

Abstract
Many estimation problems in bioinformatics are formulated as point estimation problems in a high-dimensional discrete space. In general, it is difficult to design reliable estimators for this type of problem, because the number of possible solutions is immense, which leads to an extremely low probability for every solution—even for the one with the highest probability. Therefore, maximum score and maximum likelihood estimators do not work well in this situation although they are widely employed in a number of applications. Maximizing expected accuracy (MEA) estimation, in which accuracy measures of the target problem and the entire distribution of solutions are considered, is a more successful approach. In this review, we provide an extensive discussion of algorithms and software based on MEA. We describe how a number of algorithms used in previous studies can be classified from the viewpoint of MEA. We believe that this review will be useful not only for users wishing to utilize software to solve the estimation problems appearing in this article, but also for developers wishing to design algorithms on the basis of MEA.
Key words: algorithms, alignment, RNA, secondary structure, sequence analysis
1. Introduction
In bioinformatics, there are many estimation and prediction problems, such as gene prediction from genomic sequences (Picardi and Pesole, 2010), alignment of biological sequences (Pirovano and Heringa, 2008; Pei, 2008), biological network prediction (e.g., protein-protein interaction prediction) (Skrabanek et al., 2008), phylogenetic tree estimation (Whelan, 2008), and RNA secondary structure prediction (Andersen, 2010). These problems give rise to specific point estimation problems, whose general paradigm can be stated as follows.
Problem 1 (Discrete-Space Point Estimation Problem [DSPEP])
Given data D and a discrete space Y correlated to D, find a point y in Y.
In this review, Y is called a predictive (solution) space, and it contains all the possible solutions (for the data D) of the target problem. For example, prediction of the secondary structure of an RNA sequence x is formulated as Problem 1 where 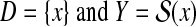 is the (discrete) space of all possible secondary structures of the RNA sequence x (see Example 3).
is the (discrete) space of all possible secondary structures of the RNA sequence x (see Example 3).
To solve this estimation problem, a score model S(y|D) (which gives a score of 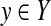 ) or a probability distribution p(y|D) (which gives a probability of y), for given data D, is often employed. In many cases, a score model S(y|D) leads to a probability distribution p(y|D) on the predictive space. For example, in RNA secondary structure prediction, the energy model (Mathews et al., 2004) leads to a probability distribution of secondary structures known as the McCaskill model (McCaskill, 1990), and in alignment, a score model of alignments (specified by a score matrix and gap open/extension costs) leads to a probability distribution of pairwise alignments known as the Miyazawa model (Miyazawa, 1995). In this study, we, therefore, make the following assumption.
) or a probability distribution p(y|D) (which gives a probability of y), for given data D, is often employed. In many cases, a score model S(y|D) leads to a probability distribution p(y|D) on the predictive space. For example, in RNA secondary structure prediction, the energy model (Mathews et al., 2004) leads to a probability distribution of secondary structures known as the McCaskill model (McCaskill, 1990), and in alignment, a score model of alignments (specified by a score matrix and gap open/extension costs) leads to a probability distribution of pairwise alignments known as the Miyazawa model (Miyazawa, 1995). In this study, we, therefore, make the following assumption.
Assumption 1
In Problem 1, a (posterior) probability distribution p(y|D) on a predictive space Y is given.
It is difficult to design reliable estimators for Problem 1. This is because there are an immense number of candidate solutions, and therefore, any point estimation, even if it is the prediction with the highest probability, is not reliable as its probability is extremely small. Hence, maximum likelihood (ML) and maximum score (minimum energy) estimators (both of which have been widely utilized) are not sufficient in those estimation problems. Moreover, as pointed out in Carvalho and Lawrence (2008), consistency, asymptotic normality, and asymptotic efficiency are not established for the ML estimator for Problem 1, although those properties have been established for the ML estimator on continuous spaces. Carvalho and Lawrence (2008) also pointed out that there is no reason for the ML estimation to be a representative solution in Y, because ML estimators do not consider the entire distribution of solutions.
When accuracy measures of a target problem are given (e.g., sensitivity, positive predictive value [PPV], Matthew's correlation coefficient [MCC], or F-score [Baldi et al., 2000]) (see Section A.1 in the Appendix), it is reasonable to design estimators that are suited to those accuracy measures. Maximizing expected accuracy (MEA) estimators, which are the main focus of this study, are able to consider both accuracy measures of the target problem and an entire distribution of solutions, and have been successfully applied to a number of estimation problems in bioinformatics (Do et al., 2006a; Sahraeian and Yoon, 2010; Lu et al., 2009; Nánási et al., 2010). In this article, we classify existing algorithms and software from the viewpoint of MEA, which will provide useful information not only for users but also for developers of such software.
This rest of this review is organized as follows. In Section 2, we explain the concepts of maximizing expected accuracy (MEA) estimation. In Section 3, we present a classification of existing algorithms from the viewpoint of MEA; therein, in Table 1, we summarize the classification. In Section 4, we discuss additional issues related to MEA estimations. In Section 5, we conclude, and in Section 6, we provide an Appendix.
Table 1.
Summary of Maximizing Expected Accuracy (MEA) Estimations in Bioinformatics
| Reference | Software | Target problem | Ya | Gain functionb | Aprc | Repd | Compe | Suitable accuracy measures |
|---|---|---|---|---|---|---|---|---|
| Kall et al. (2005) | — | Sequence feature predictionsf | L | G(label) | ✓ | DP | # of correctly predicted label | |
| Gross et al. (2007a) | CONTRAST | Gene prediction | L |  |
DP | # of correctly predicted boundary | ||
| Nánási et al. (2010) | HERD | HIV recombination prediction | L |
 g g
|
DP | — | ||
| Miyazawa (1995) | — | Pairwise alignment | B |  |
DP | Hamming distance of (un)aligned-bases | ||
| Holmes and Durbin (1998) | — | Pairwise alignment | B | 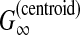 |
DP | SEN/SPS of aligned-bases | ||
| Schwartz et al. (2005) | — | Pairwise alignment | B | 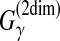 |
DP | Alignment metric accuracy (AMA) | ||
| Do et al. (2005) | ProbCons | Multiple alignment | B | 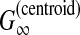 |
✓ | ✓ | DP | SEN/SPS of aligned-bases |
| Roshan and Livesay (2006) | ProbAlign | Multiple alignment | B | 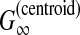 |
✓ | ✓ | DP | SEN/SPS of aligned-bases |
| Yamada et al. (2008) | PRIME | Multiple alignment | B | 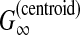 |
DP | SEN/SPS of aligned-bases | ||
| Schwartz and Pachter (2007) | AMAP | Multiple alignment | B | 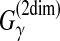 |
✓ | ✓ | SA | Alignment metric accuracy (AMA) |
| Sahraeian and Yoon (2010) | PicXAA | Multiple alignment | B | 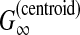 |
✓ | ✓ | DP | SEN/SPS of aligned-bases |
| Frith et al. (2010) | LAST | Genome (local) alignment | B |  |
DP | SEN/PPV of (un)aligned-bases | ||
| Ding et al. (2005) | Sfold | RNA sec. str. pred. | B |  |
SS | Hamming distance of base-pairs | ||
| Do et al. (2006a) | CONTRAfold | RNA sec. str. pred. | B | 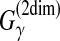 |
DP | # of correctly predicted (loop or base-pairs) positions in RNA sequence | ||
| Lu et al. (2009) | MaxExpect | RNA sec. str. pred. | B | 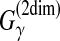 |
DP | # of correctly predicted (loop or base-pairs) positions in RNA sequence | ||
| Hamada et al. (2009a) | CentroidFold | RNA sec. str. pred. | B |  |
DP | SEN/PPV of base-pairs | ||
| Hamada et al. (2010) | CentroidFold | RNA sec. str. pred. | B | G(Acc) | DP/SS | MCC/F-score of base-pairs | ||
| Lorenz and Clote (2011) | RNAlocopt | RNA sec. str. pred. | B | 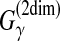 |
DP | # of correctly predicted (loop or base-pairs) positions in RNA sequence | ||
| Sato et al. (2011) | IPKnot | RNA sec. str. pred. with pseudoknot | B |  |
✓ | IP | SEN/PPV of base-pairs | |
| Hamada et al. (2009c) | CentroidHomfold | RNA sec. str. pred. with homol. seq. | B |  |
✓ | ✓ | DP | SEN/PPV of base-pairs |
| Knudsen and Hein (2003) | Pfold | RNA com. sec. str. pred. | B | 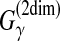 |
DP | # of correctly predicted (loop or base-pairs) positions | ||
| Bernhart et al. (2008) | RNAalifold | RNA com. sec. str. pred. | B |  |
DP | # of correctly predicted positions | ||
| Kiryu et al. (2007a) | McCaskill-MEA | RNA com. sec. str. pred. | B | 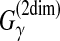 |
✓ | DP | # of correctly predicted positions | |
| Seemann et al. (2008) | PETfold | RNA com. sec. str. pred. | B | 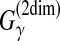 |
✓ | DP | # of correctly predicted positions | |
| Hamada et al. (2011b) | CentroidAlifold | RNA com. sec. str. pred. | B |  |
✓ | DP | SEN/PPV of base-pairs | |
| Wei et al. (2011) | RNAG | RNA com. sec. str. pred. | B |  |
GS | SEN/PPV of base-pairs | ||
| Sahraeian and Yoon (2011) | PicXAA-R | RNA multiple alignment | B | 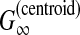 |
✓ | ✓ | DP | SPS of aligned-bases |
| Hamada et al. (2009b) | CentroidAlign | RNA multiple alignment | B |  |
✓ | ✓ | DP | SEN/PPV of aligned-bases |
| Tabei and Asai (2009) | SCARNA-LM | RNA local alignment | B |  |
DP | SEN/PPV of aligned bases | ||
| Kato et al. (2010) | RactIP | RNA-RNA interaction prediction | B |  |
IP | SEN/PPV of base-pairs/interaction base-pairs | ||
| Seemann et al. (2011) | PETcofold | RNA-RNA interaction prediction between two multiple alignments | B | 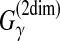 |
✓ | DP | — | |
| Hamada et al. (2011a) | — | Phylogenetic tree estimation | B |  |
✓ | — | Robinson-Foulds (RF) measure |
This table is sorted by “Target problem.”
L and B mean Y ⊂ Ln and Y ⊂{0, 1}n, respectively.
Gain function. See Section 2.3 for definitions.
The use of approximated MEA estimators in Section 2.4.2.
The use of representative MEA estimators in Section 2.4.1.
Methods for computing estimation: DP (dynamic programming), IP (integer programming), SS (stochastic sampling), GS (Gibbs sampling).
Transmembrane topology predictions, signal peptide predictions, protein secondary structure prediction, etc.
An extension of  was used.
was used.
2. Concepts of Maximizing Expected Accuracy (MEA) Estimation
2.1. Maximizing expected gain (MEG) estimator
In Problem 1 with Assumption 1, the following estimator is called a Maximum expected gain (MEG) estimator (Hamada et al., 2011a).
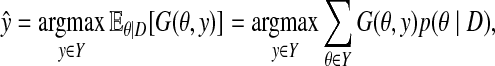 |
(1) |
where G(θ, y) on Y × Y is called a gain function, which gives higher values (gains) when θ and y are similar.
This MEG estimator is closely related to statistical decision theory, in which an estimator that minimizes expected loss is often considered (Carvalho and Lawrence, 2008). In order to facilitate the understanding of the relationship with MEA, in this review we use a gain function that should be maximized instead of minimizing a loss.
When the gain function G is designed according to the accuracy measures of the target problem (e.g., MCC, F-score, PPV and Sensitivity), the MEG estimator is called a maximum expected accuracy (MEA) estimator. (This does not mean the gain function is exactly equal to the accuracy measure.) On the other hand, when G(θ, y) is equal to the delta function, δ(θ, y), that is 1 only when θ is exactly equal to y, the estimator is called a maximum likelihood (ML) estimator. Note that it is quite unreasonable to employ the delta function as the accuracy measure, because the condition described by the delta function is too strict. ML estimators are, therefore, unsuitable as accuracy measures in many bioinformatics problems, and the gain function should be designed more carefully.
In the following two subsections, we introduce several commonly used predictive spaces and gain functions, which are used in the classification in Section 3 (and Table 1 therein).
2.2. Commonly used predictive (solution) spaces, Y
2.2.1. Y is a subset of Ln for |L| < ∞
Typically, L is a set of labels and the data D is a biological sequence with length n (e.g., DNA, RNA, or protein sequence) as in the following examples.
Example 1 (The space of protein secondary structures: 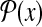 )
)
For a protein sequence x and L = {α-helix, β-strand,loop} (a set of labels for components of protein secondary structures), a protein secondary structure y (of x) can be represented as
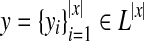 , where
, where
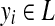 indicates the label of the i-th position in x.
indicates the label of the i-th position in x.
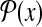 denotes the set of possible protein secondary structures of a protein sequence x.
denotes the set of possible protein secondary structures of a protein sequence x.
Example 2 (The space of gene structures: 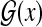 )
)
For a genome sequence x and L = {exon, intron, intergenic} (a set of labels for components of gene structures), a gene structure y can be represented as
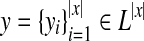 , where
, where
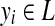 indicates the label of the i-th position in x.
indicates the label of the i-th position in x.
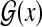 denotes the space of gene structures of a genome sequence x (Fig. 1).
denotes the space of gene structures of a genome sequence x (Fig. 1).
FIG. 1.

Example of gene prediction. The top and bottom figures are a reference gene structure θ and a predicted gene structure y, respectively. The labels X, E, and I indicate intergenic regions, exons, and introns, respectively. The vertical lines in red show boundaries (exon-intron and intergenic region-exon boundaries). We compute G(label) (θ, y) = 19 and  (θ, y)=4γ+12.
(θ, y)=4γ+12.
In general, Y is not equal to Ln but is a subset of Ln, which means that the labels of each dimension (position) in a prediction are mutually correlated and cannot be estimated independently.
2.2.2. Y is a subset of {0 1}n
Although this is a special case of the predictive space described in Subsection 2.2.1 (where L = {0, 1}), we consider it separately for convenience. In this case, 0 and 1 in a binary vector 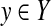 typically mean positive and negative predictions, respectively. Hence, accuracy measures (such as sensitivity, PPV, MCC, and F-score) are naturally introduced, each of which is defined by using the number of true positive, true negative, false positive, and false negative predictions (denoted as TP, TN, FP, and FN, respectively) (Baldi et al., 2000) (see Section A.1 in the Appendix).
typically mean positive and negative predictions, respectively. Hence, accuracy measures (such as sensitivity, PPV, MCC, and F-score) are naturally introduced, each of which is defined by using the number of true positive, true negative, false positive, and false negative predictions (denoted as TP, TN, FP, and FN, respectively) (Baldi et al., 2000) (see Section A.1 in the Appendix).
Example 3 (The space of secondary structures of an RNA sequence: 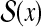 )
)
For an RNA sequence x, a secondary structure of x is represented as a upper triangular binary-valued matrix, y = {yij}1≤i≤j≤|x|, where yij = 1 means xi and xj (the i-th and j-th bases of x) form a base pair and yij = 0 means xi and xj do not form a base pair.
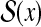 denotes the space of possible secondary structures of x.
denotes the space of possible secondary structures of x.
Example 4 (The space of alignments of two sequences: 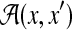
For two biological sequences x and x′, a pairwise alignment y between x and x′ is represented as a binary-valued matrix
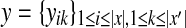 , where yik = 1 means xi aligns with
, where yik = 1 means xi aligns with
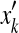 and yik = 0 means xi does not align with
and yik = 0 means xi does not align with
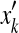 .
. 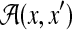 denotes the space of possible pairwise alignments of biological sequences x and x′.
denotes the space of possible pairwise alignments of biological sequences x and x′.
Note that the above predictive spaces are a subset of binary space, which means that every element in the predictive space has complicated constraints.
2.3. Commonly used gain functions
2.3.1. A gain function for Y ⊂ Ln: label gain function
For θ, 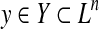 , the following gain function (originally proposed in Kall et al. [2005]) is introduced.
, the following gain function (originally proposed in Kall et al. [2005]) is introduced.
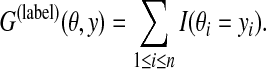 |
(2) |
Here, I(condition) is the indicator function that returns 1 only when condition is true. When θ is a correct (reference) sequence and y is a prediction, Eq. (2) is equal to the number of correctly predicted labels. The MEG estimator of this gain function, therefore, maximizes the expected number of correctly predicted labels.
Example 5 (G(label) for gene prediction)
In gene prediction from a genomic sequence, when θ is a reference sequence and y is a prediction, G(label) (θ, y) is the number of correctly predicted labels. For example, in Figure 1, G(label) (θ, y) = 19.
2.3.2. A gain function for Y ⊂ Ln: boundary gain function
For θ, 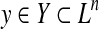 , the following gain function is introduced. (This gain function was originally proposed by Gross et al. (2007a) in the context of gene prediction.)
, the following gain function is introduced. (This gain function was originally proposed by Gross et al. (2007a) in the context of gene prediction.)
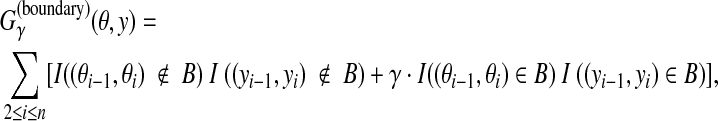 |
(3) |
where B is the list of all pairs of labels corresponding to a boundary (e.g., an exon-intron boundary for gene prediction). When θ is a correct prediction and y is a prediction, Eq. (3) is equal to a weighted sum of the number of correctly predicted boundaries and non-boundaries. The MEG estimator of this gain function is, therefore, suitable for accurate prediction of boundary of annotation (boundary accuracy).
Example 6 (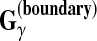 for gene prediction)
for gene prediction)
In gene prediction, when θ is a reference genomic sequence and y is a prediction,
 (θ, y) is the weighted number of correctly predicted boundaries and non-boundaries. B is the list of all pairs of labels corresponding to a boundary (e.g., an exon-intron boundary for gene prediction). Therefore, this gain function fits with exon-level or gene-level accuracy in gene prediction (Gross et al., 2007a). For example, in Figure 1,
(θ, y) is the weighted number of correctly predicted boundaries and non-boundaries. B is the list of all pairs of labels corresponding to a boundary (e.g., an exon-intron boundary for gene prediction). Therefore, this gain function fits with exon-level or gene-level accuracy in gene prediction (Gross et al., 2007a). For example, in Figure 1,
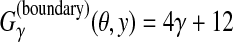 .
.
The γ in Eq. (3) is a parameter that adjusts between the sensitivity and PPV of a prediction. Using larger γ leads to more boundaries (that is, more genes) in the prediction.
2.3.3. A gain function for Y ⊂ {0, 1}n: γ-centroid gain function
For θ, 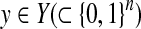 , we introduce the gain function
, we introduce the gain function
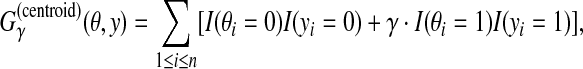 |
(4) |
where γ ≥ 0 is a weight parameter. When y is a prediction and θ is a reference sequence, this gain function is equal to a weighted sum of the number of TP and TN. (This gain function was originally proposed in the context of RNA secondary structure prediction, in Hamada et al. [2009a].)
Example 7 ( for RNA secondary structure)
for RNA secondary structure)
For two secondary structures y and θ in
 , where y is a prediction and θ is a reference structure,
, where y is a prediction and θ is a reference structure,
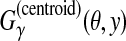 is equal to the weighted sum of the number of true-positive base pairs and true-negative base pairs. For example, in
Figure 2,
is equal to the weighted sum of the number of true-positive base pairs and true-negative base pairs. For example, in
Figure 2, 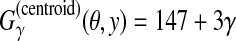 .
.
FIG. 2.

Example of RNA secondary structure prediction. The top and bottom structures are a reference θ and prediction y, respectively. (a) “p” and “l” show the correctly predicted positions of base pairs and loops, respectively, while “x” indicates wrongly predicted positions. Hence, we compute 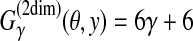 . (b) TP, TN, FP and FN are the numbers of true positive, true negative, false positive, and false negative base pairs, respectively. We, therefore, compute
. (b) TP, TN, FP and FN are the numbers of true positive, true negative, false positive, and false negative base pairs, respectively. We, therefore, compute 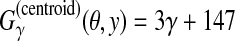 .
.
Example 8 ( for pairwise alignment)
for pairwise alignment)
For two secondary structures y and θ in
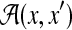 , where y is a prediction and θ is a reference structure,
, where y is a prediction and θ is a reference structure,
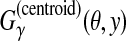 is equal to the weighted sum of the number of true-positive aligned bases and true-negative aligned bases. For example, in
Figure 3,
is equal to the weighted sum of the number of true-positive aligned bases and true-negative aligned bases. For example, in
Figure 3, 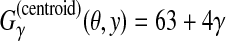 .
.
FIG. 3.

Example of pairwise alignment. The top and bottom alignments are a reference θ and prediction y, respectively. (The numbers indicate positions in the sequences and “-” indicates a gap.) (a) “p” and “l” indicate the correctly predicted positions of aligned columns, whereas “x” indicates a wrongly predicted position. Hence, we compute 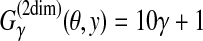 . (b) TP, TN, FP, and FN are the numbers of true positive, true negative, false positive, and false negative aligned pairs, respectively. We, therefore, compute
. (b) TP, TN, FP, and FN are the numbers of true positive, true negative, false positive, and false negative aligned pairs, respectively. We, therefore, compute 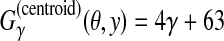 .
.
An MEG estimator with this gain function is often called a γ-centroid estimator. The parameter γ in the γ-centroid estimator can be naturally introduced based on the criterion that more true predictions and fewer false predictions are required (Hamada et al., 2011a). The parameter is used for adjusting between the sensitivity and PPV of a prediction. It is easily seen that the MEG estimator of  (1-centroid estimator) is equivalent to the centroid estimator (Carvalho and Lawrence, 2008), which minimizes the expected Hamming distance.
(1-centroid estimator) is equivalent to the centroid estimator (Carvalho and Lawrence, 2008), which minimizes the expected Hamming distance.
2.3.4. A gain function for Y ⊂ {0, 1}n: MCC/F-score
For θ, 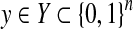 , we introduce the gain function
, we introduce the gain function
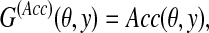 |
(5) |
where Acc is either MCC or F-score (Baldi et al., 2000), both of which are accuracy measures providing a balance between sensitivity and PPV. If G(θ, y) = MCC(θ, y) or F-score(θ, y), where θ is a reference and y is a prediction, the MEG estimator of the gain function maximizes the expected accuracy (Acc).
Example 9 (G(Acc) for RNA secondary structure prediction)
For θ,
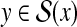 , where θ is a reference structure and y is a prediction, G(Acc) (θ, y) for Acc = MCC is equal to MCC with respect to the base pairs, which is a widely used accuracy measure. For example, in Figure 2, MCC(θ, y) = 0.661.
, where θ is a reference structure and y is a prediction, G(Acc) (θ, y) for Acc = MCC is equal to MCC with respect to the base pairs, which is a widely used accuracy measure. For example, in Figure 2, MCC(θ, y) = 0.661.
Unlike the γ-centroid estimators, the MEG estimator of this gain function does not contain any parameter. However, it is generally difficult to compute the estimator. Instead, Hamada et al. (2010) have proposed an approximate method to maximize expected MCC/F-score. In Hamada et al. (2010), the authors focused on RNA secondary structure prediction, but the method is applicable to other problems.
2.3.5. A gain function for Y ⊂ {0, 1}n × {0, 1}m
Suppose that each binary vector has two indices, that is, Y ⊂ {0, 1}n × {0, 1}m (like 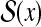 and
and 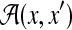 ). For θ = {θij} and y = {yij} (
). For θ = {θij} and y = {yij} (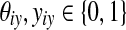 ), the gain function
), the gain function
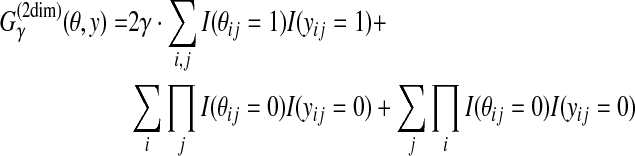 |
(6) |
is introduced. The second and third terms in the right-hand side are equal to 1 when θij = yij = 0 for all j and θij = yij = 0 for all i, respectively. If the products (Πi and Πj) are replaced by sums (Σi and Σj), the gain function is equal to (twice) the γ-centroid gain function, Eq. (4).
Interestingly, this gain function was independently proposed in Do et al. (2006a) (in the context of RNA secondary structure prediction) and in Schwartz et al. (2005) (in the context of pairwise alignment).
Example 10 (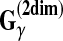 for RNA secondary structure)
for RNA secondary structure)
When Y is the space of secondary structures of a given RNA sequence x (i.e.,
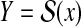 ), θ is a reference secondary structure and y is a prediction,
), θ is a reference secondary structure and y is a prediction,
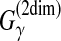 (θ,y) is equal to a (weighted) sum of the numbers of correctly and incorrectly predicted positions in the RNA sequence x. For example, in Figure 2,
(θ,y) is equal to a (weighted) sum of the numbers of correctly and incorrectly predicted positions in the RNA sequence x. For example, in Figure 2,
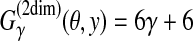 .
.
Remark 1. In RNA secondary structure prediction, the γ-centroid gain function
 is more suitable than
is more suitable than
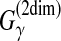 in terms of widely used accuracy measures (Hamada et al., 2009a).
in terms of widely used accuracy measures (Hamada et al., 2009a).
Example 11 (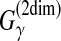 for pairwise alignment)
for pairwise alignment)
When Y is the space of possible pairwise alignments between two sequences x and x′ (i.e.,
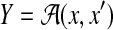 ), θ is a reference alignment and y is a predicted alignment,
), θ is a reference alignment and y is a predicted alignment,
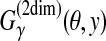 is equal to a (weighted) sum of the numbers of correctly and incorrectly predicted columns in the alignment. For example, in Figure 3,
is equal to a (weighted) sum of the numbers of correctly and incorrectly predicted columns in the alignment. For example, in Figure 3,
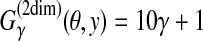 .
.
2.4. Two variants of MEG/MEA estimators
The following two variants of an MEG/MEA estimator were proposed in Hamada et al. (2011a) (in the context of a restricted class of MEA estimators, that is, the γ-centroid estimators).
2.4.1. Representative/common MEG/MEA estimator
In some cases, the data D consists of several data-points dn, for example, 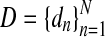 in Problem 1, and we would like to predict a common or consensus solution for these data, as described in the following examples.
in Problem 1, and we would like to predict a common or consensus solution for these data, as described in the following examples.
Example 12 (Common RNA secondary structure prediction)
Given a set of RNA sequences
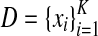 and multiple alignments of length l, predict their common secondary structure as a point in
and multiple alignments of length l, predict their common secondary structure as a point in
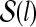 , which is the space of all the possible secondary structures of length l.
, which is the space of all the possible secondary structures of length l.
Example 13 (Sequence feature prediction in a multiple alignment)
Given a set of biological sequences
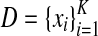 and multiple alignments of length l, predict their common sequence feature as a point in
and multiple alignments of length l, predict their common sequence feature as a point in
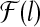 , which is the space of all the possible predictions of sequence features of length l.
, which is the space of all the possible predictions of sequence features of length l.
For those problems, the following estimator (called a representative MEG estimator) can be introduced. It gives a consensus or common prediction for probability distributions of every data point:
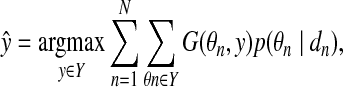 |
(7) |
where 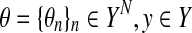 ,
, 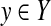 and p(θn|dn) is a probability distribution on Y, given dn.
and p(θn|dn) is a probability distribution on Y, given dn.
Example 14
For Example 12, the estimators used in McCaskill-MEA and PETfold can be considered representative estimators of the
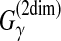 type, and the one used in CentroidAlifold can be considered representative estimator of the
type, and the one used in CentroidAlifold can be considered representative estimator of the
 type. In Example 13, Kall et al. (2005) utilized a representative estimator of G(label).
type. In Example 13, Kall et al. (2005) utilized a representative estimator of G(label).
We remark that the following example can be also considered as a similar problem by taking dn = di,k = {x(i), x(k)} for 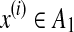 and
and 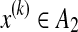 (and, therefore, a representative estimator can be introduced).
(and, therefore, a representative estimator can be introduced).
Example 15 (Pairwise alignment between two multiple alignments)
Given two multiple alignments A1 and A2, predict a pairwise alignment between A1 and A2.
We will describe further applications of representative estimators in Section 3. See also the column “Rep” in Table 1 below.
2.4.2. Approximated MEG/MEA estimator with additional information
In Problem 1, by employing additional information appropriately, it is possible to improve accuracy.
Example 16 (RNA secondary structure prediction with homologous sequences)
Given a (target) RNA sequence x and its homologous sequence h, predict a secondary structure
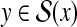 of the target sequence x by using homologous sequence information.
of the target sequence x by using homologous sequence information.
Example 17 (Pairwise alignment with homologous sequence information)
For two biological sequences x and x′ and their homologous sequence h, predict a pairwise alignment
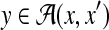 by using the homologous sequence information.
by using the homologous sequence information.
Example 18 (RNA alignment with common secondary structure information)
For two RNA sequences x and x′, predict a pairwise alignment
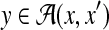 by using secondary structures that are common to x and x′.
by using secondary structures that are common to x and x′.
Ideally, a (refined) probability distribution on the predictive space Y is given by marginalizing onto 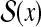 a probability distribution on a larger space Y′ given D and A (p(y′∣D, A)). In Example 16, we consider a probability distribution of possible structural alignments between x and h, and then obtain a probability distribution on
a probability distribution on a larger space Y′ given D and A (p(y′∣D, A)). In Example 16, we consider a probability distribution of possible structural alignments between x and h, and then obtain a probability distribution on 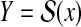 by marginalizing this distribution. In Example 17, we consider a probability distribution of multiple alignments of x, x′, and h, and then obtain a probability distribution on
by marginalizing this distribution. In Example 17, we consider a probability distribution of multiple alignments of x, x′, and h, and then obtain a probability distribution on 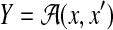 by marginalizing the distribution. In Example 18, we consider a probability distribution of possible structural alignments between x and x′, and then obtain a probability distribution on
by marginalizing the distribution. In Example 18, we consider a probability distribution of possible structural alignments between x and x′, and then obtain a probability distribution on 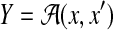 by marginalizing this distribution.
by marginalizing this distribution.
By using these marginal probability distributions on a predictive space Y, the MEG estimators are introduced directly. However, the computational cost of computing this MEG estimator is generally huge, and several heuristic methods are, therefore, employed, including a factorization of the probability distribution p(y′∣D, A). (For example, a probability distribution of possible structural alignments between x and h is factorized into the distributions of secondary structures of x and x′, and the distribution of pairwise alignments.) The factorization generally leads to a number of inconsistencies in the distribution and those inconsistencies should be resolved when the gain function is designed.
We call this type of estimator an “approximated MEA estimator” (Hamada et al., 2011a).
Example 19
For Examples 16, 17, and 18, approximated MEA estimators are employed in CentroidHomfold (Hamada et al., 2009c), ProbCons (Do et al., 2005), and CentroidAlign (Hamada et al., 2009b), respectively.
We will also describe further applications of this type of estimator in Section 3. See also the column “Apr” in Table 1 below.
2.5. Commonly used approaches to compute MEG/MEA estimators
To obtain a final prediction of MEG/MEA (and related) estimators, we need to compute the “argmax” operation in Eq. (1). There are several commonly used approaches:
Dynamic programming (DP) (Eddy, 2004)
Integer programming (IP) (Nemhauser and Wolsey, 1988)
Stochastic sampling or other stochastic approaches such as the Simulated annealing, sequence annealing (SA) (Schwartz and Pachter, 2007), or Gibbs sampling (GS)
DP algorithms are widely used in bioinformatics, including alignment and RNA secondary structure prediction (Smith and Waterman, 1981). IP is also employed in bioinformatics problems (Sato et al., 2011; Kato et al., 2010). Stochastic sampling enables us to sample directly from the posterior distribution p(y∣D). This approach has been proposed for pairwise alignments (Webb-Robertson et al., 2008), RNA secondary structure predictions (Ding et al., 2005), and structural alignments of RNA sequences (Harmanci et al., 2009).
In methods described in the next section, one of the above techniques is employed to compute a final prediction; see the “Comp” column in Table 1 below.
3. Classification of Various Estimators in Bioinformatics From The Viewpoint of MEA
In this section, we classify various estimators appearing in bioinformatics from the viewpoint of MEA. The classification considers the type of predictive space, the gain function, and the optimization method. For a summary of the classification, see Table 1.
3.1. Feature predictions in biological sequence
3.1.1. Transmembrane topology prediction and signal peptide prediction
For the prediction of sequence features like transmembrane topology, signal peptides, coil-coil structures, and protein secondary structures (which are formulated as Problem 1; for example, see Example 1), the “Optimal accuracy decoding” method used in Kall et al. (2005) can be considered as the MEA estimator of the gain function G(label) (Eq. (2)). Also, in transmembrane topology prediction and signal peptide prediction, the authors showed that this estimator achieved superior performance to the ML estimator and a (heuristic) posterior decoding method (cf. Section 4.2) proposed by Fariselli et al. (2005).
Moreover, the authors proposed an improved method for the problem which incorporated homologous sequence information (given by sequences aligned to the target sequence). This method can be considered as a representative MEA estimator (Section 2.4.1; Example 13) of the gain function G(label). In their article, the authors showed that prediction accuracy was substantially improved by employing homologous sequence information.
3.1.2. Gene prediction
Gene prediction is formulated as Problem 1 with 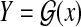 . Gross et al. (2007a) proposed the “maximum expected boundary accuracy” estimators for predicting genes in genomic sequences (the distribution p(θ∣x) on
. Gross et al. (2007a) proposed the “maximum expected boundary accuracy” estimators for predicting genes in genomic sequences (the distribution p(θ∣x) on 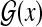 is based on a conditional random field [CRF] model in their study). It is easily seen that this is equivalent to the MEA estimator of the gain function
is based on a conditional random field [CRF] model in their study). It is easily seen that this is equivalent to the MEA estimator of the gain function  in Eq. (3) (see Example 6). In their evaluation study, “Gene Sensitivity(Sn)/Specificity(Sp)” (gene level accuracy), “Exon Sn/Sp” (exon level accuracy), and “Nucleotide Sn/Sp” (nucleotide level accuracy) were used as accuracy measures. For Gene/Exon Sn/Sp, accurate prediction of the boundaries of genes and exons is important, because, for example, exon predictions were counted as correct only if they matched the boundaries of the reference (correct) exon exactly. The MEA estimator of
in Eq. (3) (see Example 6). In their evaluation study, “Gene Sensitivity(Sn)/Specificity(Sp)” (gene level accuracy), “Exon Sn/Sp” (exon level accuracy), and “Nucleotide Sn/Sp” (nucleotide level accuracy) were used as accuracy measures. For Gene/Exon Sn/Sp, accurate prediction of the boundaries of genes and exons is important, because, for example, exon predictions were counted as correct only if they matched the boundaries of the reference (correct) exon exactly. The MEA estimator of  is, therefore, suited to those accuracy measures. Although the authors did not compare this estimator with the ML estimator or other decoding methods, they showed that it outperformed other state-of-the-art gene predictors.
is, therefore, suited to those accuracy measures. Although the authors did not compare this estimator with the ML estimator or other decoding methods, they showed that it outperformed other state-of-the-art gene predictors.
3.1.3. HIV recombination detection
For the problem of detecting recombination in the genome of the human immunodeficiency virus (HIV) with jumping hidden Markov models (HMMs) (Schultz et al., 2006), Nánási et al. (2010) proposed using the highest expected reward decoding (HERD) for the HMMs. This is a kind of MEA estimator with a special gain function that is an extension of  Eq. (3)). (Their gain function characterizes the similarity between any two annotations including boundaries.) They showed that their estimator is superior to both the ML estimator and the maximizing expected boundary accuracy estimator (see Section 3.1.2) for this problem.
Eq. (3)). (Their gain function characterizes the similarity between any two annotations including boundaries.) They showed that their estimator is superior to both the ML estimator and the maximizing expected boundary accuracy estimator (see Section 3.1.2) for this problem.
3.2. Pairwise/multiple/local alignment of biological sequences
3.2.1. Pairwise alignment
For the problem of (pairwise) alignment of two sequences x and x′ (Problem 1 with 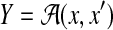 ), a posterior probability distribution of alignments of the given sequences p(θ∣x, x′) (for
), a posterior probability distribution of alignments of the given sequences p(θ∣x, x′) (for 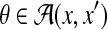 ) can be obtained by the Miyazawa model (Miyazawa, 1995), a pair HMM (Durbin et al., 1998), and the CONTRAlign model (Do et al., 2006b), which are utilized in the following MEA estimators.
) can be obtained by the Miyazawa model (Miyazawa, 1995), a pair HMM (Durbin et al., 1998), and the CONTRAlign model (Do et al., 2006b), which are utilized in the following MEA estimators.
Miyazawa (1995) proposed an estimator for pairwise alignments, which constructs alignments by using all the aligned bases whose posterior probabilities are larger than 0.5. Interestingly, a set of aligned bases whose probability is larger than 0.5 always produces a consistent alignment (Miyazawa, 1995; Carvalho and Lawrence, 2008) (i.e., one contained in 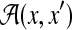 ). It is easily seen that this estimator is equivalent to the MEG estimator of
). It is easily seen that this estimator is equivalent to the MEG estimator of  (i.e., the centroid alignment) with the Miyazawa model. Miyazawa (1995) also showed that the centroid estimator is superior to the conventional maximum score estimator in computational experiments.
(i.e., the centroid alignment) with the Miyazawa model. Miyazawa (1995) also showed that the centroid estimator is superior to the conventional maximum score estimator in computational experiments.
Miyazawa's approach (Miyazawa, 1995) typically gives rise to an incomplete alignment that contains a number of unaligned residues (because all the paired residues whose posterior probability is less than 0.5 are unaligned). As an alternative, Holmes and Durbin (1998) proposed an estimator that maximizes the sum of posterior probabilities of aligned bases. This estimator is equivalent to the MEG estimator of  with a infinite γ, and is suited to the sensitivity of the aligned residues (but not to PPV).
with a infinite γ, and is suited to the sensitivity of the aligned residues (but not to PPV).
Recently, Frith et al. (2010) employed the MEG estimator of the gain function  (i.e., the γ-centroid alignment; see Example 8), in a generalization of Miyazawa (1995) and Holmes and Durbin (1998). The γ-centroid alignment is suited to accuracy measures based on (un)aligned bases. By using the parameter γ, the balance between the sensitivity and PPV with respect to (un)aligned bases is adjustable.
(i.e., the γ-centroid alignment; see Example 8), in a generalization of Miyazawa (1995) and Holmes and Durbin (1998). The γ-centroid alignment is suited to accuracy measures based on (un)aligned bases. By using the parameter γ, the balance between the sensitivity and PPV with respect to (un)aligned bases is adjustable.
On the other hand, the alignment method proposed in Schwartz et al. (2005) and Schwartz, (2007) is equivalent to the MEA estimator of the gain function 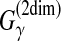 (see Example 11). In their article, they showed that the estimator maximizes the expected alignment metric accuracy (AMA), where the AMA is derived from a metric or distance between two pairwise alignments.
(see Example 11). In their article, they showed that the estimator maximizes the expected alignment metric accuracy (AMA), where the AMA is derived from a metric or distance between two pairwise alignments.
It should be emphasized that each of the above estimators can be efficiently computed by a Needleman-Wunsch-style DP algorithm in O(∣x∣∣x′∣) time. The recursive equation of the DP is written as
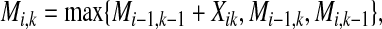 |
(8) |
where Mi,k stores the optimal value of the alignment between two sub-sequences 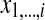 and
and 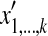 , and Xik is defined as follows.
, and Xik is defined as follows.
For the alignment method proposed by Holmes and Durbin (1998), Xik is set to be pik, the marginal probability that xi and 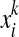 align with each other; for the MEG estimator of the gain function
align with each other; for the MEG estimator of the gain function  (γ-centroid alignment) (Frith et al., 2010), Xik is set to be (γ + 1)pik − 1; for the MEA estimator of the gain function
(γ-centroid alignment) (Frith et al., 2010), Xik is set to be (γ + 1)pik − 1; for the MEA estimator of the gain function 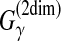 (AMA alignment) (Schwartz et al., 2005), Xik is set to be
(AMA alignment) (Schwartz et al., 2005), Xik is set to be 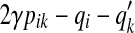 where qi (resp.
where qi (resp.  ) are the marginal probabilities that xi (resp.
) are the marginal probabilities that xi (resp.  ) aligns with a gap.
) aligns with a gap.
3.2.2. Multiple alignment of DNA/protein sequences
In most multiple alignment algorithms, pairwise alignments (according to a guide tree) are first made in order to obtain a final multiple alignment of a set of sequences S. In this step, pairwise alignment between x and x′ in S can be estimated by using the homologous sequence information of the other sequences, H = S \{x, x′} (cf. Example 17). An approximated MEA estimator of the gain function  with γ → ∞ (see Section 2.4.2) is employed in several multiple alignment problems (Hamada et al., 2011a). Interestingly, this approximated MEA estimator is equivalent to alignment methods that use a probability consistency transformation (PCT) (Do et al., 2005). The PCT was also used in ProbAlign (Roshan and Livesay, 2006) and PicXAA (Sahraeian and Yoon, 2010).
with γ → ∞ (see Section 2.4.2) is employed in several multiple alignment problems (Hamada et al., 2011a). Interestingly, this approximated MEA estimator is equivalent to alignment methods that use a probability consistency transformation (PCT) (Do et al., 2005). The PCT was also used in ProbAlign (Roshan and Livesay, 2006) and PicXAA (Sahraeian and Yoon, 2010).
In the (progressive) alignment procedure, pairwise alignment between two multiple alignments (Example 15) is employed. A representative MEA estimator has been utilized in several multiple alignment algorithms, including ProbCons (Do et al., 2005). Note that the final multiple alignment of these algorithms is obtained by using a DP algorithm.
On the other hand, the estimator used in AMAP (Schwartz and Pachter, 2007) is equivalent to the MEA estimator of the gain function 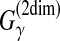 for constructing multiple alignments. The optimal alignment is computed through the stochastic approach of sequence annealing (SA).
for constructing multiple alignments. The optimal alignment is computed through the stochastic approach of sequence annealing (SA).
3.2.3. Local alignment of DNA/protein sequences
Frith et al. (2010) employed the MEA estimator of the gain function  (γ-centroid alignment; see also Section 3.2.1). It should be emphasized that the γ parameter is more important for local alignment than for global alignment, because it is used to adjust between sensitivity and PPV with respect to aligned columns in the local alignment. In fact, the authors showed that the γ-centroid alignment with an appropriate γ value greatly reduces the number of false-positive aligned bases in genome alignments compared to the conventional maximum likelihood/score alignment computed by the Viterbi algorithm.
(γ-centroid alignment; see also Section 3.2.1). It should be emphasized that the γ parameter is more important for local alignment than for global alignment, because it is used to adjust between sensitivity and PPV with respect to aligned columns in the local alignment. In fact, the authors showed that the γ-centroid alignment with an appropriate γ value greatly reduces the number of false-positive aligned bases in genome alignments compared to the conventional maximum likelihood/score alignment computed by the Viterbi algorithm.
3.3. Sequence analyses of RNAs
This field is one of the most successful applications of MEA estimation. The importance of sequence analysis of RNAs has increased due to the recent discovery of (functional) non-coding RNAs (Carninci and Hayashizaki, 2007; Mattick, 2005).
3.3.1. RNA secondary structure prediction
RNA secondary structure prediction (i.e., Problem 1 with 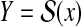 for an RNA sequence x) is a fundamental and classical problem in RNA information analysis.
for an RNA sequence x) is a fundamental and classical problem in RNA information analysis.
There exist several state-of-the-art probabilistic models for secondary structures of a given RNA sequence: (a) the McCaskill model (McCaskill, 1990) with experimentally determined energy parameters (Mathews et al., 1999), (b) the McCaskill model with Boltzmann likelihood (BL) parameters (determined by a machine learning method) (Andronescu et al., 2010, 2007), (c) the CONTRAfold model (Do et al., 2006a) based on the conditional random field (CRF) model, and (d) the stochastic context free grammar (SCFG) model (Dowell and Eddy, 2004). Those models can be utilized as the probability distribution on the predictive space 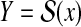 .
.
The estimator used in Sfold (Ding et al., 2005) can be considered as the MEG estimator of the gain function  (i.e., the centroid estimator) with the McCaskill model. In Sfold, the (optimal) secondary structure is computed by using a stochastic sampling technique instead of a DP algorithm. The authors showed that predictions using the centroid estimator contain fewer errors than conventional MFE predictions.
(i.e., the centroid estimator) with the McCaskill model. In Sfold, the (optimal) secondary structure is computed by using a stochastic sampling technique instead of a DP algorithm. The authors showed that predictions using the centroid estimator contain fewer errors than conventional MFE predictions.
CONTRAfold (Do et al., 2006a) utilized the MEA estimator of the gain function 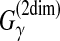 (Example 10) with the CONTRAfold model. This estimator is a pioneering work on MEA estimation in RNA secondary structure predictions and has been applied in a number of other studies of RNA sequence analysis (Lu et al., 2009; Lorenz and Clote, 2011). Computational experiments in Do et al. (2006a) showed that the MEA estimator of the gain function
(Example 10) with the CONTRAfold model. This estimator is a pioneering work on MEA estimation in RNA secondary structure predictions and has been applied in a number of other studies of RNA sequence analysis (Lu et al., 2009; Lorenz and Clote, 2011). Computational experiments in Do et al. (2006a) showed that the MEA estimator of the gain function 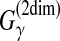 is superior to the ML estimator. More recent software, MaxExpect (Lu et al., 2009) and RNAlocopt (Lorenz and Clote, 2011), also utilized the MEA estimator of
is superior to the ML estimator. More recent software, MaxExpect (Lu et al., 2009) and RNAlocopt (Lorenz and Clote, 2011), also utilized the MEA estimator of 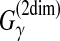 .
.
On the other hand, Hamada et al. (2009a) proved that the MEA estimator of the gain function 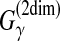 is not optimal for sensitivity, PPV, and MCC with respect to base pairs, which are the commonly used accuracy measures of secondary structure prediction. CentroidFold (Hamada et al., 2009a), therefore, utilized the MEA estimator of the gain function
is not optimal for sensitivity, PPV, and MCC with respect to base pairs, which are the commonly used accuracy measures of secondary structure prediction. CentroidFold (Hamada et al., 2009a), therefore, utilized the MEA estimator of the gain function  with various probabilistic models of secondary structures. Several computational experiments supported the theoretical result that the MEA estimator of the gain function
with various probabilistic models of secondary structures. Several computational experiments supported the theoretical result that the MEA estimator of the gain function  is better than both the MEA estimator of the gain function
is better than both the MEA estimator of the gain function 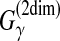 and ML estimators, when the probabilistic model of secondary structures is fixed.
and ML estimators, when the probabilistic model of secondary structures is fixed.
If we have the homologous sequences of the target RNA sequence (Example 16), the probability distribution of secondary structures of the target RNA sequence should be provided by the marginalized probability distribution of structural alignments between the target sequence and homologous sequences. An approximated MEA estimator with this probabilistic distribution has also been proposed (Hamada et al., 2009c). (The software implementing this approach is called CentroidHomfold.) In Hamada et al. (2009c, 2011c), the authors showed that the accuracy of secondary structure prediction was greatly improved by employing homologous sequence information.
The computation of most of the estimators described above is conducted by using a Nussinov-type DP algorithm (Nussinov et al., 1978) in O(∣x∣3) time:
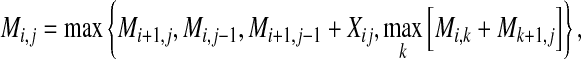 |
(9) |
where Mi,j stores the best score of the sub-sequence 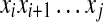 and Xij is one of the following options. Xij = (γ + 1)pij − 1 for the MEA estimator of the gain function
and Xij is one of the following options. Xij = (γ + 1)pij − 1 for the MEA estimator of the gain function  , and Xij = 2γpij − qi − qj for the MEA estimator of the gain function
, and Xij = 2γpij − qi − qj for the MEA estimator of the gain function 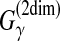 where qi is equal to loop probability of the position i. (Note that Xij = 1 when (xi, xj) form a base pair [e.g., Watson-Crick and Wobble base pairs] for Nussinov algorithm [Nussinov et al., 1978].)
where qi is equal to loop probability of the position i. (Note that Xij = 1 when (xi, xj) form a base pair [e.g., Watson-Crick and Wobble base pairs] for Nussinov algorithm [Nussinov et al., 1978].)
Although no efficient method has been reported to maximize expected Acc, where Acc is equal to MCC or F-score (i.e., the MEG estimator with the gain function G(Acc) [Eq. (5)]), Hamada et al. (2010) have recently proposed an approximate method that uses a pseudo expected MCC or F-score that is a quite good approximation to the expected MCC or F-score, respectively.
3.3.2. Common secondary structure prediction of multiple alignment of RNAs
The problem is to predict a secondary structure whose length is equal to the length of an alignment. This is often called a common or consensus secondary structure (Example 12). The RNAalifold model (Bernhart et al., 2008; Hofacker et al., 2002) and the Pfold model (Knudsen and Hein, 1999, 2003) directly provide a probability distribution p(θ∣D) for the common secondary structures of a given alignment D. Those probabilistic models are then used in the following MEA estimators.
The estimator used in the latest version of Pfold (Knudsen and Hein, 2003) is the MEA estimator of the gain function 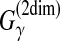 with the Pfold model. (The initial version of Pfold [Knudsen and Hein, 1999] utilized the ML-estimator with the Pfold model.)
with the Pfold model. (The initial version of Pfold [Knudsen and Hein, 1999] utilized the ML-estimator with the Pfold model.)
RNAalifold (Bernhart et al., 2008) employs the centroid estimator (the MEA estimator of the gain function  ) with the RNAalifold model as an option. (RNAalifold adopts the ML estimator with the RNAalifold model as the default.)
) with the RNAalifold model as an option. (RNAalifold adopts the ML estimator with the RNAalifold model as the default.)
McCaskill-MEA (Kiryu et al., 2007b) is deemed to be a representative MEA estimator (Section 2.4.1) of the gain function  with the McCaskill model (McCaskill, 1990). The authors showed experimentally that McCaskill-MEA was more robust to input alignment errors than RNAalifold and Pfold.
with the McCaskill model (McCaskill, 1990). The authors showed experimentally that McCaskill-MEA was more robust to input alignment errors than RNAalifold and Pfold.
The estimator used in PETfold (Seemann et al., 2008) can be considered as a representative MEA estimator of the gain function 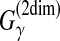 with a mixture of the distributions of the Pfold and McCaskill models. Using the mixed distribution enables us to consider both phylogenetic and free energy information.
with a mixture of the distributions of the Pfold and McCaskill models. Using the mixed distribution enables us to consider both phylogenetic and free energy information.
Recently, Hamada et al. (2011b, 2009a) also utilized a representative MEA estimator (Section 2.4.1) of the gain function  . They theoretically and experimentally showed that the estimator is superior to McCaskill-MEA, PETfold, RNAalifold, and Pfold with respect to commonly used evaluation methods of common secondary structure prediction. (The evaluation of a predicted common secondary structure is usually conducted by comparing every mapped secondary structure of the common secondary structure to the reference structure.) See Hamada et al. (2011b) for a classification of algorithms for common secondary structure prediction from the viewpoint of MEA.
. They theoretically and experimentally showed that the estimator is superior to McCaskill-MEA, PETfold, RNAalifold, and Pfold with respect to commonly used evaluation methods of common secondary structure prediction. (The evaluation of a predicted common secondary structure is usually conducted by comparing every mapped secondary structure of the common secondary structure to the reference structure.) See Hamada et al. (2011b) for a classification of algorithms for common secondary structure prediction from the viewpoint of MEA.
All the estimators described above can be computed by a DP algorithm similar to Eq. (9) (Hamada et al., 2011b).
3.3.3. Multiple alignment of RNAs
Because secondary structures are closely related to the functions of (functional) non-coding RNAs, the standard multiple alignment method (Section 3.2.2) is generally insufficient for aligning RNA sequences. Instead, structural alignment is appropriate where both consensus secondary structure and alignment are simultaneously estimated and optimized. However, it is known that the computational cost of structural alignment is high (Sankoff, 1985).
In Hamada et al. (2009b), the authors proposed a fast and accurate method for aligning multiple RNA sequences (CentroidAlign). Their estimator is equivalent to an approximate MEA estimator, which is an approximation of the MEA estimator of the gain function  with a probability distribution on usual alignments given by marginalizing the Sankoff model (cf. Example 18). Moreover, in CentroidAlign, a representative MEA estimator was also utilized when a progressive alignment is carried out. The authors showed that CentroidAlign is fast enough to deal with long RNA sequences and that it achieved favorable accuracy when compared to other algorithms.
with a probability distribution on usual alignments given by marginalizing the Sankoff model (cf. Example 18). Moreover, in CentroidAlign, a representative MEA estimator was also utilized when a progressive alignment is carried out. The authors showed that CentroidAlign is fast enough to deal with long RNA sequences and that it achieved favorable accuracy when compared to other algorithms.
3.3.4. Local alignment of RNAs
Tabei and Asai (2009) proposed a method (SCARNA-LM) for computing local alignment of RNAs. They utilized the MEA estimator with the gain function  for local alignment of RNA sequences. The probabilistic model for local alignments was based on the ProDA model (Phuong et al., 2006) (the authors incorporated secondary structure information into the model). They showed that their (MEA) estimator was better than the posterior decoding method used in ProDA (Phuong et al., 2006).
for local alignment of RNA sequences. The probabilistic model for local alignments was based on the ProDA model (Phuong et al., 2006) (the authors incorporated secondary structure information into the model). They showed that their (MEA) estimator was better than the posterior decoding method used in ProDA (Phuong et al., 2006).
3.3.5. RNA-RNA interaction prediction
RactIP (Kato et al., 2010) estimates RNA-RNA interactions, that is, joint secondary structures of two interacting RNA sequences. The method used in RactIP can be seen as an approximated MEA-based estimator with the gain function  . An approximated probability distribution of joint secondary structures of two sequences (the product of a probability distribution for secondary structures of the RNA sequence and that of the interactions between two RNA sequences) was utilized. In RactIP, the optimal prediction is solved by IP (Nemhauser and Wolsey, 1988). Although IP generally incurs a huge computational cost (NP-hard), RactIP runs very fast by using a (non-heuristic) threshold cut method (in which the base pairs whose posterior probability is less than a threshold computed from a given γ do not form base pairs) by virtue of the γ-centroid estimator. Note that a joint structure can be computed by using a DP algorithm although it incurs a relatively high computational cost (O(L5) ∼ O(L6), where L is the length of the joint structure).
. An approximated probability distribution of joint secondary structures of two sequences (the product of a probability distribution for secondary structures of the RNA sequence and that of the interactions between two RNA sequences) was utilized. In RactIP, the optimal prediction is solved by IP (Nemhauser and Wolsey, 1988). Although IP generally incurs a huge computational cost (NP-hard), RactIP runs very fast by using a (non-heuristic) threshold cut method (in which the base pairs whose posterior probability is less than a threshold computed from a given γ do not form base pairs) by virtue of the γ-centroid estimator. Note that a joint structure can be computed by using a DP algorithm although it incurs a relatively high computational cost (O(L5) ∼ O(L6), where L is the length of the joint structure).
Seemann et al. (2011) proposed an algorithm (PETcofold) to predict an RNA-RNA interaction between two multiple alignments of RNA sequences. The aim is to predict conserved interactions (and joint secondary structures) between the two multiple alignments, which is similar to the idea of predicting pairwise alignments and common secondary structure from a given multiple alignment of RNA sequences. Their algorithm can be seen as a representative MEA estimator with the gain function 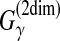 (Section 2.4.1). Like PETfold (used for common secondary structure prediction), they used a mixed distribution from the Pfold and McCaskill models in their estimator.
(Section 2.4.1). Like PETfold (used for common secondary structure prediction), they used a mixed distribution from the Pfold and McCaskill models in their estimator.
3.4. Phylogenetic tree (topology) estimation
Phylogenetic tree (topology) estimation is a classic and important problem in sequence analysis (Durbin et al., 1998). A phylogenetic tree for a given operational taxonomic unit (S) is represented as a binary vector with 2n−1 − n − 1 dimensions, where n is the number of units in S, based on partitions of S formed by cutting every edge in the tree. The topological accuracy measure for estimated trees is often based on the partitions (e.g., Robinson-Foulds [RF] measure [Robinson and Foulds, 1981]; Section 2.4 in Zhang et al. [2011]). A sampling algorithm can be used to estimate the partitioning probabilities (Metropolis et al., 1953).
Felsenstein (1985) proposed the X%-consensus tree, and the 50% consensus tree is equivalent to the tree of the centroid estimation (i.e., the centroid tree). Moreover, it is easily seen that the X%-consensus tree is equivalent to the MEG estimator with the gain function  (i.e., the γ-centroid tree) with γ = (100 − X)/X. The centroid tree is known to be suited to the topological distance (Robinson and Foulds, 1981), because it minimizes the expected topological distance. On the other hand, the γ-centroid tree is appropriate for sensitivity and PPV based on partitions of the tree (Dessimoz and Gil, 2010). However, although the γ-centroid tree with γ < 1 can be computed by selecting all the partitions (of operational taxonomic unit) whose probability is larger than 0.5 (Hamada et al., 2011a), no efficient method (such as a DP algorithm) has been reported for computing the γ-centroid tree for γ > 1.
(i.e., the γ-centroid tree) with γ = (100 − X)/X. The centroid tree is known to be suited to the topological distance (Robinson and Foulds, 1981), because it minimizes the expected topological distance. On the other hand, the γ-centroid tree is appropriate for sensitivity and PPV based on partitions of the tree (Dessimoz and Gil, 2010). However, although the γ-centroid tree with γ < 1 can be computed by selecting all the partitions (of operational taxonomic unit) whose probability is larger than 0.5 (Hamada et al., 2011a), no efficient method (such as a DP algorithm) has been reported for computing the γ-centroid tree for γ > 1.
4. Discussion
4.1. Avoiding point estimations
As described in Section 1, it is difficult to design reliable point estimators for Problem 1. Although point estimation based on the viewpoint of MEA provides a promising approach to the problem, solutions still have extremely low probability. It is, therefore, desirable to avoid point estimation if possible. When a pipeline is developed by combining several estimation algorithms, point estimation should be avoided in the middle of the pipeline even if the final prediction is a point estimation. For example, when a phylogenetic tree is estimated from several unaligned sequences, one standard approach is to predict a multiple alignment of the sequences and then estimate a phylogenetic tree from the predicted multiple alignment. This approach would not be appropriate because point estimation of multiple alignments is uncertain (i.e., results have low probability). Hence, if possible, a phylogenetic tree should be estimated considering all the possible multiple alignments. Although, in general, the computational cost might be increased by considering all the possible alignments, an approach similar to that in Section 2.4.2 is useful for reducing computational cost. It should be noted that the credibility limit of a point estimation (Webb-Robertson et al., 2008; Newberg and Lawrence, 2009) is also useful, because it is considered as a global measure of the estimation.
Another possible approach for avoiding the unreliability of point estimation for Problem 1 is to predict several suboptimal solutions (Steffen et al., 2006; Wuchty et al., 1999), giving up point estimations. It would also be useful to cluster solutions in the predictive space and estimate a solution for every cluster (Ding et al., 2004). Note that we can employ MEA-based estimators (e.g., with the gain function  ) for every cluster because a probability distribution on each cluster can be obtained by a stochastic sampling algorithm.
) for every cluster because a probability distribution on each cluster can be obtained by a stochastic sampling algorithm.
4.2. Posterior decoding methods (PDMs)
MEA/MEG estimators are considered as a special case of posterior decoding methods (PDM). In posterior decoding methods, several marginal probabilities are (heuristically) employed in order to obtain (decode) a final point estimation. Although it is often difficult to interpret PDMs from the viewpoint of MEG/MEA, we now list posterior decoding methods appearing in bioinformatics.
For sequence feature prediction (Section 3.1), Fariselli et al. (2005) proposed a posterior decoding method to predict the topology of all beta membranes proteins.
For pairwise/multiple alignment of biological sequences (Sections 3.2.1 and 3.2.2), ProDA (Phuong et al., 2006) produces local multiple alignment of protein sequences, in which a posterior decoding method with marginal probabilities for the unaligned (flanking) regions was employed. GRAPE (Lunter et al., 2008) utilizes a posterior decoding method similar to the MEA estimator of 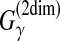 (the AMA estimator; Example 11). There are other posterior decoding methods for alignments: MORPH (Sinha and He, 2007), MSAProbs (Liu et al., 2010), and others (Koike et al., 2007; Gonnet and Lisacek, 2002).
(the AMA estimator; Example 11). There are other posterior decoding methods for alignments: MORPH (Sinha and He, 2007), MSAProbs (Liu et al., 2010), and others (Koike et al., 2007; Gonnet and Lisacek, 2002).
For RNA secondary structure prediction (Section 3.3.1), ProbKnot (Bellaousov and Mathews, 2010) uses a kind of posterior decoding method to predict secondary structure with pseudo-knots. It seems difficult to consider their estimator from the viewpoint of MEA, although the authors call their method “maximum expected accuracy.”
For (structural) RNA alignments (Section 3.3.3), PARTS (Harmanci et al., 2008), RAF (Do et al., 2008), and Murlet (Kiryu et al., 2007b) employ posterior decoding methods based on the Sankoff algorithm (Sankoff, 1985). R-coffee (Wilm et al., 2008), PicXAA-R (Sahraeian and Yoon, 2011), and MAFFT (Katoh and Toh, 2008) use a posterior decoding method similar to CentroidAlign (Hamada et al., 2009b) (Section 3.3.3) and do not produce structural alignment.
For (Bayesian) co-estimation of phylogeny and sequence alignment, Lunter et al. (2005) utilized a posterior decoding method.
4.3. Training probabilistic models from the viewpoint of MEA (MEA training)
In this review, we assumed that a probability distribution p(y∣D) on a predictive space Y is obtained beforehand in Problem 1. It is, however, important to design the probability distribution p(y∣D) itself. Distributions given by a probabilistic model such as an HMM or CRF contain a number of parameters. It would, therefore, be useful to train the parameters in the probability distribution with respect to the target accuracy measures. This type of training is called “MEA training” in general, and there have been several studies of MEA training in the field of machine learning: (Suzuki et al., 2006; Gross et al., 2007b; Jansche, 2007). There are, however, few studies applying MEA training to problems in bioinformatics (Gross et al., 2007a), and further studies in that area would be enlightening.
5. Conclusion
In this review, we have briefly described the concepts of MEA estimators, which are an alternative approach to conventional maximum likelihood or maximum score estimators. We then classified existing algorithms used in bioinformatics from the viewpoint of MEA. We believe that this review will be useful not only for users of the software mentioned in this review but also for developers wishing to design algorithms on the basis of MEA.
6. Appendix A
A.1. Accuracy measures based on TP, TN, FP, and FN
There are several measures for evaluating a prediction in estimation problems for which we have a reference (correct) prediction (Problem 1). The sensitivity (SEN), positive predictive value (PPV), Matthew's correlation coefficient (MCC), and F-score for a prediction are defined as follows:
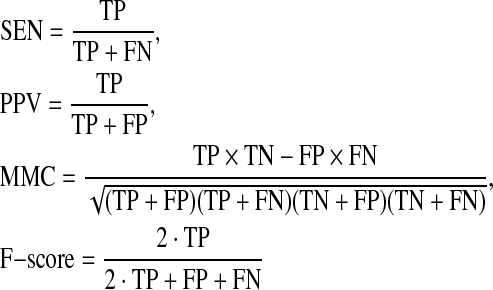 |
where TP, TN, FP, and FN are defined by
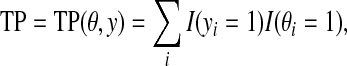 |
(10) |
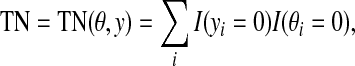 |
(11) |
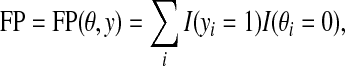 |
(12) |
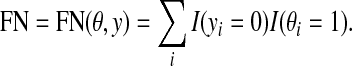 |
(13) |
where 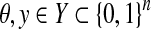 , θ is the reference and y is a prediction. It should be noted that these measures can be written as functions of TP, TN, FP, and FN. For other evaluation measures, see Baldi et al. (2000).
, θ is the reference and y is a prediction. It should be noted that these measures can be written as functions of TP, TN, FP, and FN. For other evaluation measures, see Baldi et al. (2000).
Acknowledgments
We are grateful to Dr. Martin C. Frith for commenting on the manuscript. This work was supported in part by a Grant-in-Aid for Scientific Research on Innovative Areas in Japan.
Disclosure Statement
No competing financial interests exist.
References
- Andersen E.S. Prediction and design of DNA and RNA structures. N. Biotechnol. 2010;27:184–193. doi: 10.1016/j.nbt.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Andronescu M. Condon A. Hoos H., et al. Efficient parameter estimation for RNA secondary structure prediction. Bioinformatics. 2007;23:19–28. doi: 10.1093/bioinformatics/btm223. [DOI] [PubMed] [Google Scholar]
- Andronescu M. Condon A. Hoos H.H., et al. Computational approaches for RNA energy parameter estimation. RNA. 2010;16:2304–2318. doi: 10.1261/rna.1950510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi P. Brunak S. Chauvin Y., et al. Assessing the accuracy of prediction algorithms for classification: an overview. Bioinformatics. 2000;16:412–424. doi: 10.1093/bioinformatics/16.5.412. [DOI] [PubMed] [Google Scholar]
- Bellaousov S. Mathews D.H. ProbKnot: fast prediction of RNA secondary structure including pseudoknots. RNA. 2010;16:1870–1880. doi: 10.1261/rna.2125310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhart S. Hofacker I. Will S., et al. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinform. 2008;9:474. doi: 10.1186/1471-2105-9-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P. Hayashizaki Y. Noncoding RNA transcription beyond annotated genes. Curr. Opin. Genet. Dev. 2007;17:139–144. doi: 10.1016/j.gde.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Carvalho L. Lawrence C. Centroid estimation in discrete high-dimensional spaces with applications in biology. Proc. Natl. Acad. Sci. USA. 2008;105:3209–3214. doi: 10.1073/pnas.0712329105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessimoz C. Gil M. Phylogenetic assessment of alignments reveals neglected tree signal in gaps. Genome Biol. 2010;11:R37. doi: 10.1186/gb-2010-11-4-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y. Chan C.Y. Lawrence C.E. Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res. 2004;32:135–141. doi: 10.1093/nar/gkh449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y. Chan C. Lawrence C. RNA secondary structure prediction by centroids in a Boltzmann weighted ensemble. RNA. 2005;11:1157–1166. doi: 10.1261/rna.2500605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do C. Mahabhashyam M. Brudno M., et al. ProbCons: probabilistic consistency-based multiple sequence alignment. Genome Res. 2005;15:330–340. doi: 10.1101/gr.2821705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do C. Woods D. Batzoglou S. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics. 2006a;22:e90–e98. doi: 10.1093/bioinformatics/btl246. [DOI] [PubMed] [Google Scholar]
- Do C.B. Gross S.S. Batzoglou S. Contralign: discriminative training for protein sequence alignment. Proc. RECOMB. 2006b;2006:160–174. [Google Scholar]
- Do C. Foo C. Batzoglou S. A max-margin model for efficient simultaneous alignment and folding of RNA sequences. Bioinformatics. 2008;24:i68–i76. doi: 10.1093/bioinformatics/btn177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell R. Eddy S. Evaluation of several lightweight stochastic context-free grammars for RNA secondary structure prediction. BMC Bioinform. 2004;5:71. doi: 10.1186/1471-2105-5-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin R. Eddy S. Krogh A., et al. Biological Sequence Analysis. Cambridge University Press; Cambridge, UK: 1998. [Google Scholar]
- Eddy S.R. What is dynamic programming? Nat. Biotechnol. 2004;22:909–910. doi: 10.1038/nbt0704-909. [DOI] [PubMed] [Google Scholar]
- Fariselli P. Martelli P. Casadio R. A new decoding algorithm for hidden Markov models improves the prediction of the topology of all-beta membrane proteins. BMC Bioinform. 2005;6(Suppl 4):S12. doi: 10.1186/1471-2105-6-S4-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Frith M.C. Hamada M. Horton P. Parameters for accurate genome alignment. BMC Bioinform. 2010;11:80. doi: 10.1186/1471-2105-11-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonnet P. Lisacek F. Probabilistic alignment of motifs with sequences. Bioinformatics. 2002;18:1091–1101. doi: 10.1093/bioinformatics/18.8.1091. [DOI] [PubMed] [Google Scholar]
- Gross S. Do C. Sirota M., et al. CONTRAST: a discriminative, phylogeny-free approach to multiple informant de novo gene prediction. Genome Biol. 2007a;8:R269. doi: 10.1186/gb-2007-8-12-r269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross S.S. Russakovsky O. Do C.B., et al. Training conditional random fields for maximum labelwise accuracy. In: Schölkopf B., editor; Platt J., editor; Hoffman T., editor. Advances in Neural Information Processing Systems 19. MIT Press; Cambridge, MA: 2007b. pp. 529–536. [Google Scholar]
- Hamada M. Kiryu H. Sato K., et al. Prediction of RNA secondary structure using generalized centroid estimators. Bioinformatics. 2009a;25:465–473. doi: 10.1093/bioinformatics/btn601. [DOI] [PubMed] [Google Scholar]
- Hamada M. Sato K. Kiryu H., et al. CentroidAlign: fast and accurate aligner for structured RNAs by maximizing expected sum-of-pairs score. Bioinformatics. 2009b;25:3236–3243. doi: 10.1093/bioinformatics/btp580. [DOI] [PubMed] [Google Scholar]
- Hamada M. Sato K. Kiryu H., et al. Predictions of RNA secondary structure by combining homologous sequence information. Bioinformatics. 2009c;25:i330–i338. doi: 10.1093/bioinformatics/btp228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada M. Sato K. Asai K. Prediction of RNA secondary structure by maximizing pseudo-expected accuracy. BMC Bioinform. 2010;11:586. doi: 10.1186/1471-2105-11-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada M. Kiryu H. Iwasaki W., et al. Generalized centroid estimators in bioinformatics. PLoS ONE. 2011a;6:e16450. doi: 10.1371/journal.pone.0016450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada M. Sato K. Asai K. Improving the accuracy of predicting secondary structure for aligned RNA sequences. Nucleic Acids Res. 2011b;39:393–402. doi: 10.1093/nar/gkq792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada M. Yamada K. Sato K., et al. CentroidHomfold-LAST: accurate prediction of RNA secondary structure using automatically collected homologous sequences. Nucleic Acids Res. 2011c;39:W100–W106. doi: 10.1093/nar/gkr290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmanci A. Sharma G. Mathews D. PARTS: probabilistic alignment for RNA joinT secondary structure prediction. Nucleic Acids Res. 2008;36:2406–2417. doi: 10.1093/nar/gkn043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmanci A.O. Sharma G. Mathews D.H. Stochastic sampling of the RNA structural alignment space. Nucleic Acids Res. 2009;37:4063–4075. doi: 10.1093/nar/gkp276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofacker I.L. Fekete M. Stadler P.F. Secondary structure prediction for aligned RNA sequences. J. Mol. Biol. 2002;319:1059–1066. doi: 10.1016/S0022-2836(02)00308-X. [DOI] [PubMed] [Google Scholar]
- Holmes I. Durbin R. Dynamic programming alignment accuracy. J. Comput. Biol. 1998;5:493–504. doi: 10.1089/cmb.1998.5.493. [DOI] [PubMed] [Google Scholar]
- Jansche M. A maximum expected utility framework for binary sequence labeling. Proc. ACL. 2007:736–743. [Google Scholar]
- Kall L. Krogh A. Sonnhammer E.L. An HMM posterior decoder for sequence feature prediction that includes homology information. Bioinformatics. 2005;21(Suppl 1):i251–i257. doi: 10.1093/bioinformatics/bti1014. [DOI] [PubMed] [Google Scholar]
- Kato Y. Sato K. Hamada M., et al. RactIP: fast and accurate prediction of RNA-RNA interaction using integer programming. Bioinformatics. 2010;26:i460–i466. doi: 10.1093/bioinformatics/btq372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K. Toh H. Improved accuracy of multiple ncRNA alignment by incorporating structural information into a MAFFT-based framework. BMC Bioinform. 2008;9:212. doi: 10.1186/1471-2105-9-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu H. Kin T. Asai K. Robust prediction of consensus secondary structures using averaged base pairing probability matrices. Bioinformatics. 2007a;23:434–441. doi: 10.1093/bioinformatics/btl636. [DOI] [PubMed] [Google Scholar]
- Kiryu H. Tabei Y. Kin T., et al. Murlet: a practical multiple alignment tool for structural RNA sequences. Bioinformatics. 2007b;23:1588–1598. doi: 10.1093/bioinformatics/btm146. [DOI] [PubMed] [Google Scholar]
- Knudsen B. Hein J. RNA secondary structure prediction using stochastic context-free grammars and evolutionary history. Bioinformatics. 1999;15:446–454. doi: 10.1093/bioinformatics/15.6.446. [DOI] [PubMed] [Google Scholar]
- Knudsen B. Hein J. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res. 2003;31:3423–3428. doi: 10.1093/nar/gkg614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike R. Kinoshita K. Kidera A. Probabilistic alignment detects remote homology in a pair of protein sequences without homologous sequence information. Proteins. 2007;66:655–663. doi: 10.1002/prot.21240. [DOI] [PubMed] [Google Scholar]
- Liu Y. Schmidt B. Maskell D.L. MSAProbs: multiple sequence alignment based on pair hidden Markov models and partition function posterior probabilities. Bioinformatics. 2010;26:1958–1964. doi: 10.1093/bioinformatics/btq338. [DOI] [PubMed] [Google Scholar]
- Lorenz W.A. Clote P. Computing the partition function for kinetically trapped RNA secondary structures. PLoS ONE. 2011;6:e16178. doi: 10.1371/journal.pone.0016178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z.J. Gloor J.W. Mathews D.H. Improved RNA secondary structure prediction by maximizing expected pair accuracy. RNA. 2009;15:1805–1813. doi: 10.1261/rna.1643609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G. Miklos I. Drummond A., et al. Bayesian coestimation of phylogeny and sequence alignment. BMC Bioinform. 2005;6:83. doi: 10.1186/1471-2105-6-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G. Rocco A. Mimouni N., et al. Uncertainty in homology inferences: assessing and improving genomic sequence alignment. Genome Res. 2008;18:298–309. doi: 10.1101/gr.6725608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews D.H. Sabina J. Zuker M., et al. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 1999;288:911–940. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- Mathews D. Disney M. Childs J., et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc. Natl. Acad. Sci. USA. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattick J. The functional genomics of noncoding RNA. Science. 2005;309:1527–1528. doi: 10.1126/science.1117806. [DOI] [PubMed] [Google Scholar]
- McCaskill J.S. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29:1105–1119. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- Metropolis N. Rosenbluth A. Teller M., et al. Equations of state calculations by fast computing machine. J. Chem. Phys. 1953;21:1087–1091. [Google Scholar]
- Miyazawa S. A reliable sequence alignment method based on probabilities of residue correspondences. Protein Eng. 1995;8:999–1009. doi: 10.1093/protein/8.10.999. [DOI] [PubMed] [Google Scholar]
- Nánási M. Vinař T. Brejová B. The highest expected reward decoding for hmms with application to recombination detection. Proc. CPM. 2010;10:164–176. [Google Scholar]
- Nemhauser G.L. Wolsey L.A. Integer and Combinatorial Optimization. Wiley-Interscience; New York: 1988. [Google Scholar]
- Newberg L.A. Lawrence C.E. Exact calculation of distributions on integers, with application to sequence alignment. J. Comput. Biol. 2009;16:1–18. doi: 10.1089/cmb.2008.0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussinov R. Pieczenk G. Griggs J., et al. Algorithms for loop matchings. SIAM J. Appl. Math. 1978;35:68–82. [Google Scholar]
- Pei J. Multiple protein sequence alignment. Curr. Opin. Struct. Biol. 2008;18:382–386. doi: 10.1016/j.sbi.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Phuong T.M. Do C.B. Edgar R.C., et al. Multiple alignment of protein sequences with repeats and rearrangements. Nucleic Acids Res. 2006;34:5932–5942. doi: 10.1093/nar/gkl511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E. Pesole G. Computational methods for ab initio and comparative gene finding. Methods Mol. Biol. 2010;609:269–284. doi: 10.1007/978-1-60327-241-4_16. [DOI] [PubMed] [Google Scholar]
- Pirovano W. Heringa J. Multiple sequence alignment. Methods Mol. Biol. 2008;452:143–161. doi: 10.1007/978-1-60327-159-2_7. [DOI] [PubMed] [Google Scholar]
- Robinson D.F. Foulds L.R. Comparison of phylogenetic trees. Math. Biosci. 1981;53:131–147. [Google Scholar]
- Roshan U. Livesay D. Probalign: multiple sequence alignment using partition function posterior probabilities. Bioinformatics. 2006;22:2715–2721. doi: 10.1093/bioinformatics/btl472. [DOI] [PubMed] [Google Scholar]
- Sahraeian S.M. Yoon B.J. PicXAA: greedy probabilistic construction of maximum expected accuracy alignment of multiple sequences. Nucleic Acids Res. 2010;38:4917–4928. doi: 10.1093/nar/gkq255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahraeian S.M. Yoon B.J. PicXAA-R: efficient structural alignment of multiple RNA sequences using a greedy approach. BMC Bioinform. 2011;12(Suppl 1):S38. doi: 10.1186/1471-2105-12-S1-S38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankoff D. Simultaneous solution of the RNA folding alignment and protosequence problems. SIAM J. Appl. Math. 1985;45:810–825. [Google Scholar]
- Sato K. Kato Y. Hamada M., et al. IPknot: fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics. 2011;27:i85–i93. doi: 10.1093/bioinformatics/btr215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz A.K. Zhang M. Leitner T., et al. A jumping profile Hidden Markov Model and applications to recombination sites in HIV and HCV genomes. BMC Bioinform. 2006;7:265. doi: 10.1186/1471-2105-7-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz A.S. University of California; Berkeley: 2007. Posterior decoding methods for optimization and accuracy control of multiple alignments [Ph.D. dissertation] [Google Scholar]
- Schwartz A. Pachter L. Multiple alignment by sequence annealing. Bioinformatics. 2007;23:e24–e29. doi: 10.1093/bioinformatics/btl311. [DOI] [PubMed] [Google Scholar]
- Schwartz A.S. Myers E.W. Pachter L. Alignment metric accuracy (submitted) 2005.
- Seemann S. Gorodkin J. Backofen R. Unifying evolutionary and thermodynamic information for RNA folding of multiple alignments. Nucleic Acids Res. 2008;36:6355–6362. doi: 10.1093/nar/gkn544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann S.E. Richter A.S. Gesell T., et al. PETcofold: predicting conserved interactions and structures of two multiple alignments of RNA sequences. Bioinformatics. 2011;27:211–219. doi: 10.1093/bioinformatics/btq634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S. He X. MORPH: probabilistic alignment combined with hidden Markov models of cis-regulatory modules. PLoS Comput. Biol. 2007;3:e216. doi: 10.1371/journal.pcbi.0030216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrabanek L. Saini H. K. Bader G. D., et al. Computational prediction of protein-protein interactions. Mol. Biotechnol. 2008;38:1–17. doi: 10.1007/s12033-007-0069-2. [DOI] [PubMed] [Google Scholar]
- Smith T. F. Waterman M. S. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Steffen P. Voss B. Rehmsmeier M., et al. RNAshapes: an integrated RNA analysis package based on abstract shapes. Bioinformatics. 2006;22:500–503. doi: 10.1093/bioinformatics/btk010. [DOI] [PubMed] [Google Scholar]
- Suzuki J. McDermott E. Isozaki H. Training conditional random fields with multivariate evaluation measures. Proc. ACL. 2006:217–224. [Google Scholar]
- Tabei Y. Asai K. A local multiple alignment method for detection of non-coding RNA sequences. Bioinformatics. 2009;25:1498–1505. doi: 10.1093/bioinformatics/btp261. [DOI] [PubMed] [Google Scholar]
- Webb-Robertson B. J. McCue L. A. Lawrence C. E. Measuring global credibility with application to local sequence alignment. PLoS Comput. Biol. 2008;4:e1000077. doi: 10.1371/journal.pcbi.1000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D. Alpert L.V. Lawrence C.E. RNAG: A new GIBBS sampler for predicting RNA secondary structure for unaligned sequences. Bioinformatics. 2011;27:2486–2493. doi: 10.1093/bioinformatics/btr421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan S. Inferring trees. Methods Mol. Biol. 2008;452:287–309. doi: 10.1007/978-1-60327-159-2_14. [DOI] [PubMed] [Google Scholar]
- Wilm A. Higgins D. Notredame C. R-Coffee: a method for multiple alignment of non-coding RNA. Nucleic Acids Res. 2008;36:e52. doi: 10.1093/nar/gkn174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuchty S. Fontana W. Hofacker I. L., et al. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers. 1999;49:145–165. doi: 10.1002/(SICI)1097-0282(199902)49:2<145::AID-BIP4>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Yamada S. Osamu G. Hayato Y. Improvement in speed and accuracy of multiple sequence alignment program prime. IPSJ Trans. Bioinform. (TBIO) 2008;1:2–12. [Google Scholar]
- Zhang S.B. Zhou S.Y. He J.G., et al. Phylogeny inference based on spectral graph clustering. J. Comput. Biol. 2011;18:627–637. doi: 10.1089/cmb.2009.0028. [DOI] [PubMed] [Google Scholar]