins, so called ABC transporters. In humans, 49 members of the ABC transporter family have been described. However, only a few are involved in drug efflux, namely ABCB1 (P-glycoprotein; P-gp), several members of the ABCC subfamily (multidrug-resistance protein; MRP), especially ABCC1 and ABCC2, as well as ABCG2 (breast cancer resistance protein; BCRP). In general, the substrate specificity of MDR-conferring ABC transporters overlaps. Consequently, some compounds are substrates for several ABC transporters (e.g. doxorubicin or etoposide, which are transported by ABCB1, ABCC1 as well as ABCG2 [10]).
Fig. 1.

The structures of KP1089 and other relevant thiosemicarbazones.
To enhance the anticancer activity of Triapine, the development of thiosemicarbazone derivatives with altered pharmacological characteristics is of interest. Additionally, such new derivatives can be used to gain more insights into the structural characteristics underlying the resistance against Triapine. We have recently reported the synthesis of novel gallium complexes containing α-N-heterocyclic thiosemicarbazone ligands [11,12]. These studies revealed that, in the absence of a NH2 group in the molecule, dimethylation of the terminal nitrogen strongly enhanced the cytotoxicity of α-N-heterocyclic thiosemicarbazones and their metal-containing complexes (e.g. the gallium complex KP1089) by about 1000-fold. Moreover, the increased cytotoxicity was found to be only partly dependent on the ribonucleotide reductase inhibitory ability of these new compounds implying that other yet unknown mechanisms are involved [12]. To gain more insights into the mode of action as well as to evaluate the vulnerability of terminal dimethylated α-N-heterocyclic thiosemicarbazones to drug resistance mechanisms, a new cell model with acquired resistance against the model substance KP1089 was established (Fig. 1). Our studies revealed that, in contrast to Triapine and other thiosemicarbazones which lack terminal dimethylation (TD), resistance against KP1089 (and other terminal dimethylated thiosemicarbazones) is mediated by the GSH conjugate transporter ABCC1 and the drug transporter ABCG2.
2. Materials and methods
2.1. Drugs
[Bis(2-acetylpyridine 4,4-dimethylthiosemicarbazonato-N,N,S)gallium(III)] tetrachloridogallat (KP1089), 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (Triapine), 2-acetylpyridine 4,4-dimethylthiosemicarbazone (KP1550), [bis(2-acetylpyridine thiosemicarbazonato-N,N,S)gallium(III)] nitrat (KP1657), [bis(3-aminopyridine-2-carboxaldehyde 4,4-dimethylthiosemicarbazonato-N,N,S)gallium(III)] hexafluorophosphate (KP1719), [bis(2-formylpyridine 4,4-dimethylthiosemicarbazonato-N,N,S)gallium(III)] hexafluorophosphate (KP1740), were prepared as described previously [11–13]. The compounds were dissolved in DMSO and diluted into the culture media at the concentrations indicated (DMSO concentrations were always below 0.1%). Verapamil was purchased from Abbott (Vienna, Austria), cyclosporin A from Sandoz (Basel, Switzerland), BCNU from Bristol-Myers Squibb (Munich, Germany), paclitaxel from Rhone-Poulenc RORER (Essex, GB), gallium nitrate octahydrate from Merck (Darmstadt, Germany). All other substances were purchased from Sigma–Aldrich (Vienna, Austria). All solutions were freshly prepared before usage.
2.2. Cell culture
The following human cell lines and their chemoresistant sublines were used in this study [14,15]: the epidermal carcinoma-derived cell line KB-3-1 and its ABCB1-overexpressing subline KBC-1 (generously donated by D.W. Shen, Bethesda, USA) [16]; the promyelocytic leukemia cell line HL60 and its ABCC1-overexpressing subline HL60/adr (from M. Center, Kansas State University, USA), the small cell lung carcinoma cell line GLC-4 and its ABCC1- and LRP-overexpressing subline GLC-4/adr (from E.G. deVries, Groningen, The Netherlands); the breast adenocarcinoma cell line MDA-MB-231 with its respective ABCG2(R482T)-transfected subclone MDA-MB-231/bcrp (from D. Ross, University of Maryland, Greenbaum Cancer Centre, USA), the non-small cell lung carcinoma cell line SW1573 with its ABCC1- and LRP-overexpressing subline 2R120 as well as its ABCB1- and ABCC1-overexpressing subline 2R160 (H. Broxterman, Department of Medical Oncology, Free University Hospital, Amsterdam, The Netherlands). The ABC transporter expression levels of the used cell lines were checked by Western blotting (Fig. S1). All cell lines were grown in RPMI 1640 supplemented with 10% fetal bovine serum with the exception of SW1573 cells, which were grown in DMEM with 10% serum. Cultures were regularly checked for Mycoplasma contamination.
2.3. Cytotoxicity assays
Cells were plated (2 × 103 cells/well for KB, MDA-MB-231, SW1573 cells; 5 × 103 cells/well for HL60 cells, and 4 × 103 cells/well for GLC-4, 2R120, and 2R160 cells) in 100 μl per well in 96-well plates. After a recovery period of 24 h, drugs were added in another 100 μl growth medium and cells were exposed for 72 h. The proportion of viable cells was determined by MTT assay following the manufacturer's recommendations (EZ4U, Biomedica, Vienna, Austria). Cytotoxicity was calculated using the Graph Pad Prism software (La Jolla, USA) (using a point-to-point function) and was expressed as IC50 values calculated from full dose-response curves (drug concentrations inducing a 50% reduction of cell number in comparison to untreated control cells cultured in parallel).
2.4. Western blot analyses
Cell fractionation, protein separation and Western blotting were performed as described [17]. The following antibodies were used: anti-P-gp monoclonal mouse C219 (Signet, Dedham, USA), dilution: 1:100; anti-LRP monoclonal mouse clone 42 (Transduction Lab., Lexington, USA), 1:1000; anti-BCRP monoclonal mouse MAB4146 (Chemicon, Temicola, USA), 1:500, anti-MRP1 monoclonal rat MRPr1 (Sanbio, Uden, The Netherlands), 1:40; anti-MRP2 monoclonal mouse C250 (Alexis Corp., Lausen, Switzerland), 1:50; anti-MRP3 monoclonal mouse M3II-9 (Alexis Corp., Lausen, Switzerland), 1:40. All secondary, peroxidase-labeled antibodies from Szabo-Scandic were used at working dilutions of 1:10,000.
2.5. Cytogenetic analyses
Genomic DNA was isolated using the QIAamp DNA Blood Mini Kit (Qiagen GmbH, Austria) following the manufacturer's instructions. Comparative genomic hybridization (CGH), fluorescence in situ hybridization (FISH) were performed as described previously [18,19]. For DNA amplification, linker–adapter PCR was used as described [18]. For the detection of the MRP1 locus the BAC clone CTD-2504F3 supplied by Sanger Institute (Hinxton, Cambridge, UK) was used.
2.6. ArrayCGH (aCGH) analyses
aCGH was performed using 4 × 44 K human whole genome oligonucleotide-based arrays (Agilent Technologies Österreich GmbH, Austria) as published [20]. Labeling and hybridization procedures were done according to the instructions provided by Agilent. Briefly, 500 ng of tumor DNA and reference DNA (human male genomic DNA, Promega Corporation, Madison, USA) were digested with AluI and RsaI (both from Promega), then differentially labeled by random priming with cyanine 5- and cyanine 3-dUTP (Perkin-Elmer, MA, USA), respectively, using the BioPrime Array CGH Genomic Labeling Kit (Life Technologies Corporation, Invitrogen, Paisley, UK). After purification with Amicon Ultra Centrifugal Filters (MILLIPORE GmbH, Vienna, Austria) the two labeled products together with blocking agent, Hybridization Buffer (both included in the Oligo aCGH/Chip-on-Chip Hybridization Kit, Agilent Technologies), and human cot-DNA (Roche Austria GmbH, Vienna, Austria) were combined and hybridized onto 4 × 44 K oligonucleotide arrays. Hybridization was carried out for 48 h at 65 °C in a hybridization oven. Afterwards, slides were washed according to the protocol and scanned with a G2505B Micro Array Scanner (Agilent Technologies). Feature extraction and data analyses were carried out using the Feature Extraction (version 10.7.3.1) and DNA Analytics software (version 4.0.81), respectively.
2.7. Binding of KP1550 to GSH
A solution containing 1-chloro-2,4-dinitrobenzene (CDNB; 100 μM in DMF) or KP1550 (100 μM in DMF) and GSH (100 μM in H2O) was incubated with 1.7 units glutathione-S-transferase (GST from equine liver; 1 unit conjugates 1.0 μmol of CDNB with GSH per min) in NaHCO3 buffer pH 6.5 for 1 h at 37 °C. The samples were centrifuged using Amicon Ultra Centrifugal Filters with a 10 K membrane to separate the protein and the solutions were measured by electrospray ionization mass spectrometry (ESI-MS; Bruker esquire3000 ion trap mass spectrometer).
2.8. Quantification of the intracellular GSH levels
Glutathione (≥98%) and glutathione–glycine–13C2,15N (GSH IS) standards were purchased from Sigma Aldrich, Vienna, Austria. Stock solutions containing 1 g/L GSH and the working solutions were prepared daily in ultrapure water. Cell extracts were diluted 1:100 prior to measurement. For LC–MS measurements a Capillary Pump 1100 series, an m-wellplate sampler and a column oven from Agilent Technologies were used. For separation a 150 mm × 2.1 mm ZIC-HILIC column (3.5 μm particle size) equipped with a 20 mm × 2.1 mm ZIC-HILIC guard column (5 μm particle size) from Merck, Darmstadt, Germany was used.
LC–MS conditions were as follows: flow rate: 100 μL/min; injection volume: 5 μL, column temperature: 45 °C. For the gradient eluent A (98% (v/v) water, 1% (v/v) ACN, 1% (v/v) formic acid) and eluent B (98% (v/v) ACN, 1% (v/v) water, 1% (v/v) formic acid) were used according to the following timetable: 60% B for 3 min, followed by a reduction of B to 10% within 5 min, reconstitution of the starting conditions (60% B) within 1 min and re-equilibration at the starting conditions for 10 min. Total analysis time: 19 min. For MS ion trap detection an Agilent 6430 Ion Trap together with an ESI source from Agilent Technologies was used. Source parameters were as follows: positive ionization mode, MS–MS mode (drying gas temperature 350 °C, drying gas flow 10 L/min, nebulizer pressure 25 psi and capillary voltage 4000 V.
2.9. Calcein-AM efflux assays
Cells were trypsinised and incubated as single cell suspensions (3 × 105 cells/sample) in serum-containing medium with the known ABCC1 inhibitor cyclosporin A (1 μM) for 30 min at 37 °C. Then the fluorescent ABCC1 substrate calcein-AM (0.25 μM) was added and the samples were incubated for another 30 min. Subsequently, cells were analyzed by flow cytometry with an excitation of 494 nm and an emission of 517 nm bandpass filter.
2.10. Pheophorbide A (PhA) efflux assays
Cells were trypsinised and incubated as single cell suspensions (3 × 105 cells/sample) in serum-containing medium with the known ABCG2 inhibitor fumitremorgin C (10 μM) for 30 min at 37 °C. Then the chlorophyll catabolite PhA (10 μM) was added and the samples were incubated for another hour. Cells were washed in cold medium and analyzed by flow cytometry with a 488 nm argon laser and a 585 nm bandpass filter [21].
3. Results
3.1. Selection of a KP1089-resistant cell model and characterization of the cross-resistance profile
The KP1089-resistant KB cell line was generated by continuous exposure of KB-3-1 cells to increasing concentrations of KP1089 (starting point 0.25 nM; end point 50 nM) over a period of one year. The gallium compound was administered to KB-3-1 cells twice a week at the day after passage, when cells had attached to the culture flasks. KP1089-resistant cells were termed KB-1089(50 nM) and displayed a more than 20-fold resistance to the selection drug (Table 1, Fig. 2A) as compared to the parental cell line. As shown in Table 1, KB-1089(50 nM) cells were distinctly cross-resistant to several (natural) chemotherapeutics including vincristine (16-fold), anthracyclines (≫6-fold), as well as etoposide (4.6-fold). No cross-resistance to taxol, the alkylating agents BCNU and temozolomide, or to platinum drugs was observed. Moreover, KB-1089(50 nM) cells were collaterally sensitive to the topoisomerase I inhibitor camptothecin (0.6-fold) as well as to the ribonucleotide reductase inhibitors hydroxyurea (0.5-fold), and gallium nitrate (0.6-fold). In addition, the KP1089-resistant cell line was found to be especially sensitive (≪0.1-fold) to depletion of the intracellular GSH levels by the gamma-glutamylcysteine synthetase inhibitor buthionine sulfoximine (BSO). In contrast, no difference in sensitivity to the glutathione-S-transferase inhibitor ethacrynic acid was observed. With regard to other thiosemicarbazones (Table 2), the resistance of KB-1089(50 nM) cells was independent from the presence of a gallium center in the molecular structure. However, TD was found to be essential for the resistance as KB-1089(50 nM) cells were distinctly cross-resistant against all thiosemicarbazone derivatives, which are terminal dimethylated (KP1550, KP1740 and KP1719), while no cross-resistance against thiosemicarbazones lacking TD (KP1657 and Triapine) was found.
Table 1.
Cross-resistance pattern of KB-1089(50 nM) cells: cancer chemotherapeutics and GSH-modifying drugs. The significance of resistance in comparison to the parental line has been calculated by Student's t-test using Graph Pad Prism Software.
| Drug | KB-3-1 |
KB-1089(50 nM) |
|||
|---|---|---|---|---|---|
| IC50 | ±SD | IC50 | ±SD | Relative resistance | |
| KP1089 (nM) | 4.9 | ±0.8 | ≫100 | – | ≫20-fold* |
| Vincristine (nM) | 1.1 | ±0.6 | 17.85 | ±0.2 | 16-fold* |
| Adriamycin (nM) | 36.8 | ±0.9 | ≫250 | – | ≫7-fold* |
| Daunomycin (μM) | 24.0 | ±1.2 | ≫150 | – | ≫6-fold* |
| Etoposide (nM) | 0.5 | ±0.01 | 2.3 | ±1.2 | 4.6-fold |
| Taxol (nM) | 4.8 | ±0.03 | 4.9 | ±0.06 | 1-fold |
| BCNU (μM) | 9.7 | ±2.0 | 13.3 | ±4.0 | 1-fold |
| Temozolomide (mM) | 0.45 | ±0.06 | 0.64 | ±1.3 | 1-fold |
| Cisplatin (μM) | 1.9 | ±0.7 | 1.5 | ±1.7 | 1-fold |
| Oxaliplatin (μM) | 1.1 | ±0.4 | 1.0 | ±0.2 | 1-fold |
| Ethacrynic acid (μM) | 24.3 | ±1.3 | 22.3 | ±1.7 | 1-fold |
| Ga(NO3)3 (μM) | 343.7 | ±12.0 | 205.2 | ±12.5 | 0.6-fold* |
| Camptothecin (nM) | 226.4 | ±1.1 | 137.1 | ±19.1 | 0.6-fold** |
| Hydroxyurea (mM) | 1.67 | ±0.07 | 0.81 | ±0.03 | 0.5-fold* |
| BSO (μM) | ≫50 | – | 3.5 | ±0.4 | <0.1-fold* |
p < 0.05.
p < 0.01.
***p < 0.001.
Fig. 2.

KP1089 resistance and genomic characterization of KB-1089(50 nM) cells. (A) Anticancer activity of KP1089 was tested in KB-3-1 and KB-1089(50 nM) cells by MTT assay after 72 h drug incubation. Values given are means ± SD of three experiments performed in triplicates. (B) Comparative genome hybridization (CGH) profiles of chromosomes 16. (C) Array CGH results for total chromosome 16 of KB-1089(50 nM) (gray) in comparison to KB-3-1 (black) cells are shown in the left panel. The middle panel gives the gene view of the 16p region specifically amplified in KB-1089 cells. Each dot indicates the position of one oligonucleotide and gray boxes the positions of genes within this region. The right panel gives the quantification of those oligonucleotide probes residing in the highly amplified region. All values are the log 2 ratios of means as compared to normal control DNA.
Table 2.
Cross-resistance pattern of KB-1089(50 nM) cells: thiosemicarbazones. The significance of resistance in comparison to the parental line has been calculated by Student's t-test using Graph Pad Prism Software.
| Formula of the ligand | Drug (nM) | KB-3-1 |
KB-1089 |
|||
|---|---|---|---|---|---|---|
| IC50 | ±SD | IC50 | ±SD | Relative resistance | ||
| KP1089 Ga-complex |
4.9 | ±0.8 | ≫100 | – | ≫20-fold* | |
| KP1550 metal-free ligand |
26.7 | ±2.6 | ≫100 | – | ≫3.7-fold* | |
| KP1740 Ga-complex |
360 | ±25 | ≫2500 | – | 7-fold* | |
| KP1657 Ga-complex |
5300 | ±60 | 6700 | ±20 | 1-fold | |
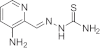 |
Triapine metal-free ligand |
2800 | ±20 | 2300 | ±100 | 1-fold |
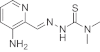 |
KP1719 metal-free ligand |
390 | ±13 | ≫2500 | – | 6.4-fold* |
p < 0.001.
3.2. Genomic characterization of KB-1089(50 nM) cells
To gain more insights into the cytogenetic alterations induced by selection with KP1089, CGH experiments were performed. In accordance to published data [22], KB-3-1 harbored multiple gains and losses such as a loss of 4q, a gain in 1q12-21, and a very prominent gain of 5p. Notably, KB-1089(50 nM) cells had additional changes only in 2 chromosomes compared to KB-3-1 cells: loss of the complete chromosome 10 (data not shown) and strong rearrangements of chromosome 16 (Fig. 2B). These changes were exactly confirmed by indirect CGH hybridization of a 1:1 mix of KB-1089(50 nM) and KB-3-1 DNA on normal metaphase chromosomes (data not shown). To investigate the changes with higher resolution, array CGH (aCGH) analyses were performed. In accordance to the CGH data, only a few and very distinct alterations were observed in KB-1089(50 nM) as compared to KB-3-1 cells. A reduced copy number of almost the whole chromosome 10 in KB-1089(50 nM) cells with the exception of the very terminal telomeric p-arm region was confirmed (data not shown). With regard to chromosome 16, the aCGH analysis revealed that one copy of the complete q-arm was lost in the KP1089-resistant cell line. Moreover, the very strong and distinct amplicon was verified in the p-arm (Fig. 2C). The amplicon consisted of two parts concerning the copy number alterations (log two values are given in the right panel of Fig. 2C), one distal part with an about 16-fold increase in the copy numbers of the MPV17L and parts of the NDE1 gene and one high level amplified part (up to >100-fold) harboring the MYH11, ABCC1, and ABCC6 genes (Fig. 2C). To confirm this amplicon in the KB-1089(50 nM) model, FISH studies were performed on metaphase preparations of parental and resistant cells. Parental KB-3-1 cells displayed an unstable hyperdiploid to triploid karyotype with the ABCC1 BAC probe localizing two to four copies of the ABCC1 gene. Three of these copies localized to the respective region at chromosomes 16 while the occasionally occurring additional copy was part of a marker chromosome containing only a small amount of material from chromosome 16 (Fig. 3A). In the KP1089-resistant subline multiple small DNA particles were observed by DAPI staining indicative for double minute chromosomes. FISH analysis demonstrated that the extrachromosomal DNA contained amplified sequences of ABCC1 as evidenced by colocalization with the ABCC1 BAC probe (Fig. 3A). In accordance to these data, enhanced ABCC1 expression was also detected on protein level as determined by Western blot analyses (Fig. 3B). Furthermore, selection with KP1089 was found to induce the expression of ABCC2 and ABCG2 (probably via epigenetic mechanisms), while drug selection had no effects on ABCB1 and ABCC5 levels (data not shown).
Fig. 3.

ABCC1 amplification and overexpression induced by KP1089 selection. (A) Cytogenetic preparations of KB-3-1 and KB-1089(50 nM) cells were subjected to FISH analysis with whole chromosome 16 paint (red) and an ABCC1 BAC probe (green). In the lower panel interphase nuclei of KB-1089(50 nM) cells are shown. DNA was counterstained with DAPI. Expression of ABCC1 (B), ABCB1, ABCC2, and ABCG2 (C) in membrane-enriched fractions of the indicated cell lines was determined by Western blotting. Ponceau-staining proofing equal lane loading is shown in the Supplementary material (Fig. S1). One of the two immunoblots delivering comparable results is shown representatively.
3.3. Identification of KP1089 as substrate of ABCC1 and ABCG2 but not of ABCB1
As selection with KP1089 led to strong amplification of the ABCC1 gene and induced expression of ABCC1, ABCC2, and ABCG2, it was investigated whether overexpression of these transporter proteins is associated with resistance to KP1089. To this end, the anticancer activity of KP1089 was tested in several cell lines, which are characterized by specific ABC transporter (over)expression, in comparison to their ABC transporter-negative chemosensitive parental lines. As shown in Table 3, ABCC1 expression was associated with significantly increased IC50 values of the respective resistance cell models GLC-4/adr (5.3-fold), HL60/adr (5.8-fold), SW1573/2R120 (≫17.8-fold; high level ABCC1 expression), and SW1573/2R160 (2.5-fold; low level ABCC1 expression). Also ABCG2-transfected MDA-MB-231/bcrp cells displayed 3.8-fold resistance in comparison to the vector control-transfected MDA-MB-231/vc cells. With regard to ABCB1, no significant impact was found on the anticancer activity of KP1089. To confirm that the resistance of the KP1089-selected cell line is dependent on the ABCC1 and ABCG2 transporter activity, the known ABCC1 inhibitor cyclosporine A (CSA) and the ABCG2 modulator fumitremorgin C (FTC) [17,23,24] were used. As shown in Fig. 4A, inhibition of ABCC1 and ABCG2 distinctly sensitized KB-1089(50 nM) cells to the anticancer activity of KP1089. Furthermore, combination of CSA with FTC led to complete re-sensitation of KB-1089(50 nM) cells. In cells specifically overexpressing ABCC1 (an experiment using HL60/adr cells is shown as an example in Fig. 4B), ABCC1 inhibition was already sufficient for full reversal of the KP1089 resistance. In accordance with these results, KB-1089(50 nM) cells displayed reduced accumulation of the fluorescent ABCC1 substrate Calcein-AM and the ABCG2 substrate PhA, which could be significantly increased by coincubation with the inhibitors CSA and FTC (Fig. 4C).
Table 3.
Impact of ABC-transporter-mediated drug resistance mechanisms on the activity of KP1089. The significance of resistance in comparison to the parental line has been calculated by Student's t-test using Graph Pad Prism Software.
| Cell line | IC50 KP1089 (nM) | ±SD | Relative resistance | Resistance mechanism |
|---|---|---|---|---|
| GLC-4 | 3.2 | ±1.2 | – | Parental |
| GLC-4/adr | 16.9 | ±0.8 | 5.3-fold* | ABCC1, MVP |
| HL60 | 0.6 | ±0.06 | – | Parental |
| HL60/adr | 3.5 | ±1.0 | 5.8-fold** | ABCC1, ABCC5 |
| SW1573 | 2.8 | ±0.7 | – | Parental |
| SW1573/2R120 | ≫50 | ±0.9 | ≫17.8-fold* | ABCC1 |
| SW1573/2R160 | 7.0 | ±1.0 | 2.5-fold** | ABCB1, ABCC1 |
| MDA-MB-231/vc | 2.0 | ±1.0 | – | Parental |
| MDA-MB-231/bcrp | 7.5 | ±0.9 | 3.8-fold** | ABCG2 |
| KB-3-1 | 4.6 | ±1.8 | – | Parental |
| KBC-1 | 3.9 | ±0.8 | 0.8-fold | ABCB1 |
p < 0.05.
p < 0.01.
***p < 0.001.
Fig. 4.

Impact of MDR modulators on the cytotoxic potential of KP1089. (A) KB-1089(50 nM) and KB-3-1 cells were treated with KP1089 in combination with the ABCC1 inhibitor CSA (1 μM), the ABCG2 modulator FTC (10 μM) or a combination of CSA and FTC. Significance of difference to the respective KP1089-treated groups was calculated by 2-way-ANOVA (with Bonferroni post correction) using Graph Pad Prism Software. (B) HL60/adr, and HL60 cells were incubated with KP1089 and co-administered with the ABCC1 inhibitors verapamil (10 μM) and CSA (1 μM). After 72 h drug treatment vitality was determined by MTT assay. Values given are means ± SD of two experiments performed in triplicate. Significance of difference to the respective KP1089-treated groups was calculated by 2-way-ANOVA (with Bonferroni post correction) using Graph Pad Prism Software. (C) The ABCC1 and ABCG2 transport activity in KB-1089(50 nM) cells was determined by Calcein-AM and PhA assays, respectively. CSA (1 μM) and FTC (10 μM) were used as respective inhibitors. *Significant difference from untreated KB-1089(50 nM) cells is p < 0.05 calculated by Student's t-test using Graph Pad Prism Software.
3.4. Role of GSH in the resistance against KP1089
Drug transport of the ABCC family members is known to be GSH-dependent [25]. As shown in Fig. 5A, KB-1089(50 nM) cells were characterized by distinctly lower basal GSH levels (2.6-fold) in comparison to parental KB-3-1 cells. In both cell lines incubation with BSO led to distinct depletion of the intracellular GSH pools [in case of KB-1089(50 nM) even below the limit of detection (LOD)]. To assess the importance of the intracellular GSH pools for the resistance of KB-1089(50 nM) cells, the impact of GSH depletion on the activity of KP1089 was tested in 72 h vitality assays. Reduction of GSH led to distinct re-sensitation of KB-1089(50 nM) as well as HL60/adr cells to the anticancer activity of KP1089 (Fig. 5B and C). Notably, BSO treatment also significantly sensitized chemosensitive KB-3-1 cells (Fig. 5D), which lack high ABC transporter expression (compare Fig. 3B), against KP1089. This is in contrast to the known ABCC1 substrate vinblastine where BSO treatment sensitized KB-1089(50 nM) cells, only, but had no effect in KB-3-1 cells (Fig. 5E), corresponding to reports on other ABCC1-overexpressing cell models [26,27]. This indicates that, in contrast to vinblastine, the GSH pools are not only relevant for KP1089 transport by ABCC1 but might ABC transporter-independently regulate the KP1089 responsiveness of cancer cells.
Fig. 5.

The role of GSH in the anticancer activity of KP1089. (A) Intracellular GSH levels of KB-3-1 and KB-1089 were detected by LC–MS in cytosolic lysates of untreated cells and after 20 h exposure to BSO (25 μM) as described in the Material and Methods Section. (B) HL60/adr, KB-1089(50 nM) (C), and KB-3-1 cells were treated with KP1089 after 20 h preincubation with the gamma-glutamylcysteine synthetase inhibitor BSO. The BSO-induced increase in sensitivity (-fold change) to KP1089 at the IC50 concentrations is given in the brackets Values given are means ± SD of two experiments performed in triplicate. Significance of difference to the respective KP1089-treated groups was calculated by 2-way-ANOVA (with Bonferroni post correction) using Graph Pad Prism Software. (D) Impact of GSH depletion on the anticancer activity of vincristine against KB-1089(50 nM) and KB-3-1 cells was determined as described above. Significance of difference to the respective vincristine-treated groups was calculated by 2-way-ANOVA (with Bonferroni post correction) using Graph Pad Prism Software.
A possible conjugation of the KP1089 ligand with GSH in the presence of GST was investigated using electrospray ionization mass spectrometry (ESI-MS). Basically, the conjugation can take place via a GST-catalyzed oxidation of the thiosemicarbazone sulfur atom with a GSH molecule leading to the formation of a disulfide bond. CDNB was used as a positive control for GSH conjugation by GST [28]. After incubation of CDNB with GSH and GST the ESI-MS spectra in the negative ion mode showed a distinct peak at 472 m/z according to the conjugation of CDNB with GSH. In contrast, no KP1550-GSH conjugate (peak would be around 526 m/z) was observable in these experiments (data not shown). This suggests that GST is not able to catalyze the oxidative conjugation of KP1089 and GSH, which is in accordance to the lack of impact of the GST inhibitor EA on the anticancer activity of KP1089 (Fig. S2).
4. Discussion
Thiosemicarbazones are known for their promising anticancer activity, which resulted in clinical studies of Triapine against cancer [1,2]. However, despite promising activity against leukemia [1,2], Triapine has proven ineffective against a variety of solid malignancies [3–5]. Although an impact of multidrug-resistance proteins (especially ABCB1) was suggested [6–8], the reasons for this inactivity are widely unknown. Moreover, the impact of intrinsic and acquired drug resistance on α-N-heterocyclic thiosemicarbazones in general is so far rather unexplored and in case of the highly cytotoxic terminal dimethylated derivatives completely missing.
In this study, the new gallium thiosemicarbazone complex KP1089 was identified as substrate for ABCC1 and ABCG2 but not for ABCB1. Moreover, we discovered that the ABCC1- and ABCG2-mediated resistance is based on TD of the thiosemicarbazone structure. Thus, no cross-resistance of KP1089-selected cells against thiosemicarbazones lacking TD was observed. This is especially of interest, as terminal dimethylated thiosemicarbazones exhibit an about 1000-fold increased anticancer activity in comparison to thiosemicarbazones without TD [11,12]. So far, the mechanisms underlying this enhanced activity are widely unknown and the matter of ongoing evaluations. Such, KP1089 has been recently tested against a panel of 60 cell lines as part of the in vitro anticancer-screening services provided by the National Cancer Institute (NCI) and, subsequently, has been selected by the NCI for further in vivo evaluation due to its interesting activity profile which differs from other thiosemicarbazones (unpublished results). Also the here presented impact of ABCC1 indicates that KP1089 distinctly differs from other thiosemicarbazones lacking TD such as KP1657 or Triapine.
In general, the influence of ABCC1 on KP1089 in contrast to thiosemicarbazones without TD is rather unexpected and difficult to explain as the physico-chemical properties of these molecules are expected to be widely similar [12,29]. A possible explanation might be enhanced lipophilicity of terminal dimethylated thiosemicarbazones in comparison to compounds without TD as it has been shown in case of dipyridylketone thiosemicarbazones [30]. Enhanced lipophilicity might be associated with stronger affinity to hydrophobic binding sites in the ABCC1 molecule [31] and, thus, more effective drug export. Another explanation might be a difference in the metabolization of terminal dimethylated thiosemicarbazones. ABCC subfamily members mainly transport organic anions after their conjugation to hydrophilic ligands such as GSH, glucoronic acid, or sulfate [25] and, such, are essential in the excretion of phase II detoxification products [32]. Consequently, ABCC1 might not transport KP1089 directly but one of its metabolites. The transport of KP1089 conjugates would be comparable to observations with other metal drugs (e.g. cisplatin, arsenic trioxide), where transport by ABCC family members was shown for their GSH conjugates only [9]. Also in case of KP1089, the intracellular GSH pools turned out to be important. Such, depletion of the intracellular GSH levels sensitized cells against KP1089 in ABCC1-overexpressing cell models (but also to a lesser extent in chemo-sensitive parental cells) and KB-1089(50 nM) cells were per se very sensitive to GSH depletion. This might be explained by the lower intracellular GSH pools of KB-1089(50 nM) cells in comparison to KB-3-1 cells, which has been also reported for other ABCC1-overexpressing cell models such as GLC-4/adr [21] or HL60/adr cells [33]. In addition, collateral sensitivity to BSO treatment was shown for other ABCC1-overexpressing cell lines [34]. This further supports the assumption that the ABCC1-mediated resistance against KP1089 is GSH-dependent. However, in cell-free settings no GSH-KP1089 conjugate formation was observed (even in the presence of GST) and, accordingly, a GST inhibitor did not re-sensitize KB-1089(50 nM) cells to KP1089 treatment. Consequently, a possible GSH conjugation to KP1089 has to be mediated by other mechanisms. Alternatively, GSH conjugation is not always necessary for ABCC-mediated drug transport. Lipophilic compounds can be co-transported together with unbound GSH as it has been reported for vincristine or doxorubicin [25,31]. To gain more insights into the interaction of KP1089 with GSH further studies are of interest and mass spectrometry-based assays are currently established to monitor the intracellular metabolism of KP1089 in resistant and chemosensitive cancer cells.
With regard to other thiosemicarbazones, the knowledge on resistance development is, as already mentioned above, rather sparse. So far, only one murine L1210 cell model with acquired resistance to 4-methyl-5-amino-1-formylisoquinoline thiosemicarbazone (MAIQ; which has no TD; Fig. 1) has been reported [6,35]. This cell line, which was cross-resistant to Triapine as well as etoposide, daunomycin, and vinblastine, was shown to harbor several resistance-mediating mechanisms including (1) enhanced activity of the thiosemicarbazone target ribonucleotide reductase, (2) multiple metabolic alterations such as overexpression of disulfide isomerases, and (3) overexpression of the mdr1, mdr3 as well as the mrp gene [6,35]. Furthermore, Rappa et al. reported resistance of mdr1- as well as mrp-transfected murine cancer cells against Triapine [6]. Also in clinical trials an impact of the patients’ mdr genotype on the clinical outcome was suggested [7,8]. The observation of ABCC1-mediated resistance by Rappa et al. [6] is of interest, as we did not observe any cross-resistance of KB-1089(50 nM) cells against Triapine, which suggests that the activity of this thiosemicarbazone is not affected by ABCC1 and ABCG2 expression. With respect to ABCB1, it is noteworthy that in course of a bioinformatic evaluation recently performed on the NCI screening dataset several thiosemicarbazones came in the focus of interest because of their enhanced activity in ABCB1-positive background [36–38]. Detailed structure-activity-studies on isatin-β-thiosemicarbazones further revealed that these ABCB1-targeting properties are most prominent in derivatives with a para-substituted terminal 4′-phenyl moiety like 1-isatin-4-(4′-methoxyphenyl)-3-thiosemicarbazone (NSC73306; Fig. 1) [37]. In addition to ABCB1, NSC73306 was shown to modulate the activity of ABCG2 (although this transporter did not protect cells from NSC73306 activity) but not the activity of ABCC1, ABCC4, and ABCC5 [39].
Overall, our data indicate that in case of thiosemicarbazones small structural modifications can have very pronounced effects on the resistance profile as well as the interaction of this compound class with ABC transporters. Consequently, a more detailed investigation of thiosemicarbazones with respect to ABC transporters might be not only of interest for the clinical usability of these compounds but also for the improvement of the understanding of ABC transporter-mediated drug resistance in general.
In conclusion, this study represents the first report on the pronounced impact of TD on the resistance profile of α-N-heterocyclic thiosemicarbazones and their gallium complexes. Moreover, we were able to demonstrate that, although terminal dimethylated thiosemicarbazones are substrates for ABCC1 and ABCG2, these compounds are still more active in cells harboring these efflux pumps than thiosemicarbazones lacking TD. This makes the terminal dimethylated thiosemicarbazones of interest for further (pre)clinical evaluation.
Acknowledgments
We are indebted to Vera Bachinger for the skilful handling of cell culture, Christian Balcarek for competent technical assistance and Samuel Meier for measurement of mass spectra. This work was supported by the Austrian Science Fond (FWF) grants L212-B11 and P22072-B11 (to W. Berger).
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bcp.2012.03.004.
Appendix A. Supplementary data
References
- 1.Giles F.J., Fracasso P.M., Kantarjian H.M., Cortes J.E., Brown R.A., Verstovsek S. Phase I and pharmacodynamic study of Triapine, a novel ribonucleotide reductase inhibitor, in patients with advanced leukemia. Leuk Res. 2003;27:1077–1083. doi: 10.1016/s0145-2126(03)00118-8. [DOI] [PubMed] [Google Scholar]
- 2.Karp J.E., Giles F.J., Gojo I., Morris L., Greer J., Johnson B. A phase I study of the novel ribonucleotide reductase inhibitor 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine) in combination with the nucleoside analog fludarabine for patients with refractory acute leukemias and aggressive myeloproliferative disorders. Leuk Res. 2008;32:71–77. doi: 10.1016/j.leukres.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Attia S., Kolesar J., Mahoney M.R., Pitot H.C., Laheru D., Heun J. A phase 2 consortium (P2C) trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP) for advanced adenocarcinoma of the pancreas. Invest New Drug. 2008 doi: 10.1007/s10637-008-9123-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma B., Goh B.C., Tan E.H., Lam K.C., Soo R., Leong S.S. A multicenter phase II trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, Triapine((R))) and gemcitabine in advanced non-small-cell lung cancer with pharmacokinetic evaluation using peripheral blood mononuclear cells. Invest New Drug. 2008;26:169–173. doi: 10.1007/s10637-007-9085-0. [DOI] [PubMed] [Google Scholar]
- 5.Knox J.J., Hotte S.J., Kollmannsberger C., Winquist E., Fisher B., Eisenhauer E.A. Phase II study of Triapine in patients with metastatic renal cell carcinoma: a trial of the National Cancer Institute of Canada Clinical Trials Group (NCIC IND.161) Invest New Drug. 2007;25:471–477. doi: 10.1007/s10637-007-9044-9. [DOI] [PubMed] [Google Scholar]
- 6.Rappa G., Lorico A., Liu M.C., Kruh G.D., Cory A.H., Cory J.G. Overexpression of the multidrug resistance genes mdr1, mdr3, and mrp in L1210 leukemia cells resistant to inhibitors of ribonucleotide reductase. Biochem Pharmacol. 1997;54:649–655. doi: 10.1016/s0006-2952(97)00210-4. [DOI] [PubMed] [Google Scholar]
- 7.Traynor A.M., Lee J.W., Bayer G.K., Tate J.M., Thomas S.P., Mazurczak M. A phase II trial of triapine (NSC# 663249) and gemcitabine as second line treatment of advanced non-small cell lung cancer: Eastern Cooperative Oncology Group Study 1503. Invest New Drug. 2010;28:91–97. doi: 10.1007/s10637-009-9230-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Attia S., Kolesar J., Mahoney M.R., Pitot H.C., Laheru D., Heun J. A phase 2 consortium (P2C) trial of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP) for advanced adenocarcinoma of the pancreas. Invest New Drug. 2008;26:369–379. doi: 10.1007/s10637-008-9123-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heffeter P., Jungwirth U., Jakupec M., Hartinger C., Galanski M., Elbling L. Resistance against novel anticancer metal compounds: differences and similarities. Drug Resist Update. 2008;11:1–16. doi: 10.1016/j.drup.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 10.Szakacs G., Paterson J.K., Ludwig J.A., Booth-Genthe C., Gottesman M.M. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 11.Kowol C.R., Berger R., Eichinger R., Roller A., Jakupec M.A., Schmidt P.P. Gallium(III) and iron(III) complexes of alpha-N-heterocyclic thiosemicarbazones: synthesis, characterization, cytotoxicity, and interaction with ribonucleotide reductase. J Med Chem. 2007;50:1254–1265. doi: 10.1021/jm0612618. [DOI] [PubMed] [Google Scholar]
- 12.Kowol C.R., Trondl R., Heffeter P., Arion V.B., Jakupec M.A., Roller A. Impact of metal coordination on cytotoxicity of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (triapine) and novel insights into terminal dimethylation. J Med Chem. 2009 doi: 10.1021/jm900528d. [DOI] [PubMed] [Google Scholar]
- 13.Arion V.B., Jakupec M.A., Galanski M., Unfried P., Keppler B.K. Synthesis, structure, spectroscopic and in vitro antitumour studies of a novel gallium(III) complex with 2-acetylpyridine (4)N-dimethylthiosemicarbazone. J Inorg Biochem. 2002;91:298–305. doi: 10.1016/s0162-0134(02)00419-1. [DOI] [PubMed] [Google Scholar]
- 14.Korynevska A., Heffeter P., Matselyukh B., Elbling L., Micksche M., Stoika R. Mechanisms underlying the anticancer activities of the angucycline landomycin E. Biochem Pharmacol. 2007;74:1713–1726. doi: 10.1016/j.bcp.2007.08.026. [DOI] [PubMed] [Google Scholar]
- 15.Heffeter P., Jakupec M.A., Korner W., Chiba P., Pirker C., Dornetshuber R. Multidrug-resistant cancer cells are preferential targets of the new antineoplastic lanthanum compound KP772 (FFC24) Biochem Pharmacol. 2007;73:1873–1886. doi: 10.1016/j.bcp.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen D.W., Cardarelli C., Hwang J., Cornwell M., Richert N., Ishii S. Multiple drug-resistant human KB carcinoma cells independently selected for high-level resistance to colchicine, adriamycin, or vinblastine show changes in expression of specific proteins. J Biol Chem. 1986;261:7762–7770. [PubMed] [Google Scholar]
- 17.Heffeter P., Pongratz M., Steiner E., Chiba P., Jakupec M.A., Elbling L. Intrinsic and acquired forms of resistance against the anticancer ruthenium compound KP1019 [indazolium trans-[tetrachlorobis(1H-indazole)ruthenate (III)] (FFC14A) J Pharmacol Exp Ther. 2005;312:281–289. doi: 10.1124/jpet.104.073395. [DOI] [PubMed] [Google Scholar]
- 18.Pirker C., Raidl M., Steiner E., Elbling L., Holzmann K., Spiegl-Kreinecker S. Whole genome amplification for CGH analysis: linker–adapter PCR as the method of choice for difficult and limited samples. Cytometry A. 2004;61:26–34. doi: 10.1002/cyto.a.20060. [DOI] [PubMed] [Google Scholar]
- 19.Pirker C., Holzmann K., Spiegl-Kreinecker S., Elbling L., Thallinger C., Pehamberger H. Chromosomal imbalances in primary and metastatic melanomas: over-representation of essential telomerase genes. Melanoma Res. 2003;13:483–492. doi: 10.1097/00008390-200310000-00007. [DOI] [PubMed] [Google Scholar]
- 20.Pirker C., Lotsch D., Spiegl-Kreinecker S., Jantscher F., Sutterluty H., Micksche M. Response of experimental malignant melanoma models to the pan-Aurora kinase inhibitor VE-465. Exp Dermatol. 2010;19:1040–1047. doi: 10.1111/j.1600-0625.2010.01182.x. [DOI] [PubMed] [Google Scholar]
- 21.Dornetshuber R., Heffeter P., Sulyok M., Schumacher R., Chiba P., Kopp S. Interactions between ABC-transport proteins and the secondary Fusarium metabolites enniatin and beauvericin. Mol Nutr Food Res. 2009;53:904–920. doi: 10.1002/mnfr.200800384. [DOI] [PubMed] [Google Scholar]
- 22.Wang J., Tai L.S., Tzang C.H., Fong W.F., Guan X.Y., Yang M. 1p31, 7q21 and 18q21 chromosomal aberrations and candidate genes in acquired vinblastine resistance of human cervical carcinoma KB cells. Oncol Rep. 2008;19:1155–1164. [PubMed] [Google Scholar]
- 23.Duckett D.R., Cameron M.D. Metabolism considerations for kinase inhibitors in cancer treatment. Expert Opin Drug Metab Toxicol. 2010;6:1175–1193. doi: 10.1517/17425255.2010.506873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou S.F., Wang L.L., Di Y.M., Xue C.C., Duan W., Li C.G. Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug development. Curr Med Chem. 2008;15:1981–2039. doi: 10.2174/092986708785132870. [DOI] [PubMed] [Google Scholar]
- 25.Cole S.P., Deeley R.G. Transport of glutathione and glutathione conjugates by MRP1. Trends Pharmacol Sci. 2006;27:438–446. doi: 10.1016/j.tips.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 26.Akan I., Akan S., Akca H., Savas B., Ozben T. Multidrug resistance-associated protein 1 (MRP1) mediated vincristine resistance: effects of N-acetylcysteine and Buthionine sulfoximine. Cancer Cell Int. 2005;5:22. doi: 10.1186/1475-2867-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chuman Y., Chen Z.S., Seto K., Sumizawa T., Furukawa T., Tani A. Reversal of MRP-mediated vincristine resistance in KB cells by buthionine sulfoximine in combination with PAK-104P. Cancer Lett. 1998;129:69–76. doi: 10.1016/s0304-3835(98)00083-4. [DOI] [PubMed] [Google Scholar]
- 28.Habig W.H., Jakoby W.B. Assays for differentiation of glutathione S-transferases. Methods Enzymol. 1981;77:398–405. doi: 10.1016/s0076-6879(81)77053-8. [DOI] [PubMed] [Google Scholar]
- 29.Enyedy E.A., Primik M.F., Kowol C.R., Arion V.B., Kiss T., Keppler B.K. Interaction of Triapine and related thiosemicarbazones with iron(III)/(II) and gallium(III): a comparative solution equilibrium study. Dalton Trans. 2011;40:5895–5905. doi: 10.1039/c0dt01835j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernhardt P.V., Sharpe P.C., Islam M., Lovejoy D.B., Kalinowski D.S., Richardson D.R. Iron chelators of the dipyridylketone thiosemicarbazone class: precomplexation and transmetalation effects on anticancer activity. J Med Chem. 2009;52:407–415. doi: 10.1021/jm801012z. [DOI] [PubMed] [Google Scholar]
- 31.Chang X.B. A molecular understanding of ATP-dependent solute transport by multidrug resistance-associated protein MRP1. Cancer Metastasis Rev. 2007;26:15–37. doi: 10.1007/s10555-007-9041-7. [DOI] [PubMed] [Google Scholar]
- 32.Keppler D. Multidrug resistance proteins (MRPs, ABCCs): importance for pathophysiology and drug therapy. Handb Exp Pharmacol. 2011;29:9–32. doi: 10.1007/978-3-642-14541-4_8. 3. [DOI] [PubMed] [Google Scholar]
- 33.Lutzky J., Astor M.B., Taub R.N., Baker M.A., Bhalla K., Gervasoni J.E., Jr. Role of glutathione and dependent enzymes in anthracycline-resistant HL60/AR cells. Cancer Res. 1989;49:4120–4125. [PubMed] [Google Scholar]
- 34.Laberge R.M., Karwatsky J., Lincoln M.C., Leimanis M.L., Georges E. Modulation of GSH levels in ABCC1 expressing tumor cells triggers apoptosis through oxidative stress. Biochem Pharmacol. 2007;73:1727–1737. doi: 10.1016/j.bcp.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 35.Crenshaw T.R., Cory J.G. Overexpression of protein disulfide isomerase-like protein in a mouse leukemia L1210 cell line selected for resistance to 4-methyl-5-amino-1-formylisoquinoline thiosemicarbazone, a ribonucleotide reductase inhibitor. Adv Enzyme Regul. 2002;42:143–157. doi: 10.1016/s0065-2571(01)00028-0. [DOI] [PubMed] [Google Scholar]
- 36.Ludwig J.A., Szakacs G., Martin S.E., Chu B.F., Cardarelli C., Sauna Z.E. Selective toxicity of NSC73306 in MDR1-positive cells as a new strategy to circumvent multidrug resistance in cancer. Cancer Res. 2006;66:4808–4815. doi: 10.1158/0008-5472.CAN-05-3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall M.D., Salam N.K., Hellawell J.L., Fales H.M., Kensler C.B., Ludwig J.A. Synthesis, activity, and pharmacophore development for isatin-beta-thiosemicarbazones with selective activity toward multidrug-resistant cells. J Med Chem. 2009;52:3191–3204. doi: 10.1021/jm800861c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang Y., Blower P.E., Yang C., Barbacioru C., Dai Z., Zhang Y. Correlating gene expression with chemical scaffolds of cytotoxic agents: ellipticines as substrates and inhibitors of MDR1. Pharmacogenomics J. 2005;5:112–125. doi: 10.1038/sj.tpj.6500297. [DOI] [PubMed] [Google Scholar]
- 39.Wu C.P., Shukla S., Calcagno A.M., Hall M.D., Gottesman M.M., Ambudkar S.V. Evidence for dual mode of action of a thiosemicarbazone, NSC73306: a potent substrate of the multidrug resistance linked ABCG2 transporter. Mol Cancer Ther. 2007;6:3287–3296. doi: 10.1158/1535-7163.MCT-07-2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.