

Abstract
Aims: Hydrogen sulfide (H2S) is a signaling molecule, which influences many physiological processes. While H2S is produced and degraded in many cell types, the kinetics of its turnover in different tissues has not been reported. In this study, we have assessed the rates of H2S production in murine liver, kidney, and brain homogenates at pH 7.4, 37°C, and at physiologically relevant cysteine concentrations. We have also studied the kinetics of H2S clearance by liver, kidney, and brain homogenates under aerobic and anaerobic conditions. Results: We find that the rate of H2S production by these tissue homogenates is considerably higher than background rates observed in the absence of exogenous substrates. An exponential decay of H2S with time is observed and, as expected, is significantly faster under aerobic conditions. The half-life for H2S under aerobic conditions is 2.0, 2.8, and 10.0 min with liver, kidney, and brain homogenate, respectively. Western-blot analysis of the sulfur dioxygenase, ETHE1, involved in H2S catabolism, demonstrates higher steady-state protein levels in liver and kidney versus brain. Innovation: By combining experimental and simulation approaches, we demonstrate high rates of tissue H2S turnover and provide estimates of steady-state H2S levels. Conclusion: Our study reveals that tissues maintain a high metabolic flux of sulfur through H2S, providing a rationale for how H2S levels can be rapidly regulated. Antioxid. Redox Signal. 17, 22–31.
Introduction
H2S is an endogenously produced signaling molecule that modulates varied physiological processes. It induces relaxation of vascular smooth muscle cells by opening ATP-sensitive potassium channels (47), enhances NMDA receptor-sensitivity to neurotransmitters and facilitates the induction of long-term potentiation in hippocampal neurons (1), protects from myocardial ischemia-reperfusion injury (9), inhibits cell proliferation (46), and reduces mitochondrial respiration by reversibly inhibiting cytochrome c oxidase and induces a suspended animation-like metabolic state (3, 4). H2S is a potent inhibitor of cytochrome c oxidase and therefore, toxic for aerobic life at high concentrations (4). Because of its signaling role and toxicity, tissue H2S levels must be tightly regulated. However, the mechanisms underlying H2S homeostasis (i.e., its production and clearance) are not well understood.
H2S is primarily generated by two PLP-dependent enzymes in the transsulfuration pathway (Fig. 1), cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE), which employ cysteine and homocysteine as substrates (20, 33). A third enzyme proposed to contribute to H2S production, is 3-mercaptopyruvate sulfur transferase (MST). However, this reaction requires a reducing agent to release H2S from the enzyme-bound persulfide formed from the substrate, mercaptopyruvate (20, 29).
FIG. 1.

Scheme showing H2S metabolism in mammalian cells. CAT, cysteine aminotransferase.
Innovation.
The technical challenges associated with H2S measurement have led to widely varying reports of steady-state concentrations spanning a range over five orders of magnitude. Similarly, measures of H2S biogenesis and removal rates, which determine the steady-state concentrations in tissues, have yielded widely varying values. For H2S to exert a signaling role, mechanisms for rapid regulation of its concentration in response to environmental/cellular cues (e.g., oxygen concentration) must exist. However, the virtual absence of kinetic data has limited insights into H2S turnover in tissues, and therefore, strategies for its regulation. We demonstrate that robust rates of H2S production in murine liver, kidney, and brain at physiologically relevant cysteine concentrations and pH. The high H2S generating flux is countered by rapid H2S clearance under aerobic conditions accounting for very low steady-state tissue H2S levels. Consequently, even small deviations in the rates of H2S production and/or clearance are expected to lead to rapid and several fold changes in H2S levels. The kinetics of H2S metabolism thus provides for fast and effective regulation of H2S levels in tissues, an essential feature for a signaling molecule.
H2S catabolism occurs via a mitochondrial sulfide oxidation pathway (Fig. 1) and is coupled to energy production (15, 17). In the first step, H2S is oxidized by a mitochondrial membrane-bound flavoprotein, sulfide quinone oxidoreductase (SQR), which forms a protein-bound persulfide while the electrons are transferred to ubiquinone. In the second step, a sulfur dioxygenase (ETHE1) oxidizes SQR-bound sulfane sulfur to sulfite, which is subsequently converted to thiosulfate by the transfer of a second persulfide equivalent catalyzed by the sulfur transferase, rhodhanese. In some organisms, thiosulfate is metabolized to sulfate by thiosulfate reductase (27). Alternatively, sulfite can be oxidized to sulfate by sulfite oxidase.
In tissues, an unknown fraction of H2S exists in the bound state, generated by nucleophilic attack of the hydrosulfide anion on cysteine disulfide or sulfenic acid, forming cysteine persulfide. While the resulting persulfides are stable under oxidizing conditions, in the reducing cellular milieu, they need to be sequestered from disulfide exchange reactions initiated by proximal cysteine residues on proteins or by exogenous thiolates (e.g., cysteine or glutathione) or antioxidant enzymes (e.g., thioredoxin). In brain, protein-bound sulfide constitutes a significant proportion of the sulfide pool that may: 1) serve to sequester H2S, thereby protecting cells from its toxic effect, and 2) function as a stored H2S pool that can be mobilized to generate free H2S upon demand (19).
An additional sink for H2S in cells is heme-containing proteins. In invertebrates that live in sulfide-rich environments, globins bind H2S with high affinity and carry bound sulfide to symbiotic sulfide-oxidizing bacteria, thereby avoiding toxicity associated with high H2S concentrations (41). Depending on the polarity of the hemoglobin active site, heme-bound sulfide can either dissociate reversibly (from nonpolar active sites), or reduce ferric to ferrous heme and release a sulfide radical (HS•, from polar active sites) (32). However, the affinity of mammalian heme proteins for sulfide is low (30, 31), making it unlikely that globins represent a significant sink for H2S.
Steady state H2S levels in tissues is governed by the rates of its production and removal and are very low; 14–17 nM in liver and brain (12). The literature on H2S production rates by mammalian tissues is confusing since the data have been obtained under a variety of conditions including nonphysiological pH (12, 25), high substrate concentrations (8, 12, 21, 25), or in samples fixed with 10% trichloracetic acid (1, 47), which releases sulfide from iron–sulfur cluster-containing proteins, leading to overestimation. The rate of H2S production by tissue extracts decreases dramatically under aerobic versus anaerobic conditions even at a very high concentration of cysteine (10 mM), indicating efficient oxygen-dependent H2S consumption (12).
Homocysteine concentration in mammalian tissues is estimated to be ∼1–10 μM (18), while the concentration of cysteine is ∼100 μM in liver and brain, and up to 1.0 mM in kidney (37, 40). Kinetic data and simulation analyses indicate that at physiologically relevant substrate concentrations (10 μM homocysteine and 100 μM cysteine), CBS generates H2S mainly via condensation of cysteine and homocysteine while CSE generates H2S primarily via α,β-replacement of cysteine (7, 34). Since CSE is significantly more abundant than CBS in murine liver and kidney (21), one can conclude that cysteine is the primary substrate for H2S production in these tissues.
H2S oxidation capacity has been reported for different tissues and cells (8, 13, 15, 22, 23, 42). In some of these studies, H2S degradation was monitored indirectly [i.e., by formation of thiosulfate and sulfate (13) or by oxygen consumption (15, 23)]. Reports on direct measurements of H2S degradation are limited to cultured rat aorta smooth muscle cells (8), rat aorta tissue (22, 42), and blood from several vertebrate species (42). Thus, to our knowledge, direct measurements of the kinetics of H2S clearance in several major mammalian tissues (e.g., liver, kidney, and brain) are not available. The highest H2S oxidation rate was found in colon, which needs to detoxify large quantities of H2S produced by colonic bacteria rapidly. By comparison, H2S oxidation activity of liver, muscle, and erythrocytes is ∼4, 20, and 60-fold lower, respectively, than colon (13).
In this study, we have compared the kinetics of H2S production and clearance by murine liver, kidney, and brain homogenates at physiologically relevant substrate concentrations. Our study provides evidence for high rates of H2S turnover and provides estimates of steady-state H2S levels in different tissues.
Results
Dependence of H2S production rates on cysteine concentration and at pH 5.8 versus 7.4
A pH of 5.8 was previously used for tissue H2S analysis (12, 24) since at this pH, the equilibrium between the ionized (HS-) and gaseous (H2S) forms is shifted to gas, facilitating H2S quantitation. However, the effect of this relatively low pH on the balance between H2S production and clearance rates is not known and could potentially perturb the tissue H2S production rates that were measured. We therefore examined the effect of pH 5.8 versus 7.4 on H2S production and clearance in murine tissues. H2S was analyzed by gas chromatography with a sulfur chemiluminescence detector, which provides very high sensitivity and reliable detection of H2S even in very dilute samples (12).
Detectable rates of H2S production were observed at physiologically relevant cysteine concentrations (0.1–1.0 mM) at pH 7.4 using mouse liver, kidney, and brain homogenates (Fig. 2) and H2S accumulation is linear with time in this concentration range (not shown). At any cysteine concentration, the rate of H2S production by liver homogenate is significantly higher than for kidney or brain. The rates of H2S production are comparable for kidney and brain at cysteine concentrations between 0.1 and 1.0 mM, but are higher in kidney at 5.0 mM cysteine (Figs. 2B and 2C, insets). In all cases, a low rate of H2S production was observed even in the absence of exogenous cysteine and probably resulted from the presence of low endogenous cysteine in the tissue samples. The background rate of H2S generation was 21±14 (n=7), 13±2 (n=3), and 12±2 (n=5) μmoles h−1 kg tissue−1 in liver, kidney, and brain, respectively. In liver and brain, even at 0.1 mM cysteine, the rate of H2S production was significantly higher compared to the background rate (p<0.002) and values of 484±271 (n=7) and 29±7 (n=4) μmoles h−1 kg tissue−1 respectively, were obtained. In kidney, a statistically significant (p<0.03) increase in the H2S production over the background rate was observed only at ≥0.5 mM cysteine; a value of 104±44 (n=3) μmoles h−1 kg tissue−1 was obtained at 0.5 mM cysteine.
FIG. 2.

Dependence of H2S production rates on cysteine concentrations. H2S generation was monitored at 37°C in 100 mM HEPES buffer, pH 7.4 (closed circles) or 100 mM sodium phosphate buffer, pH 5.8 (open circles) using murine liver (A), kidney (B), and (C) brain homogenates. Insets show the dependence of H2S production over an extended cysteine concentration range. Data represent the mean±SD of 3–7 independent experiments. In liver and brain homogenates at pH 7.4, the rates of H2S production were significantly higher (p<0.002) at all concentrations of cysteine compared to the background rates in the absence of exogenous cysteine. In kidney homogenate at cysteine concentrations ≥0.5 mM, the rates of H2S production were significantly higher (p<0.03) compared to background rates, while at 0.2 mM cysteine, the rate of H2S production was not significantly different from the background rate (p>0.07).
For comparison, the rate of H2S production was tested at pH 5.8 with liver homogenate and found to be significantly lower and in fact, negligible at physiologically relevant cysteine concentrations. The background rate of H2S formation was 9.0±1.2 (n=5) μmoles h−1 kg tissue−1. H2S production rates of 21.0±6.3 μmoles h−1 kg tissue−1 (n=3) and 56.2±26.3 μmoles h−1 kg tissue−1 (n=5) were obtained at 0.5 and 1.0 mM cysteine concentrations, respectively. We used 100 mM HEPES buffer for the pH 7.4 measurements since our previous in vitro kinetic studies on H2S production by purified CBS and CSE were conducted under those conditions (7, 34). To ensure that the low H2S production rate at pH 5.8 was not due to a difference in buffers employed at the two pH values, the analysis was repeated at pH 7.4 with 100 mM potassium phosphate buffer. The rate of H2S production was found to be independent of buffer but dependent on pH (not shown).
H2S clearance in the presence of oxidants
In control experiments (i.e., in buffer lacking tissue homogenate), we found that the concentration of H2S decreased relatively slowly. Surprisingly, the kinetics of H2S disappearance in buffer under anaerobic and aerobic conditions were comparable over a 20-min time period (Fig. 3). Addition of 1.0 mM glutathione disulfide (GSSG) or 0.5 mM cystine to the buffer did not alter the kinetics of H2S disappearance (Fig. 3). Interestingly, significant activation of H2S degradation was observed with buffer containing 1.0 mM cystamine (Fig. 3).
FIG. 3.

Kinetics of H2S disappearance in the presence of small-molecule oxidants. The kinetics of H2S disappearance was monitored in control samples containing 500 μl of 100 mM HEPES buffer, pH 7.4. The sample contained in a total volume of 20 ml, 10 ml of H2S (40 ppm) in nitrogen and either 9.5 ml of nitrogen (filled symbols) or 9.5 ml of air (open symbols). The rate of disappearance of H2S was monitored at 25°C in presence of either buffer alone (circles) or buffer containing 1 mM GSSG (triangles), or 0.5 mM cystine (inverted triangles), or 1 mM cystamine (squares). The lines represent exponential fits to the data sets. The data represent means±SD of 3–4 independent experiments.
H2S clearance in the presence of murine tissues
In the presence of tissue homogenate, H2S disappeared rapidly under aerobic conditions with the kinetics being fast in liver and kidney and slower in brain (Fig. 4). As expected, the exponential decay in H2S levels occurred more rapidly under aerobic versus anaerobic conditions. Under aerobic conditions, H2S levels tended to zero with half-lives of 2.0, 2.8, and 10.0 min for liver, kidney, and brain homogenates, respectively. The kinetics of H2S clearance were not affected by the presence of a metal chelator [50 μM DTPA (diethylenetriamine pentaacetic acid)] in either 100 mM HEPES, pH 7.4 or 100 mM sodium phosphate, pH 5.8 (Supplementary Fig. S1; supplementary data are available online at www.liebertonline.com/ars).
FIG. 4.

Kinetics of H2S disappearance in the presence of murine tissue homogenates. The kinetics of H2S disappearance was monitored at 25°C in a total volume of 20 ml containing 500 μl murine liver (A), kidney (B), and brain (C) homogenates, 10 ml of H2S (40 ppm) in nitrogen and 9.5 ml of nitrogen (filled symbols) or air (open symbols). With the liver samples, the kinetics of H2S disappearance was measured at pH 7.4 (circles) and pH 5.8 (triangles). The lines represent exponential fits to the experimental data sets. The data represent means±SD of 5–10 independent experiments.
Under anaerobic conditions, the exponential fits yielded non-zero values for final H2S concentrations. In kidney and brain samples, the final H2S concentrations were similar and relatively high (64%–68% of the original value with half-lives corresponding to 1.8 and 2.9 min, respectively), but lower in liver samples (16% with half-life of 5.4 min). A high degree of variability was consistently observed with liver, for reasons that are presently unclear. The effect of pH was studied with liver extract (Fig. 4A). In comparison to pH 7.4, the rate of H2S disappearance was only slightly lower at pH 5.8 and this was not due to the difference between HEPES versus phosphate buffer used at pH 7.4 versus 5.8, respectively (data not shown).
To exclude oxygen more rigorously, the kinetics of H2S removal by liver and kidney samples were determined under strictly anaerobic conditions inside an OMNI-LAB anaerobic chamber (0.2–0.5 ppm O2 concentration). The slow disappearance of H2S with buffer was again observed under these conditions (Table 1) as seen previously (Fig. 3). With kidney homogenate, H2S levels were ∼20% higher than with buffer alone. In contrast, H2S levels decreased significantly in the presence of liver homogenates compared to control samples, although the decrease was not as large as under standard anaerobic conditions. Flushing liver homogenates with CO for 10 min did not alter H2S levels, while addition of 2 mM dithiothreitol slightly decreased the rate of H2S removal under strictly anaerobic conditions.
Table 1.
Disappearance of H2S Under Aerobic and Anaerobic Conditions
| Sample/conditions | Percent H2S left after 20 min incubation (mean±SD) | Number of experiments | Statistical significance |
|---|---|---|---|
| Aerobic buffer | 84.8±5.1 | 6 | The difference between all the buffer data is not significant p>0.1 |
| Anaerobic buffer | 81.4±2.8 | 7 | |
| “Strictly” anaerobic buffer | 82.7±2.2 | 7 | |
| “Strictly” anaerobic kidney homogenate | 100.1±7.9 | 3 | The difference between kidney and all the buffer data is significant p<0.01 |
| “Strictly” anaerobic liver homogenate | 51.8±21.5 | 6 | The difference between all the liver data is not significant p>0.1 The difference between all the liver and buffer data is significant p<0.02 |
| “Strictly” anaerobic liver homogenate flushed with CO | 63.1±12.8 | 3 | |
| “Strictly” anaerobic liver homogenate+2 mM DTT | 71.0±8.9 | 4 |
H2S levels were monitored after 20 min at 25°C, in the following samples: aerobic buffer, anaerobic buffer, buffer in an anaerobic chamber (“strictly” anaerobic), kidney homogenate in anaerobic chamber, liver homogenate in anaerobic chamber, liver homogenate flushed with CO in anaerobic chamber, liver homogenate+2 mM DTT in anaerobic chamber.
H2S clearance by fresh versus frozen liver homogenates
To assess the role of mitochondrial integrity on H2S clearance, the kinetics of aerobic H2S degradation was compared in homogenates prepared from freshly-isolated versus snap-frozen liver. Exponential decay kinetics were observed in both samples and t1/2 values of 0.8 and 1.5 min were obtained for homogenates prepared from fresh and frozen liver, respectively (Supplementary Fig. S2). However, if homogenate from freshly-isolated liver was stored on ice, H2S decay kinetics slowed with time, and following a 1 h incubation, coincided with the kinetics observed with homogenate from frozen liver (Fig. S2). In contrast, the kinetics of H2S degradation obtained with homogenates prepared from frozen liver were stable over a 1–2 h period.
Expression of ETHE1 protein in murine tissues
Western blot analysis revealed two bands that cross react with purified anti-human ETHE1 antibody in murine liver and kidney samples, while a laddering of bands was observed in brain samples (Fig. 5). The lowest band has a molecular mass of ∼25 kDa and corresponds to purified recombinant human ETHE1 lacking the mitochondrial targeting sequence. The intensity of this band in liver and kidney samples is approximately equal, but it is barely visible in the brain sample. The upper band has a molecular mass of ∼37 kDa and is more intense than the lower band in the brain sample. It is not known whether this band represent a cross-reacting protein or a modified form of ETHE1.
FIG. 5.

Western blot analysis of ETHE1 expression in murine liver, kidney and brain. Tissue extracts (100 μg of protein/lane) was separated on a denaturing 12% polyacrylamide gel and detected with anti-human ETHE1 antibody as described under Methods. A sample of purified recombinant human ETHE1 (5 ng) lacking the mitochondrial leader sequence was used as a positive control. The positions of molecular weight standards at 25, 37, and 50 kDa are indicated.
Discussion
The study of H2S production and clearance kinetics is important since the balance between these rates determine steady-state H2S levels in tissues. Furthermore, regulation of either one of these processes, or both, represents a mechanism for modulating H2S levels. The rate of H2S production in different mammalian tissues remains a subject of discussion. Very low rates of H2S production were reported for liver and brain tissue even at unphysiologically high (10 mM) cysteine concentrations (12). In fact, production of H2S by liver homogenates could not be detected below 10 mM cysteine in these studies (12, 25). However, these experiments were conducted at pH 5.8 (to enhance protonation of the sulfide anion to generate H2S) and/or under aerobic conditions (12, 25). In our study, we report that the rate of H2S production by mammalian tissues is significantly lower at pH 5.8 versus 7.4. This is expected since the pH optima of enzymes responsible for H2S biogenesis are reported to be 8.5 for CBS (11), 8.2 for CSE (35), and 9.0–10.0 for MST (38, 44). Furthermore, since the mitochondrial pathway for H2S catabolism is oxygen dependent, aerobic conditions are expected to increase H2S consumption competing with the rate of H2S production.
Significant rates of H2S production by liver and brain homogenates were detected under anaerobic conditions at pH 7.4 with 0.1 mM cysteine, which represents a physiologically relevant concentration for these tissues (37, 40). In kidney, where steady-state cysteine levels can be as high as 1.0 mM (36, 40), H2S production was detected only at ≥0.5 mM cysteine at pH 7.4. At pH 5.8, measurable H2S production by liver homogenate was detected only at ≥0.5 mM cysteine. The low H2S accumulation observed in the absence of exogenous cysteine likely derives from endogenous cysteine present in samples. During preparation of homogenates, the tissues were diluted 10-fold (mass/volume) with buffer and the resulting cysteine concentrations were expected to be ∼0.01 mM in liver and brain samples and 0.1 mM in the kidney sample. Release of H2S from the bound sulfide pool is unlikely to occur at pH 7.4 and in the absence of reductants.
H2S production rates were also measured under anaerobic conditions, where H2S consumption should be suppressed. However, even under anaerobic conditions, significant H2S clearance was observed (Fig. 4) and H2S concentrations decreased ∼30%–40% in kidney and brain homogenates over the observation time course, and even further in liver homogenates (Fig. 4). Thus, the rates of H2S production obtained in our experiments represent an underestimation.
One mechanism for regulating H2S levels is via sulfide catabolism, which is catalyzed by an oxygen-dependent mitochondrial pathway (Fig. 1). Under aerobic conditions and in the absence of tissue samples, oxidation of H2S in buffer was negligible over a 20-min period (Fig. 3). Since the rate of H2S depletion in the absence of tissue is similar under aerobic and anaerobic conditions, we attribute the observed loss to leakage from the syringe and/or to interactions of sulfide with the plastic ware. In principle, depletion of H2S in the presence of tissue homogenates can occur by oxidation or via reaction with disulfides such as GSSG or cystine, producing persulfide. In liver and kidney, GSSG concentrations are relatively high (in the hundreds of micromolar range (40)) while cystine levels are very low in liver and brain but ∼100 μM in kidney (6). Thus, these metabolites could affect the rate of tissue H2S concentration. However, neither 1.0 mM GSSG nor 0.5 mM cystine in buffer at pH 7.4 had a measurable effect on the rate of H2S loss (Fig. 3). Based on the reported rates of reaction of H2S with oxidized thiols (10), <1% of the total H2S is expected to be consumed in reaction with GSSG under our experimental conditions.
Interestingly, a decrease in H2S levels was observed in the presence of 1.0 mM cystamine, the oxidized form of cysteamine, which is produced during coenzyme A catabolism. The tissue concentration of cystamine however, is low (in the micromolar range), making this an unlikely sink for H2S.
The rate of aerobic H2S clearance by liver homogenates observed in our experiments is similar to the rate of liver sulfate plus thiosulfate production reported previously (13). In all three tissue homogenates, loss of H2S occurs more rapidly under aerobic conditions due to the coupling of the first enzyme, SQR to the electron transport chain and the direct dependence on oxygen of ETHE1, the second enzyme (Fig. 1). Liver and kidney mitochondria possess high SQR activity, in contrast to brain mitochondria where the activity is low (23). Our Western blot data (Fig. 5) reveal high ETHE1 expression levels in liver and kidney, while in brain, the 25 kD band corresponding to ETHE1 is very weak. The identity of the 37 kDa cross-reacting band is presently not known. One possibility is that it represents modified forms of ETHE1. Even if we assume that all cross-reacting bands correspond to active ETHE1, it is clear that the sum of band intensities in liver or kidney samples is significantly higher than in brain sample. The reported tissue abundance of other enzymes involved in H2S degradation [i.e., rhodanese and sulfite oxidase in mammals (28, 45)] corresponds to the relative aerobic H2S degradation activity observed in our study.
The weak pH dependence of the observed H2S degradation rate was surprising. To our knowledge, the pH dependencies of the activities of mammalian SQR and ETHE1 have not been reported. SQR from the archaeon Acidianus ambivalens shows maximal activity at pH 6.0 and ∼30% activity at pH 7.4 (5). Murine rhodanese exhibits maximal activity between pH 5–6 and ∼25% activity at pH 7.4 (39) and human sulfite oxidase has maximal activity in a wide pH range from 7.0–9.5 with 50% activity at pH 6.0 (43). Thus, based on the available pH dependence data, the activities of the enzymes involved in mitochondrial sulfide catabolism might be expected to be weakly dependent on pH in the 5.8–7.4 range. Furthermore, the interaction of the enzymes in the sulfide oxidation pathway might also modulate their pH dependence.
Since H2S turnover was studied in homogenates prepared from snap-frozen tissues, it is possible that during thawing, mitochondria might have been damaged. The kinetics of aerobic H2S degradation in homogenates prepared from freshly isolated versus snap-frozen and thawed liver was compared (Fig. S2). A ∼2-fold higher rate of H2S clearance was observed together with a time-dependent decrease in the H2S degradation rate upon incubation of the homogenate from freshly isolated liver on ice. Thus, mitochondrial integrity is not required for the activity of the aerobic sulfide-catabolism pathway.
The slower loss of H2S in the presence of tissue homogenates under anaerobic conditions is probably due to the incomplete removal of oxygen from the samples. However, the explanation is likely to be more complex since it does not account for the tissue-dependent differences in H2S disappearance rates under anaerobic conditions (Fig. 4). Reaction of H2S with disulfides or metals present in tissue samples might be another mechanism for H2S depletion. However, we did not observe significant reaction of H2S with disulfides in control experiments (Fig. 3). Furthermore, the presence of the metal chelator, DTPA, did not affect the rate of H2S clearance, indicating that protein-bound versus free metals are more likely to contribute to H2S binding. Rapid disappearance of H2S has been reported in the presence of 5% solution of BSA under anaerobic conditions (42). Thus, under anaerobic conditions H2S can disappear at least in part due to reaction with proteins in tissue homogenates. This is supported by our observation that under “strictly” anaerobic conditions, DTT decreases the rate of H2S degradation (Table 1). It is likely that the partitioning of H2S between clearance pathways differ between liver, kidney, and brain since H2S removal by kidney samples was not observed under strictly anaerobic conditions (Table 1). Rather, an increase in H2S levels was observed compared to controls, probably due to H2S production from endogenous tissue cysteine (Fig. 4). While H2S produced from endogenous cysteine is rapidly degraded under aerobic conditions, under “strictly” anaerobic conditions where the oxygen concentration is very limited, H2S production in kidney homogenate (containing the ∼10-fold higher cysteine levels compared to the other tissues) might be sufficiently high so that H2S levels are maintained steady under our experimental conditions (Table 1). The lower endogenous cysteine levels in liver homogenate, together with higher H2S degradation activity, accounts for the observed decrease in H2S levels in this sample even under “strictly” anaerobic conditions.
Based on the tissue H2S production and clearance rates at physiologically relevant concentrations of cysteine determined in this study, an estimate of steady-state tissue H2S levels can be obtained. The exponential decay of H2S in the presence of tissue homogenate indicates that the rate of H2S clearance is a linear function of its concentration (Equation 1).
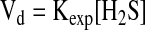 |
[Eq.1] |
Here Vd denotes the rate of H2S clearance and Kexp is a reaction constant under our experimental conditions. This constant can be calculated from the half-time (T1/2) of H2S, as shown in Equation 2.
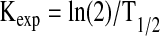 |
[Eq.2] |
At pH 7.4 and in the presence of air (Fig. 4), the T1/2 value for liver homogenate is 2.0 min and Kexp=0.347 min−1. Normalizing for tissue dilution (the sample contained 50 mg tissue in 20 ml of total volume, which is equivalent to 2.5 g tissue in 1 L of total volume or 0.25% of liver density), a KL of (0.347 min−1 x 400) or 138.8 min−1 is obtained. Based on a Q10 of ∼2, the rate constant is estimated to be 277 min−1 at 37°C.
The rate of H2S clearance by liver, VcL, is described by Equation 3.
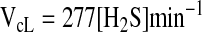 |
[Eq.3] |
Under steady-state conditions, the rate of H2S clearance must be equal to the rate of H2S production (Vp), that is, 8.1 μmoles min−1 kg tissue−1. Substituting this value for VcL in Equation 3, an estimate of 29 nmoles kg tissue−1 for the steady-state concentration of H2S in murine liver is obtained. Similarly, the steady-state concentration of H2S can be estimated for kidney (at 0.5 mM cysteine) and brain (at 0.1 mM cysteine). Values of 8.7 nmoles kg tissue−1 and 18 nmoles kg tissue−1 for kidney and brain, respectively, are obtained from this analysis. Using the value for the H2S clearance rate by fresh liver homogenate, we obtain a T1/2 of 0.8 min and an estimated steady-state H2S concentration in liver of 12 nmoles kg tissue−1. These estimates are consistent with the low nanomolar tissue concentrations of H2S that have been reported (12) and emphasize the importance of careful sample preparation for correct analysis of tissue H2S levels.
Materials and Methods
Materials
Cysteine, cystine dihydrochloride, cystamine dihydrochloride, diethylenetriamine pentaacetic acid, and GSSG were from Sigma. HEPES and sodium phosphate (monobasic) were from Fisher Scientific. Buffers were prepared with deionized water obtained with Milli-Q Advantage A 10 System (Millipore). The pH of buffers was adjusted with sodium hydroxide (Acros Organics). Nitrogen, carbon monoxide (Ultra High Purity grade) and H2S standard (40 ppm (1.785 μmole L−1)) in nitrogen were obtained from Cryogenic Gases (Detroit, MI). Polypropylene syringes (Norm-Ject) were obtained from Fisher Scientific, plastic 3-way stopcocks were obtained from Smiths Medical, sleeve stopper septa were from Sigma, and Tedlar gas sample bags, used for H2S sample handling were obtained from Jensen Inert.
Animals and tissue collection
Male Balb/c mice (2-month-old) were purchased from the Jackson Laboratory (Bar Harbor, ME). Animals were sacrificed in a CO2 atmosphere, and liver, kidney, and brain were collected immediately, frozen in liquid nitrogen and stored at −80 oC until further use. All procedures for animal handling were performed in accordance with the protocols approved by the University's Committee on Use and Care of Animals.
H2S production assays
Frozen mouse tissue was disrupted at 0°C using a glass homogenizer in 100 mM HEPES buffer, pH 7.4, or in 100 mM sodium phosphate buffer, pH 5.8, to obtain a tissue concentration of 100 mg/ml. Reactions for H2S production were prepared in polypropylene syringes as described previously (12). Tissue homogenate (400 μl), buffer, and cysteine (stock solutions prepared in buffer) were mixed in 20 ml syringe barrels in a total liquid volume of 0.5 ml to give final cysteine concentrations of 0.0, 0.1, 0.2, 0.5, 1.0, and 5.0 mM. Syringes were sealed with plungers and made anaerobic by flushing the headspace with nitrogen five times using a 3-way stopcock, and then left with nitrogen in a final total volume (liquid+gas) of 20 ml. Syringes were placed in an incubator at 37°C with gentle shaking (75 rpm) for 20 min. Control reactions in which buffer replaced tissue homogenate, were prepared in parallel. Aliquots of 200 μl from the gas phase of the reaction syringes were collected using gas tight syringes through a septum attached to the stopcock, and injected into an HP 6890 gas chromatograph (GC) (Hewlett Packard) equipped with a DB-1 column (30 m x 0.53 mm x 1.0 μm). The carrier gas (helium) flow rate was 2 ml/min and a temperature gradient ranging from 30–110°C over 20 min was employed. For samples containing high H2S concentrations, a 20-fold dilution with nitrogen was used to prevent column overloading. H2S was detected using a 355 sulfur chemiluminescence detector (Agilent).
The H2S standard from Cryogenic Gases had a stock concentration of 40 ppm (1.785 μmole L−1) in nitrogen. Aliquots (200 μl) of varying H2S dilutions in nitrogen were injected into the GC to generate a standard curve. The amount of H2S in the injected sample was calculated from the peak areas using the calibration coefficient obtained from the standard curve. Accounting for the gas fraction in reaction volume (19.5 ml), gives the total amount of H2S in the gas phase of the reaction syringe. Note that total H2S was distributed between gas and liquid (homogenate) phases, and dissolved H2S is present as a mixture of the ionized form (HS-) and protonated form (H2S), which in turn, is in equilibrium with H2S in the gas phase. The amount of dissolved H2S was calculated by multiplying the concentration of H2S in the gas phase by 1.6, the ratio between the concentration of H2S in the gas and liquid phases at 37°C (12) and by the liquid volume (0.5 ml). The HS- ion concentration in the liquid phase was calculated using a pKa of 6.8 for ionization of H2S in water at 37°C (16). The total amount of H2S in the reaction mixture was calculated as the sum of H2S in the gas phase and of dissolved H2S and SH-. The sum of H2S in both phases produced in blank reactions lacking tissue was subtracted from that of reactions containing tissue. The resulting H2S concentration was then used to obtain the specific activity for H2S production by dividing the net H2S produced by the incubation time and by the amount of tissue. The rate of H2S production was expressed as mmol h−1 kg of tissue−1.
The dependence of the H2S pKa on ionic strength in concentrated solutions has been reported (2, 16, 26). According to these reports, the pKa for H2S at 37°C decreases from 6.8 in water to 6.7–6.6 at physiological salt concentrations. To our knowledge, reliable experimental data on the pKa of H2S at physiologically relevant ionic conditions has not been reported. Under our experimental conditions, with a gas to liquid volume ratio of 39:1, most of the H2S in the reaction mixture is present in the gas phase even at pH 7.4 (∼80%). Thus, our results are not very sensitive to the pKa value used for H2S and a value of 6.8 was employed in our calculations. Based on our estimates, a change in the pKa value from 6.8 to 6.6 would lead to a <10% difference in the H2S amount under our experimental conditions.
H2S degradation assays
Tissue homogenates were prepared as described above; 0.5 ml of homogenate was placed in a polypropylene syringe barrel, and the syringe was sealed with a plunger. For anaerobic assays, the syringe headspace was flushed five times with nitrogen using a 3-way stopcock, and then filled with nitrogen to a total volume (liquid+gas) of 10 ml. Then, 10 ml of 40 ppm H2S in nitrogen was added to the syringe to give a final volume of 20 ml. For aerobic assays, the syringe headspace was filled with air to a total volume (liquid+gas) of 10 ml, and 10 ml of 40 ppm H2S in nitrogen was added, using a 3-way stopcock, to give a total final volume of 20 ml. The total initial amount of H2S in the syringes was 17.9 nmoles. The syringes were incubated at room temperature (25°C) with stirring and 200 μl aliquots were collected from the gas phase for H2S analysis after 1, 2, 3, 5, 10, 15, and 20 min of incubation. Levels of H2S in the samples were measured as described above. The amount of H2S in the syringes was calculated using the ratio between the concentration of H2S in the liquid and gas phases of 2.0 at 25°C (12) and a pKa of 7.0 for ionization of H2S in water at 25°C (16).
In several separate experiments, we compared aerobic H2S degradation by liver homogenates, prepared with regular buffers (HEPES pH 7.4, or sodium phosphate pH 5.8) or with buffers containing the metal chelator, DTPA (8, 22). For this, frozen liver was divided to two pieces, and the pieces were homogenized in buffer±50 μM DTPA. Aerobic H2S degradation was analyzed in both homogenates as described above.
To analyze the influence of mitochondrial integrity on H2S degradation kinetics, liver was divided into two equal sections immediately following collection; one part was immediately frozen in liquid nitrogen and stored on dry ice. The second part was gently disrupted in 100 mM HEPES buffer, pH 7.4 at 0°C using a glass homogenizer to obtain a tissue concentration of 100 mg ml−1. A 0.5 ml aliquot of homogenate was immediately used for analysis of the kinetics of aerobic H2S degradation as described above while the rest of homogenate was stored on ice for 30 or 60 min prior to being used for analysis of H2S clearance rate. The frozen liver section was homogenized with 100 mM HEPES buffer, pH 7.4, and used for analysis of the kinetics of aerobic H2S degradation as described above.
For strictly anaerobic assays, buffer, tissue homogenates, and the reaction mixtures in syringes were prepared in OMNI-LAB anaerobic chamber (VAC, Hawthorne, CA) filled with nitrogen. The oxygen levels were ∼0.2–0.5 ppm. Under strictly anaerobic conditions, samples for H2S analysis were only collected after 20 min incubation. In control experiments, 500 μl of buffer (100 mM HEPES, pH 7.4) with or without different thiol metabolites were placed in a syringe instead of tissue homogenate.
Antibody production and purification
Polyclonal anti-human ETHE1 chicken antibodies were developed in Aves Labs (Tigard, OR) against recombinant human protein purified in our laboratory. The antigen, which was >95% pure, was separated on a denaturing 12% polyacrylamide gel and the protein band was excised and used for antibody production. Purified antibodies were obtained using an affinity resin, prepared by coupling recombinant human ETHE1 purified in house to Actigel ALT (Sterogene, Carlsbad, CA) according to vendor's protocol. Purified antibodies were used for Western blot analysis in dilution of 1:1000.
Western blot analysis
Frozen tissues were powdered in liquid nitrogen using a porcelain mortar and pestle and the powder was suspended in lysis buffer (20 mM HEPES, pH 7.5, 25 mM KCl, 0.5% NONIDET P-40, 10 μl/ml protease inhibitor cocktail for mammalian cells and tissue extracts (Sigma), 25 μg/ml tosyllysine chloromethylketone, and 17 μg/ml phenylmethylsulfonyl fluoride). The protein concentration in tissue extracts was determined using the Bradford assay and bovine serum albumin as standard as described previously (14). Proteins were resolved on 12% SDS-polyacrylamide gels and transferred to PVDF membranes. Blots were probed with polyclonal anti-human ETHE1 chicken antibodies. Secondary anti-chicken antibodies conjugated with horseradish peroxidase (Aves Labs) were used (1:250,000 dilution) and signals were visualized using the chemiluminescent peroxidase substrate kit (SuperSignal® West Dura (Thermo Scientific)).
Statistical analysis and kinetic fits
Statistical analysis and exponential fits of kinetic data were performed using Origin 7 software (OriginLab, Northampton, MA). Data are presented as mean±SD.
Supplementary Material
Abbreviations Used
- CBS
cystathionine β-synthase
- CSE
cystathionine γ-lyase (γ-cystathionase)
- DTPA
diethylenetriamine pentaacetic acid
- ETHE1
sulfur dioxygenase
- GC
gas chromatograph
- GSSG
glutathione oxidized
- MST
mercaptopyruvate sulfur transferases
- SQR
sulfide: quinine oxidoreductase
Acknowledgments
The authors gratefully acknowledge Drs. Michael Levitt, Julie Furne (VA Medical Center, MN), and David Linden (Mayo Clinic, MN) for help with setting up the GC assay for H2S. This work was supported by grants from the National Institutes of Health (DK64959 and HL58984).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Abe K. Kimura H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci. 1996;16:1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almgren T. Dyrssen D. Elgquist B. Dissociation of hydrogen sulphide in seawater and comparison of pH scales. Marine Chem. 1976;4:289–297. [Google Scholar]
- 3.Blackstone E. Morrison M. Roth MB. H2S induces a suspended animation-like state in mice. Science. 2005;308:518. doi: 10.1126/science.1108581. [DOI] [PubMed] [Google Scholar]
- 4.Bouillaud F. Blachier F. Mitochondria and sulfide: A very old story of poisoning, feeding and signaling? Antioxid Redox Signal. 2011;15:379–391. doi: 10.1089/ars.2010.3678. [DOI] [PubMed] [Google Scholar]
- 5.Brito JA. Sousa FL. Stelter M. Bandeiras TM. Vonrhein C. Teixeira M. Pereira MM. Archer M. Structural and functional insights into sulfide:quinone oxidoreductase. Biochemistry. 2009;48:5613–5622. doi: 10.1021/bi9003827. [DOI] [PubMed] [Google Scholar]
- 6.Cherqui S. Sevin C. Hamard G. Kalatzis V. Sich M. Pequignot MO. Gogat K. Abitbol M. Broyer M. Gubler MC. Antignac C. Intralysosomal cystine accumulation in mice lacking cystinosin, the protein defective in cystinosis. Mol Cell Biol. 2002;22:7622–7632. doi: 10.1128/MCB.22.21.7622-7632.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiku T. Padovani D. Zhu W. Singh S. Vitvitsky V. Banerjee R. H2S biogenesis by cystathionine gamma-lyase leads to the novel sulfur metabolites, lanthionine and homolanthionine, and is responsive to the grade of hyperhomocysteinemia. J Biol Chem. 2009;284:11601–11612. doi: 10.1074/jbc.M808026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doeller JE. Isbell TS. Benavides G. Koenitzer J. Patel H. Patel RP. Lancaster JR., Jr. Darley-Usmar VM. Kraus DW. Polarographic measurement of hydrogen sulfide production and consumption by mammalian tissues. Anal Biochem. 2005;341:40–51. doi: 10.1016/j.ab.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 9.Elrod JW. Calvert JW. Morrison J. Doeller JE. Kraus DW. Tao L. Jiao X. Scalia R. Kiss L. Szabo C. Kimura H. Chow CW. Lefer DJ. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA. 2007;104:15560–15565. doi: 10.1073/pnas.0705891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Francoleon NE. Carrington SJ. Fukuto JM. The reaction of H(2)S with oxidized thiols: Generation of persulfides and implications to H(2)S biology. Arch Biochem Biophys. 2011;516:146–153. doi: 10.1016/j.abb.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 11.Frank N. Kent JO. Meier M. Kraus JP. Purification and characterization of the wild type and truncated human cystathionine beta-synthase enzymes expressed in E. coli. Arch Biochem Biophys. 2008;470:64–72. doi: 10.1016/j.abb.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furne J. Saeed A. Levitt MD. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am J Physiol Regul Integr Comp Physiol. 2008;295:R1479–1485. doi: 10.1152/ajpregu.90566.2008. [DOI] [PubMed] [Google Scholar]
- 13.Furne J. Springfield J. Koenig T. DeMaster E. Levitt MD. Oxidation of hydrogen sulfide and methanethiol to thiosulfate by rat tissues: A specialized function of the colonic mucosa. Biochem Pharmacol. 2001;62:255–259. doi: 10.1016/s0006-2952(01)00657-8. [DOI] [PubMed] [Google Scholar]
- 14.Garg SK. Yan Z. Vitvitsky V. Banerjee R. Differential dependence on cysteine from transsulfuration versus transport during T cell activation. Antioxid Redox Signal. 2010;15:39–47. doi: 10.1089/ars.2010.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goubern M. Andriamihaja M. Nubel T. Blachier F. Bouillaud F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007;21:1699–1706. doi: 10.1096/fj.06-7407com. [DOI] [PubMed] [Google Scholar]
- 16.Hershey JP. Plese T. Millero FJ. The pK1 for the dissociation of H2S in various ionic media. Geochimica Cosmochimica Acta. 1988;52:2047–2051. [Google Scholar]
- 17.Hildebrandt TM. Grieshaber MK. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008;275:3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x. [DOI] [PubMed] [Google Scholar]
- 18.Hirche F. Schroder A. Knoth B. Stangl GI. Eder K. Effect of dietary methionine on plasma and liver cholesterol concentrations in rats and expression of hepatic genes involved in cholesterol metabolism. Br J Nutr. 2006;95:879–888. doi: 10.1079/bjn20061729. [DOI] [PubMed] [Google Scholar]
- 19.Ishigami M. Hiraki K. Umemura K. Ogasawara Y. Ishii K. Kimura H. A source of hydrogen sulfide and a mechanism of its release in the brain. Antioxid Redox Signal. 2009;11:205–214. doi: 10.1089/ars.2008.2132. [DOI] [PubMed] [Google Scholar]
- 20.Kabil O. Banerjee R. The redox biochemistry of hydrogen sulfide. J Biol Chem. 2010;285:21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kabil O. Vitvitsky V. Xie P. Banerjee R. The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid Redox Signal. 2011;15:363–372. doi: 10.1089/ars.2010.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koenitzer JR. Isbell TS. Patel HD. Benavides GA. Dickinson DA. Patel RP. Darley-Usmar VM. Lancaster JR., Jr. Doeller JE. Kraus DW. Hydrogen sulfide mediates vasoactivity in an O2-dependent manner. Am J Physiol Heart Circ Physiol. 2007;292:H1953–1960. doi: 10.1152/ajpheart.01193.2006. [DOI] [PubMed] [Google Scholar]
- 23.Lagoutte E. Mimoun S. Andriamihaja M. Chaumontet C. Blachier F. Bouillaud F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim Biophys Acta. 2010;1797:1500–1511. doi: 10.1016/j.bbabio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 24.Levitt MD. Abdel-Rehim MS. Furne J. Free and acid-labile hydrogen sulfide concentrations in mouse tissues: Anomalously high free hydrogen sulfide in aortic tissue. Antioxid Redox Signal. 2011;15:373–378. doi: 10.1089/ars.2010.3525. [DOI] [PubMed] [Google Scholar]
- 25.Linden DR. Sha L. Mazzone A. Stoltz GJ. Bernard CE. Furne JK. Levitt MD. Farrugia G. Szurszewski JH. Production of the gaseous signal molecule hydrogen sulfide in mouse tissues. J Neurochem. 2008;106:1577–1585. doi: 10.1111/j.1471-4159.2008.05502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Millero FJ. The thermodynamics and kinetics of the hydrogen sulfide system in natural waters. Marine Chem. 1986;18:121–147. [Google Scholar]
- 27.Muller FH. Bandeiras TM. Urich T. Teixeira M. Gomes CM. Kletzin A. Coupling of the pathway of sulphur oxidation to dioxygen reduction: Characterization of a novel membrane-bound thiosulphate:quinone oxidoreductase. Mol Microbiol. 2004;53:1147–1160. doi: 10.1111/j.1365-2958.2004.04193.x. [DOI] [PubMed] [Google Scholar]
- 28.Nagahara N. Ito T. Kitamura H. Nishino T. Tissue and subcellular distribution of mercaptopyruvate sulfurtransferase in the rat: confocal laser fluorescence and immunoelectron microscopic studies combined with biochemical analysis. Histochem Cell Biol. 1998;110:243–250. doi: 10.1007/s004180050286. [DOI] [PubMed] [Google Scholar]
- 29.Nagahara N. Yoshii T. Abe Y. Matsumura T. Thioredoxin-dependent enzymatic activation of mercaptopyruvate sulfurtransferase. An intersubunit disulfide bond serves as a redox switch for activation. J Biol Chem. 2007;282:1561–1569. doi: 10.1074/jbc.M605931200. [DOI] [PubMed] [Google Scholar]
- 30.Nguyen BD. Zhao X. Vyas K. La Mar GN. Lile RA. Brucker EA. Phillips GN., Jr. Olson JS. Wittenberg JB. Solution and crystal structures of a sperm whale myoglobin triple mutant that mimics the sulfide-binding hemoglobin from Lucina pectinata. J Biol Chem. 1998;273:9517–9526. doi: 10.1074/jbc.273.16.9517. [DOI] [PubMed] [Google Scholar]
- 31.Nicoletti FP. Comandini A. Bonamore A. Boechi L. Boubeta FM. Feis A. Smulevich G. Boffi A. Sulfide binding properties of truncated hemoglobins. Biochemistry. 2010;49:2269–2278. doi: 10.1021/bi901671d. [DOI] [PubMed] [Google Scholar]
- 32.Pietri R. Lewis A. Leon RG. Casabona G. Kiger L. Yeh SR. Fernandez-Alberti S. Marden MC. Cadilla CL. Lopez-Garriga J. Factors controlling the reactivity of hydrogen sulfide with heme proteins. Biochemistry. 2009;48:4881–4894. doi: 10.1021/bi801738j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh S. Banerjee R. PLP-dependent H2S biogenesis. Biochim Biophys Acta. 2011;1814:1518–1527. doi: 10.1016/j.bbapap.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh S. Padovani D. Leslie RA. Chiku T. Banerjee R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J Biol Chem. 2009;284:22457–22466. doi: 10.1074/jbc.M109.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steegborn C. Clausen T. Sondermann P. Jacob U. Worbs M. Marinkovic S. Huber R. Wahl MC. Kinetics and inhibition of recombinant human cystathionine gamma-lyase. Toward the rational control of transsulfuration. J Biol Chem. 1999;274:12675–12684. doi: 10.1074/jbc.274.18.12675. [DOI] [PubMed] [Google Scholar]
- 36.Stipanuk MH. Metabolism of sulfur-containing amino acids. Annu Rev Nutr. 1986;6:179–209. doi: 10.1146/annurev.nu.06.070186.001143. [DOI] [PubMed] [Google Scholar]
- 37.Stipanuk MH. Londono M. Lee JI. Hu M. Yu AF. Enzymes and metabolites of cysteine metabolism in nonhepatic tissues of rats show little response to changes in dietary protein or sulfur amino acid levels. J Nutr. 2002;132:3369–3378. doi: 10.1093/jn/132.11.3369. [DOI] [PubMed] [Google Scholar]
- 38.Vachek H. Wood JL. Purification and properties of mercaptopyruvate sulfur transferase of Escherichia coli. Biochim Biophys Acta. 1972;258:133–146. doi: 10.1016/0005-2744(72)90973-4. [DOI] [PubMed] [Google Scholar]
- 39.Vazquez E. Polo C. Stedile G. Schebor C. Karahanian E. Batlle A. Isolation and partial purification of mitochondrial and cytosolic rhodanese from liver of normal and p-dimethylaminoazobenzene treated mice. Int J Biochem Cell Biol. 1995;27:523–529. doi: 10.1016/1357-2725(94)00089-t. [DOI] [PubMed] [Google Scholar]
- 40.Vitvitsky V. Dayal S. Stabler S. Zhou Y. Wang H. Lentz SR. Banerjee R. Perturbations in homocysteine-linked redox homeostasis in a murine model for hyperhomocysteinemia. Am J Physiol Regul Integr Comp Physiol. 2004;287:R39–46. doi: 10.1152/ajpregu.00036.2004. [DOI] [PubMed] [Google Scholar]
- 41.Weber RE. Vinogradov SN. Nonvertebrate hemoglobins: Functions and molecular adaptations. Physiol Rev. 2001;81:569–628. doi: 10.1152/physrev.2001.81.2.569. [DOI] [PubMed] [Google Scholar]
- 42.Whitfield NL. Kreimier EL. Verdial FC. Skovgaard N. Olson KR. Reappraisal of H2S/sulfide concentration in vertebrate blood and its potential significance in ischemic preconditioning and vascular signaling. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1930–1937. doi: 10.1152/ajpregu.00025.2008. [DOI] [PubMed] [Google Scholar]
- 43.Wilson HL. Wilkinson SR. Rajagopalan KV. The G473D mutation impairs dimerization and catalysis in human sulfite oxidase. Biochemistry. 2006;45:2149–2160. doi: 10.1021/bi051609l. [DOI] [PubMed] [Google Scholar]
- 44.Wlodek L. Ostrowski WS. 3-Mercaptopyruvate sulphurtransferase from rat erythrocytes. Acta Biochim Pol. 1982;29:121–133. [PubMed] [Google Scholar]
- 45.Woo WH. Yang H. Wong KP. Halliwell B. Sulphite oxidase gene expression in human brain and in other human and rat tissues. Biochem Biophys Res Commun. 2003;305:619–623. doi: 10.1016/s0006-291x(03)00833-7. [DOI] [PubMed] [Google Scholar]
- 46.Yang G. Cao K. Wu L. Wang R. Cystathionine gamma-lyase overexpression inhibits cell proliferation via a H2S-dependent modulation of ERK1/2 phosphorylation and p21Cip/WAK-1. J Biol Chem. 2004;279:49199–49205. doi: 10.1074/jbc.M408997200. [DOI] [PubMed] [Google Scholar]
- 47.Zhao W. Zhang J. Lu Y. Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.