

Abstract
Inhibitory neurotransmission ensures normal brain function by counteracting and integrating excitatory activity. γ-Aminobutyric acid (GABA) is the main inhibitory neurotransmitter in the mammalian central nervous system, and mediates its effects via two classes of receptors: the GABAA and GABAB receptors. GABAA receptors are heteropentameric GABA-gated chloride channels and responsible for fast inhibitory neurotransmission. GABAB receptors are heterodimeric G protein coupled receptors (GPCR) that mediate slow and prolonged inhibitory transmission. The extent of inhibitory neurotransmission is determined by a variety of factors, such as the degree of transmitter release and changes in receptor activity by posttranslational modifications (e.g., phosphorylation), as well as by the number of receptors present in the plasma membrane available for signal transduction. The level of GABAB receptors at the cell surface critically depends on the residence time at the cell surface and finally the rates of endocytosis and degradation. In this review we focus primarily on recent advances in the understanding of trafficking mechanisms that determine the expression level of GABAB receptors in the plasma membrane, and thereby signaling strength.
Keywords: GABAB receptors, Neuron, Trafficking, Endocytosis, Recycling, Degradation
FUNCTIONS OF GABAB RECEPTORS
Metabotropic GABAB receptors are widely distributed throughout the central nervous system where they mediate slow, prolonged inhibition to control neuronal excitation, and contribute to synaptic plasticity[1].
GABAB receptors are present at pre- and postsynaptic sites of both inhibitory and excitatory neurons. Electron microscopy revealed that GABAB receptors are located predominantly at areas close to neurotransmitter release sites and at peri- and extrasynaptic areas of spines and dendrites, but only rarely directly at active zones or postsynaptic densities[2-7]. This location of GABAB receptors implies that they are not directly activated by synaptically released GABA. One mechanism to activate GABAB receptors requires intense neuronal activity, resulting in a spill-over of synaptically released GABA[8]. However, there are also other sources that may increase the ambient level of GABA, such as activity-dependent release of GABA from dendrites and glia cells[9-11]. Recently, it has been shown that basal synaptic activity generates a sufficient concentration of ambient GABA to tonically induce a low level of presynaptic GABAB receptor activation, which results in the control of transmitter release[12].
Binding of GABA to the GABAB receptor activates Gi/o-type G proteins[13-18], which in turn modulate three major effector systems: adenylyl cyclases, voltage-sensitive Ca2+ channels and inwardly-rectifying K+ channels (Figure 1).
Figure 1.

Structural organization of GABAB receptors. Functional GABAB receptors are heterodimers composed of the two subunits GABAB1 and GABAB2. Both subunits are heptahelical membrane proteins with a large extracellular located N-terminal domain containing a “Venus flytrap” module and a large intracellular C-terminal domain containing a coiled-coil protein-protein interaction module. GABAB1 and GABAB2 heterodimerize via their “Venus flytrap” and coiled-coiled domains. An endoplasmic reticulum (ER) retention/retrieval signal is present distal to the coiled-coil domain in GABAB1 and prevents ER exit of GABAB1 unless it is masked by heterodimerization with GABAB2. The “Venus flytrap” module of GABAB1 constitutes the GABA binding site, whereas that of GABAB2 is inactive and not involved in ligand binding. Instead, the heptahelical domain of GABAB2 contains a binding site for allosteric modulators, which affects the affinity of ligands binding to the GABA site. Binding of GABA results in the recruitment and activation of Gαi/o proteins via GABAB2. The activated Gαi/o subunit inhibits the adenylyl cyclase, resulting in lowered cAMP levels, while the Gβγ dimer activates K+ channels and inhibits Ca2+ channels, leading in either case to neuronal inhibition. There exist two isoforms of GABAB1, named GABAB1a and GABAB1b, which are generated by alternative promoter usage. They only differ by the additional presence of two so-called “sushi repeats” (protein-protein interaction modules) in the N-terminal domain of GABAB1a. GABA: γ-Aminobutyric acid; ATP: Adenosine-5'-triphosphate; cAMP: 3'-5'-cyclic adenosine monophosphate.
The α subunit of the activated G protein inhibits adenylyl cyclase activity, which decreases cellular 3'-5'-cyclic adenosine monophosphate (cAMP) levels and affects the activity of cAMP-dependent processes. Unfortunately, the contribution of GABAB receptor-induced lowering of cAMP levels to physiological processes is poorly investigated. So far it has been shown that it retards synaptic vesicle recruitment during sustained activity, which reduces transmitter release[19]. In addition, GABAB receptor-mediated Gαi/o effects may be important for long-term adaptations involving regulation of protein kinase activity and gene transcription[20-22].
However, the most well established GABAB receptor actions are mediated via the βγ dimer of the activated G protein. At presynaptic sites, voltage-sensitive P/Q- and N-type Ca2+ channels are the predominant effectors of GABAB receptors[23-27]. GABAB receptor activated Gβγ inhibits Ca2+ channel activity by slowing their current activation kinetics[28], which eventually results in reduced transmitter release. Postsynaptically, GABAB receptor effects are mainly mediated by the family of G protein-gated inwardly rectifying K+ channels (GIRK1-4 also called Kir3.1-3.4)[29,30]. Gβγ directly binds to GIRK channels[31,32] and activates them[33,34], resulting in an outward K+ current. This hyperpolarizes the membrane and consequently inhibits neuronal activity. However, there is no strict mechanistic segregation of pre- (Ca2+ channels) and postsynaptic (K+ channels) effector systems. There is accumulating evidence that GABAB receptors also activate K+ channels at presynaptic sites, which assists inhibition of transmitter release[35-37]. Conversely, there is also data for GABAB receptor mediated inhibition of postsynaptic Ca2+ channels[38-41]. This provides an additional mechanism for controlling the excitability of dendrites and spines. Thus, the current data is consistent with a complex pattern of regulating the activity of multiple G protein-gated inwardly rectifying K+ channels and voltage-sensitive Ca2+ channels, both at pre- and postsynaptic sites, resulting in the inhibition of neuronal activity.
To ensure efficient activation of the effector system, GABAB receptors are localized in close proximity to their effector channels[36,42] and may even constitute signaling complexes by physical interaction[36,43].
MOLECULAR ORGANIZATION OF GABAB RECEPTORS
Although the GABAB receptor was discovered in 1980[44], its molecular identity and characterization was delayed for almost 20 years until the first constituent of the receptor was cloned. This delay was due to the fact that all biochemical attempts to purify the receptor failed and expression cloning proved unsuccessful. The development of high-affinity antagonists eventually permitted the successful screening of expression libraries yielding two cDNAs derived from a single gene, GABAB1a and GABAB1b[45]. GABAB1a and GABAB1b are generated by differential promoter usage[46] and differ solely by the presence of an additional N-terminal sequence in GABAB1a coding for two protein-protein interaction domains, so-called “sushi domains”. GABAB1a and GABAB1b show all the characteristics of class III G protein-coupled receptors (e.g., a very large extracellular domain, seven transmembrane-spanning (heptahelical) sequences and a large intracellular located C-terminal domain) (Figure 1). So far, no functional differences among GABAB receptors containing GABAB1a and GABAB1b have been detected. The cloning of these first GABAB receptor constituents provided the basis for numerous research efforts analyzing the molecular characterization and function of GABAB receptors. It soon became clear that functional GABAB receptors are obligatory heterodimers composed of GABAB1 (either GABAB1a or GABAB1b) and a second heptahelical membrane protein named GABAB2, sharing about 35% sequence identity with GABAB1[47-51]. Both subunits serve distinct functions within the heterodimeric receptor complex. GABAB1 contains the agonist and antagonist binding site in the large N-terminal extracellular domain, which is most likely arranged in a Venus flytrap-like structure[52-54]. Association with GABAB2 is necessary to keep the GABA binding site in a high affinity state[55,56]. On the other hand, GABAB2 contains a binding site for allosteric modulators, which is not however associated with the N-terminal Venus flytrap domain, but is located in the heptahelical domain[57]. Binding of ligands to this site does not directly activate the GABAB receptor but instead affects the affinity of orthosteric agonists and antagonists to GABAB1[58]. Finally, GABAB2 is responsible for G protein activation[56,59-63] and plays an important role in cell surface trafficking of the heterodimerized receptor complex by masking an arginine-based endoplasmic reticulum (ER) retention/retrieval (RXR) signal present in the C-terminal domain of GABAB1[64-68].
THE ROLE OF DESENSITIZATION AND PHOSPHORYLATION ON THE AVAILABILITY OF FUNCTIONAL GABAB RECEPTORS
Prolonged exposure of G protein coupled receptors (GPCR) to agonists generally leads to a complex series of events in order to attenuate or terminate signal transduction, protecting the cell from overstimulation. Signal transduction is often attenuated by desensitization of the receptors (i.e., abrogating signaling), although the agonist is still present[69,70]. Desensitization of many GPCRs involves phosphorylation-dependent uncoupling of the receptor from the G proteins, followed by internalization of the receptor. Activated GPCRs are usually phosphorylated by G protein-coupled receptor kinase (GRKs) at serine and/or threonine residues residing in the carboxyl-terminal tail- or intracellular loop regions, which rapidly attenuates receptor responses. Phosphorylation leads to the recruitment of arrestins, which is thought to sterically prohibit signaling to G proteins and induces internalization of the receptor by linking it to components (clathrin, AP2 complex) of the endocytosis machinery[69,70]. Internalized receptors are then either degraded in lysosomes or are dephosphorylated and subsequently recycled to the plasma membrane, where they are again available for signaling.
It is well known that prolonged activation of GABAB receptors commonly leads to their desensitization. Recent studies suggest that there might be more than one mechanism for desensitization of GABAB receptors[71-75], which does not follow the classical desensitization pattern of GPCRs described above (Figure 2). Distinct desensitization mechanisms for GABAB receptors may be operative in different neuronal populations.
Figure 2.
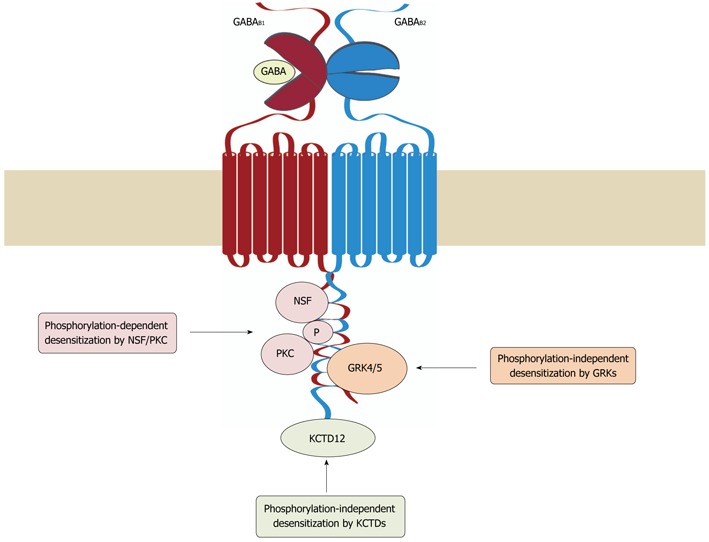
Mechanisms of GABAB receptor desensitization. Three distinct mechanisms have been so far implicated in the desensitization of GABAB receptors. In cerebellar granule cells, G protein receptor kinase (GRK) 4 and 5 associate with GABAB receptors and induce desensitization of the receptors in a phosphorylation-independent manner. In cortical and hippocampal neurons, desensitization of the receptors involves the interaction of NEM-sensitive fusion protein (NSF) with GABAB1 and GABAB2, which is thought to prime the receptor for phosphorylation by protein kinase C (PKC). Association of potassium channel tetramerization domain-containing (KCTD) proteins 12 and 12b with the C-terminus of GABAB2 appears to render the receptor complex competent for desensitization. GABA: γ-Aminobutyric acid.
A study conducted in mouse cerebellar granule cells showed that GRK4, which is mainly expressed in testes and cerebellum[69,76,77], promotes agonist-induced desensitization of GABAB receptors via direct association, but does not involve GABAB receptor phosphorylation[78]. These findings were confirmed by Kainaide et al[75], who demonstrated that the association of GABAB2 with GRK4 or GRK5, but not GRK2, -3 or -6, leads to agonist-induced receptor desensitization in Xenopus oocytes and baby hamster kidney cells. Interestingly, GRK4 and -5-mediated desensitization was partially suppressed by application of S(+)-ketamine, which leads to inhibition of the GABAB receptors/GRK complex formation by an as yet unidentified mechanism[79]. It is currently not understood how GRK4 and -5 mediate desensitization of GABAB receptors. However, it might well be that the binding of GRK4 and -5 disrupts GABAB receptor/G-protein interaction.
On the other hand, for cortical and hippocampal neurons, a phosphorylation-dependent desensitization mechanism of GABAB receptors was reported[74]. This mechanism is based on the direct interaction of NEM-sensitive fusion (NSF) protein with the C-terminal domains of GABAB1 and GABAB2, which primes the receptor for recruitment of protein kinase C (PKC). The data indicate that the association of GABAB receptors with NSF is a prerequisite for recruiting PKC to the receptor upon agonist activation. PKC phosphorylates the receptor leading to its desensitization, and induces dissociation of NSF from the receptors. The precise roles of NSF and PKC in this complex process remain to be determined. NSF might be required for unmasking phosphorylation sites of the receptor or involved in PKC activation. In addition, it is unclear whether NSF dissociates from the receptor before desensitization occurs or whether releasing NSF initiates recovery of the receptor from desensitization.
Another factor determining desensitization of GABAB receptors was recently discovered by functional proteomics[80]. Members of the potassium channel tetramerization domain-containing (KCTD) protein family were found to interact as tetramers with the C-terminus of GABAB2, generating high-molecular mass protein complexes. Depending on the co-expressed KCTD subtype, distinct parameters of GABAB receptor function were affected, such as agonist potency, signaling onset or desensitization. Interestingly, only when GABAB receptors were co-expressed with KCTD-12 or -12b desensitization of GABAB receptors was observed, whereas in the presence of KCTD-8 or -16 the receptors displayed no desensitization[80]. This finding may explain the observation that GABAB receptor desensitization varies among different neuronal populations. One striking example is the ventral tegmental area (VTA). GABAB receptors expressed in GABAergic neurons of the VTA display baclofen-induced, largely non-desensitizing, currents, whereas in dopaminergic neurons of the VTA baclofen elicited desensitizing currents[81]. These findings suggest that the general ability of GABAB receptors to desensitize may be determined by the associated KCTD subtype, which may then recruit distinct desensitization mechanisms depending on the neuronal population.
Interestingly, PKA-dependent phosphorylation appears to counteract desensitization of GABAB receptors. Couve et al[71] showed that PKA exclusively phosphorylates serine 892 (S892) in the C-terminal domain of GABAB2, resulting in reduced receptor desensitization. This effect on desensitization can be overcome by activation of the receptors, which results in inhibition of adenylyl cyclases, reduced cAMP levels and consequently diminished PKA activity and GABAB2-S892 phosphorylation. The precise mechanism as to how PKA phosphorylation of GABAB2-S892 affects desensitization of GABAB receptors remains unclear. There is an indication that it stabilizes cell surface GABAB receptors and thereby increases effector coupling[71,82]. This is, however, unlikely because it is now well accepted that prolonged agonist exposure does not trigger increased internalization of cell surface receptors[78,82-85]. However, GABAB2-S892 phosphorylation provides a mechanism for regulating the extent of GABAB receptor desensitization by the activity of Gαs-coupled GCPRs that enhance PKA activity.
Another kinase that is involved in regulating GABAB receptor activity is the 5’AMP-dependent protein kinase (AMPK). AMPK directly binds to the C-terminus of GABAB1 and phosphorylates S917 and S783 in the C-terminal domains of GABAB1 and GABAB2, respectively[86]. Functional analysis revealed that phosphorylation of S783 resulted in a stabilization of baclofen-induced K+ currents[86]. This effect has been shown to be of particular relevance in limiting neuronal cell death in experimental ischemia. Anoxic or ischemic conditions are associated with neuronal over-excitation, a decline in cellular adenosine-5'-triphosphate (ATP) and a rise in Ca2+ and AMP levels, which are all factors activating AMPK[87,88]. Under such conditions, increased phosphorylation of GABAB2-S783 was detected along with an over-expression of a GABAB2 mutant that cannot be phosphorylated at this site associated with increased neuronal death[86]. These findings support a mechanism in which AMPK functions as a metabolic sensor that detects severe cellular stress and phosphorylates, amongst others, GABAB receptors. This is thought to result in enhanced GABAB receptor signaling that counteracts over-excitation of the neuron and limits neuronal death.
REGULATION OF GABAB RECEPTORS BY TRAFFICKING
The lifecycle of a plasma membrane protein like the GABAB receptor starts with its synthesis at the rough ER where the nascent protein is co-translationally incorporated into the ER membrane. After folding, initial posttranslational processing, and assembly, the receptor is exported to the Golgi apparatus where it is further processed and finally transported via the trans-Golgi network to the plasma membrane. After a certain time span of function, the receptor is internalized and recycled back into the plasma membrane for another cycle of function, or is eventually degraded into lysosomes. To ensure a constant number of receptors in the plasma membrane for signaling, these trafficking events need to be precisely coordinated. On the other hand, regulation of each of the different trafficking steps permits adjusting the number of cell surface receptors, and thus signaling strength, according to the physiological requirements.
ER export of GABAB receptors
Little is known about the early stages in the lifecycle of GABAB receptors. So far it is clear that exit of heterodimeric GABAB receptors from the ER is controlled by an arginine-based ER retention/retrieval signal (RXR) present in the C-terminal domain of GABAB1[64-66]. The mechanism that prevents cell surface trafficking of GABAB1 appears to involve the coat protein complex I (COP I ), which plays a central role in the retrograde transport of proteins from the Golgi apparatus back to the ER[89]. COP I binds to the ER retention/retrieval signal of GABAB1 and shuttles monomeric GABAB1 that reached the cis-Golgi apparatus back to the ER. Heterodimerization with GABAB2 masks the ER retention/retrieval signal and permits forward transport[64-68]. In contrast to GABAB1, monomeric GABAB2 can leave the ER and reach the cell surface. However, it is assumed that the GABAB2 expression level in the ER is a limiting factor for ER exit of the heterodimeric GABAB receptors. This mechanism is thought to ensure that only correctly folded and assembled (i.e., functional) receptors are exported to the cell surface.
Endocytosis of GABAB receptors
There are two principal mechanisms by which GPCRs are internalized from the plasma membrane, constitutive endocytosis and agonist-induced endocytosis. Constitutive endocytosis constantly removes receptors from the cell surface, whereas agonist-induced endocytosis initiates removal of receptors from the plasma membrane upon activation of the receptors and ensures fast termination of signaling. It is now well established that GABAB receptors undergo constitutive endocytosis, whereas the presence of agonist-induced internalization of the receptors is less clear.
Heterologously expressed, as well as neuronal, GABAB receptors display fast constitutive internalization, as evidenced by distinct experimental approaches including immunofluorescence staining and microscopy, live cell imaging and cell surface biotinylation methods[83,84,90-93]. Constitutive internalization of GABAB receptors is a fast process, as shown by the rapid loss of labeled receptors from the cell surface, which reaches a plateau after 10-30 min (40% of labeled receptors remain at the cell surface), with rates of internalization of ranging from 2-10 min[92-94]. GABAB receptors internalize as heterodimers and are not dissociated into its subunits prior to endocytosis[84,90,92,95]. The rate of internalization appears to be determined by GABAB2. GABAB1, which contains an inactivated ER retention signal so that it is exported to the plasma membrane, displays a considerably faster rate of internalization than the GABAB1,2 heterodimer[92]. This is due to a dileucine motif within the coiled-coil domain of GABAB1, which gets masked upon assembly with GABAB2.
The data so far suggest that for endocytosis, GABAB receptors are recruited to clathrin-coated pits and internalized in a dynamin-dependent manner[83,90,95]. Clathrin-coated pits are composed of clathrin heavy and light chains that form a polymeric lattice and contain numerous adaptor and endocytic accessory proteins. For endocytosis, the cargo-loaded clathrin-coated pit invaginates and is eventually released from the plasma membrane in a GTP-dependent reaction mediated by dynamin[96]. There is evidence based on colocalization and immunoprecipitation data that GABAB receptors interact with the AP2 adaptor, which is one of the adaptor complexes that recruit membrane proteins to clathrin-coated pits[83,84,90].
Colocalization studies with marker proteins for various endosomal compartments revealed that endocytosed GABAB receptors first enter early endosomes and are then either sorted to Rab4 or Rab11-positive recycling endosomes, or to Rab7-positive late endosomes, and finally to lysosomes for degradation[84,90,92,95,97,98].
In addition to the colocalization data, there is also functional evidence that endocytosed GABAB receptors constitutively recycle back to the cell surface. Using immunofluorescence staining and tagged GABAB receptors transfected into hippocampal neurons Vargas et al[90] showed that a significant fraction of endocytosed receptors recycle back to the cell surface. Quantitative cell surface biotinylation and immunofluorescence-based methods indicate that the vast majority of native GABAB receptors in cortical neurons are rapidly recycled to the plasma membrane. After 15 min, about half of the internalized receptors have recycled back to the cell surface, and after 30 min this has increased to the majority of the receptors[84,94].
In summary, the current data indicate that GABAB receptors constitutively internalize at a high rate via the classical clathrin-dependent pathway and rapidly recycle back to the cell surface. Since endocytosis and recycling are highly energy-consuming processes, this mechanism is most likely of significant physiological relevance. The most obvious explanation is that a high rate of constitutive internalization and recycling generates a pool of intracellular receptors that can be immediately inserted into the plasma membrane to increase the cell surface number of receptors by increasing the rate of recycling while leaving the rate of internalization constant. In the case of synaptic AMPA receptors, such a mechanism has been proposed to contribute to increasing the level of the receptors during the early phase of long-term potentiation, which is thought to underlie learning and memory formation[99].
Degradation of GABAB receptors
Most cell surface receptors are eventually degraded in lysosomes, the major catabolic cellular compartment. After endocytosis, the endocytic vesicles carrying the receptors fuse with early endosomes, which then mature to late endosomes containing the material destined for degradation. Mature late endosomes are competent to fuse with lysosomes that contain a variety of hydrolases for the breakdown of all kinds of macromolecules[100].
There is now solid data that, at the end of their lifetime, GABAB receptors are endocytosed and degraded in lysosomes. This is evidenced by the intracellular accumulation of internalized GABAB receptors upon inhibition of lysosomal function[83,84,101] and the colocalization of intracellular GABAB receptors with marker proteins for late endosomes and lysosomes[84,92]. GABAB receptors are most likely sorted by the ESCRT (endosomal sorting complex required for transport) machinery to lysosomes, because the knockdown of tumor susceptibility gene 101 (TSG101), an integral component of the ESCRT machinery, prevents degradation of the receptors[101]. Three distinct ESCRT complexes sequentially target mono- and K63-linked polyubiquitinated membrane proteins to late endosomes[102]. However, it remains to be shown whether GABAB receptors are ubiquitinated and whether ubiquitination serves as a lysosomal sorting signal.
Another unresolved issue is how the decision is made as to whether a receptor is sorted to the degradation pathway. As discussed above, the vast majority of endocytosed GABAB receptors recycle back to the plasma membrane and only few are degraded. However, pharmacological inhibition of recycling leads to rapid lysosomal degradation of the receptors (about 50% of the total receptor population within 30 min)[84]. This indicates that recycling and degradation of GABAB receptors is tightly controlled, and decreasing the rate of recycling constitutes a mechanism to rapidly reduce the receptor number (discussed below).
Regulation of cell surface GABAb receptors by glutamatergic excitatory activity
GABAB receptors control glutamate signaling via presynaptic and postsynaptic mechanisms. They are abundantly expressed at glutamatergic synapses[2-5,103] where they are activated by GABA spillover from adjacent GABAergic terminals and inhibit glutamate release[8,104-106]. This limits activation of postsynaptically located excitatory glutamate receptors (AMPA/kainate and NMDA receptors). Although GABAB receptors are also located in close proximity to AMPA and NMDA receptors they do not appear to directly modulate AMPA and NMDA receptor excitatory postsynaptic currents (EPSCs)[105]. However, activation of postsynaptic GABAB receptors seem to limit Ca2+-influx through NMDA receptors by inhibition of the cAMP/PKA signaling pathway, which normally enhance NMDA receptor Ca2+ conductance[105].
Besides the prominent regulation of glutamate signaling by GABAB receptors, there is now evidence that glutamatergic activity, in return, may affect GABAB receptor expression to attenuate inhibitory control (Figure 3). Application of glutamate to cultured neurons dramatically down-regulates cell surface GABAB receptors and GABAB receptor-activated currents[90,94,97,98]. Specific activation of AMPA receptors[94] or NMDA receptors[97,98] was sufficient to induce the down-regulation of GABAB receptors. Interestingly, the kinetics of AMPA-induced down-regulation of GABAB receptors was significantly slower than that induced by glutamate and was accelerated upon co-activation of group I metabotropic glutamate receptors[94]. These findings indicate that beside the ionotropic AMPA and NMDA receptors, metabotropic glutamate receptors also contribute to the glutamate-induced down-regulation of GABAB receptors. The underlying mechanism of this rapid down-regulation of GABAB receptors is a shift of the recycling/degradation equilibrium towards lysosomal degradation[94,97]. Glutamate application reduced the rate of GABAB receptor recycling without altering the rate of their internalization and was fully restored after inhibition of lysosomal degradation. The precise intracellular signaling cascade leading to the glutamate-induced shift in sorting the GABAB receptors preferentially to the degradation pathway is currently not fully resolved. It is clear that the down-regulation of GABAB receptors depends on the influx of Ca2+[94,98], which is most likely mediated by L-type voltage-gated Ca2+ channels[94]. Two downstream effector systems were identified to be involved in the down-regulation of GABAB receptors (Figure 3). One depends on phosphorylation of serine 867 (S867) in GABAB1 by Ca2+/calmodulin-dependent protein kinase II (CaMKII)[98]. The other involves phosphorylation of serine 783 (S783) in GABAB2 by AMP kinase and subsequent dephosphorylation by protein phosphatase 2A (PP2A)[97]. Mutational inactivation of each phosphorylation site prevented glutamate-induced down-regulation of GABAB receptors. However, while the cell surface expression of the receptors containing the mutant GABAB1(S867A) was normal[98], the mutant GABAB2(S783A) was expressed to a significantly lesser level in the plasma membrane[97]. This suggests that phosphorylation of S783 in GABAB2 is involved in sorting the receptors to the recycling pathway, while phosphorylation of S867 in GABAB1 may constitute a direct signal for sorting the receptors to lysosomal degradation. Alternatively, phosphorylation of GABAB1(S867) may be required for dephosphorylation of S783 in GABAB2, for instance by recruiting PPA2 to the receptor. In this respect it would be very interesting to test whether phosphorylation of GABAB1(S867) by CaMKII is required for dephosphorylation of GABAB2(S783) by PPA2.
Figure 3.
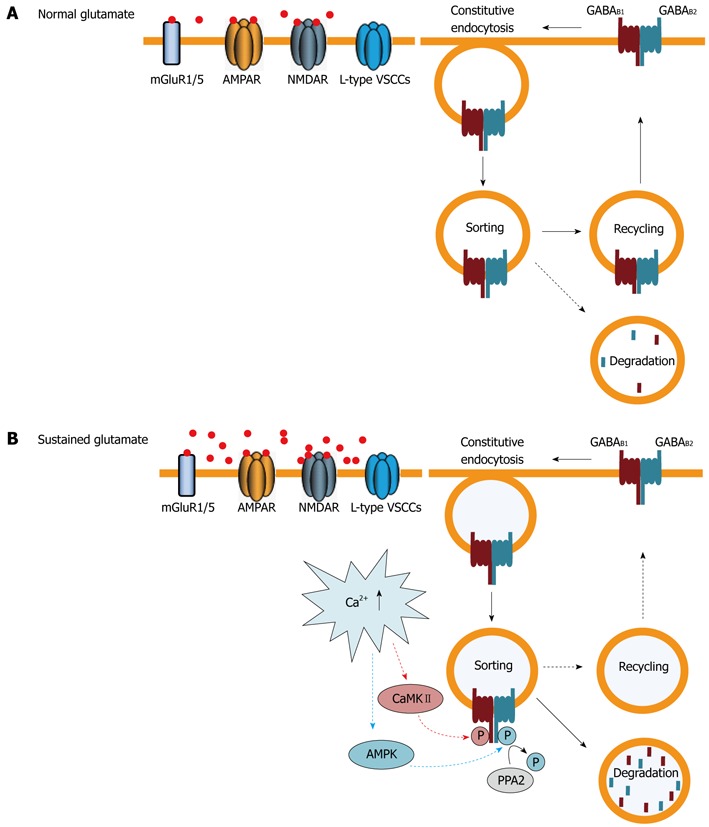
Regulation of cell surface GABAB receptors by trafficking. A: Under normal conditions GABAB receptors are constitutively internalized and recycled back to the plasma membrane. Only a small fraction of receptors are sorted to lysosomes for degradation; B: Sustained activation of glutamate receptors [primarily 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl) propanoic acid (AMPA) and N-methyl-D-aspartic acid (NMDA) receptors] and L-type voltage-gated Ca2+ channels raises intracellular Ca2+ levels. This induces phosphorylation of GABAB1 at serine 867 by calmodulin-dependent protein kinaseII (CaMKII) and of GABAB2 at serine 783 by adenosine monophosphate (AMP) kinase, followed by slow dephosphorylation, by protein phosphatase 2 (PPA2). These events shift the recycling/degradation equilibrium towards degradation so that the majority of GABAB receptors are no longer recycled, but instead degraded in lysosomes. Since constitutive endocytosis of the receptors remains unaffected, this mechanism results in a rapid down-regulation of GABAB receptors. AMPK: 5’AMP-dependent protein kinase; GABA: γ-Aminobutyric acid; VSCCs: Voltage-sensitive calcium channels.
What is the physiological relevance of this mechanism? Since glutamate-induced down-regulation of GABAB receptors has so far only been studied in cultured neurons, the role of this process in vivo remains to be shown. However, there are physiological, as well as pathological, conditions involving sustained activity of glutamate receptors where this mechanism might be operative. Under pathological conditions associated with excessive activation of glutamate receptors, such as ischemia, down-regulation of GABAB receptors results in diminished inhibitory control and may further enhance excitotoxicity and neuronal cell death. This view is supported by an in vitro model of ischemia where total GABAB2 protein levels were found to be strongly reduced 60 min after the ischemic insult[107]. Likewise, in an in vivo model of hypoxia/ischemia, significantly reduced levels of GABAB receptors were detected[108].
Under normal physiological conditions, glutamate-induced down-regulation of GABAB receptors may contribute to the process of long-term potentiation, which is thought to be the molecular basis for learning and memory formation, as long-term potentiation is associated with sustained activity of glutamate receptors[109]. In this scenario, enhanced glutamatergic activity would induce the down-regulation of GABAB receptors and consequently relieve the synapses from inhibition, resulting in a further increase of synaptic excitability.
CONCLUSION
Trafficking events play a pivotal role in the cell surface availability of receptors and largely determine their signaling strength. Currently, we are only beginning to identify and understand the trafficking mechanisms of GABAB receptors and how cell surface expression of the receptors is regulated. In particular, we almost completely lack knowledge on forward trafficking of GABAB receptors from the ER via the Golgi network to the plasma membrane. In addition, mechanisms on the targeting of the receptors to specific sites in the neuron are unknown. There is an initial indication that GABAB1 may be transported independent of GABAB2 within the ER into dendrites and are then assembled and exported to the plasma membrane[110]. This finding implies that heterodimerization of GABAB receptors is a spatially and temporally controlled mechanism, and would provide an additional level to regulate cell surface expression of the receptors. It is now clear that GABAB receptors are constitutively endocytosed via the clathrin and dynamin-dependent pathway, and are predominantly recycled back to the plasma membrane with only a minor fraction being degraded in lysosomes. The equilibrium of sorting the receptors to the recycling and degradation pathway appears to be controlled by phosphorylation/dephosphorylation events and regulated by changes in neuronal activity associated with increased influx of Ca2+. It will be a major future effort to unravel the mechanisms involved in trafficking, sorting and degradation of GABAB receptors and how they are regulated by physiological and pathological stimuli. It is now well established that receptor trafficking regulates signal transduction and that disturbances in these mechanisms may contribute to disease states[111]. Since GABAB receptors have been implicated in a variety of neurological disorders-ranging from epilepsy, addiction, schizophrenia, depression, anxiety to chronic pain-it is likely that altered GABAB receptor trafficking is involved, at least to some extent, in these diseases. We expect that a deeper knowledge of the trafficking mechanisms of GABAB receptors under physiological and pathological conditions will provide the basis for the development of novel and highly selective future therapeutic interventions.
Footnotes
Supported by The Swiss Science Foundation Grant, 31003A_121963
Peer reviewers: Francisco Ciruela, Dr., Deptartment de Patologia i Terapèutica Experimental, Pavelló de Govern, L’Hospitalet de Llobregat 08907, Spain; Sung H Kim, Profossor, College of Oriental Medicine, Kyunghee University, Cancer Preventive Material Research Center, Seoul 130-701, South Korea; Joe B Blumer, Assistant Professor, Department of Pharmacology, Medical University of South Carolina, 114 Doughty St., STB331, Charleston, SC 29425, United States; Saobo Lei, Associate Professor, Department of Pharmacology, Physiology and Therapeutics, University of North Dakotam, 501 N Columbia Road, Grand Forks, ND 58203, United States
S- Editor Cheng JX L- Editor Rutherford A E- Editor Zhang DN
References
- 1.Ulrich D, Bettler B. GABA(B) receptors: synaptic functions and mechanisms of diversity. Curr Opin Neurobiol. 2007;17:298–303. doi: 10.1016/j.conb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Luján R, Shigemoto R, Kulik A, Juiz JM. Localization of the GABAB receptor 1a/b subunit relative to glutamatergic synapses in the dorsal cochlear nucleus of the rat. J Comp Neurol. 2004;475:36–46. doi: 10.1002/cne.20160. [DOI] [PubMed] [Google Scholar]
- 3.Kulik A, Vida I, Luján R, Haas CA, López-Bendito G, Shigemoto R, Frotscher M. Subcellular localization of metabotropic GABA(B) receptor subunits GABA(B1a/b) and GABA(B2) in the rat hippocampus. J Neurosci. 2003;23:11026–11035. doi: 10.1523/JNEUROSCI.23-35-11026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lacey CJ, Boyes J, Gerlach O, Chen L, Magill PJ, Bolam JP. GABA(B) receptors at glutamatergic synapses in the rat striatum. Neuroscience. 2005;136:1083–1095. doi: 10.1016/j.neuroscience.2005.07.013. [DOI] [PubMed] [Google Scholar]
- 5.Chen L, Boyes J, Yung WH, Bolam JP. Subcellular localization of GABAB receptor subunits in rat globus pallidus. J Comp Neurol. 2004;474:340–352. doi: 10.1002/cne.20143. [DOI] [PubMed] [Google Scholar]
- 6.Boyes J, Bolam JP. The subcellular localization of GABA(B) receptor subunits in the rat substantia nigra. Eur J Neurosci. 2003;18:3279–3293. doi: 10.1111/j.1460-9568.2003.03076.x. [DOI] [PubMed] [Google Scholar]
- 7.Luján R, Shigemoto R. Localization of metabotropic GABA receptor subunits GABAB1 and GABAB2 relative to synaptic sites in the rat developing cerebellum. Eur J Neurosci. 2006;23:1479–1490. doi: 10.1111/j.1460-9568.2006.04669.x. [DOI] [PubMed] [Google Scholar]
- 8.Scanziani M. GABA spillover activates postsynaptic GABA(B) receptors to control rhythmic hippocampal activity. Neuron. 2000;25:673–681. doi: 10.1016/s0896-6273(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 9.Zilberter Y, Kaiser KM, Sakmann B. Dendritic GABA release depresses excitatory transmission between layer 2/3 pyramidal and bitufted neurons in rat neocortex. Neuron. 1999;24:979–988. doi: 10.1016/s0896-6273(00)81044-2. [DOI] [PubMed] [Google Scholar]
- 10.Angulo MC, Le Meur K, Kozlov AS, Charpak S, Audinat E. GABA, a forgotten gliotransmitter. Prog Neurobiol. 2008;86:297–303. doi: 10.1016/j.pneurobio.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Vélez-Fort M, Audinat E, Angulo MC. Central Role of GABA in Neuron-Glia Interactions. Neuroscientist. 2011:Epub ahead of print. doi: 10.1177/1073858411403317. [DOI] [PubMed] [Google Scholar]
- 12.Laviv T, Riven I, Dolev I, Vertkin I, Balana B, Slesinger PA, Slutsky I. Basal GABA regulates GABA(B)R conformation and release probability at single hippocampal synapses. Neuron. 2010;67:253–267. doi: 10.1016/j.neuron.2010.06.022. [DOI] [PubMed] [Google Scholar]
- 13.Nishikawa M, Hirouchi M, Kuriyama K. Functional coupling of Gi subtype with GABAB receptor/adenylyl cyclase system: analysis using a reconstituted system with purified GTP-binding protein from bovine cerebral cortex. Neurochem Int. 1997;31:21–25. doi: 10.1016/s0197-0186(96)00138-6. [DOI] [PubMed] [Google Scholar]
- 14.Morishita R, Kato K, Asano T. GABAB receptors couple to G proteins Go, Go* and Gi1 but not to Gi2. FEBS Lett. 1990;271:231–235. doi: 10.1016/0014-5793(90)80413-d. [DOI] [PubMed] [Google Scholar]
- 15.Menon-Johansson AS, Berrow N, Dolphin AC. G(o) transduces GABAB-receptor modulation of N-type calcium channels in cultured dorsal root ganglion neurons. Pflugers Arch. 1993;425:335–343. doi: 10.1007/BF00374184. [DOI] [PubMed] [Google Scholar]
- 16.Odagaki Y, Koyama T. Identification of galpha subtype(s) involved in gamma-aminobutyric acid(B) receptor-mediated high-affinity guanosine triphosphatase activity in rat cerebral cortical membranes. Neurosci Lett. 2001;297:137–141. doi: 10.1016/s0304-3940(00)01692-x. [DOI] [PubMed] [Google Scholar]
- 17.Campbell V, Berrow N, Dolphin AC. GABAB receptor modulation of Ca2+ currents in rat sensory neurones by the G protein G(0): antisense oligonucleotide studies. J Physiol. 1993;470:1–11. doi: 10.1113/jphysiol.1993.sp019842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mannoury la Cour C, Herbelles C, Pasteau V, de Nanteuil G, Millan MJ. Influence of positive allosteric modulators on GABA(B) receptor coupling in rat brain: a scintillation proximity assay characterisation of G protein subtypes. J Neurochem. 2008;105:308–323. doi: 10.1111/j.1471-4159.2007.05131.x. [DOI] [PubMed] [Google Scholar]
- 19.Sakaba T, Neher E. Direct modulation of synaptic vesicle priming by GABA(B) receptor activation at a glutamatergic synapse. Nature. 2003;424:775–778. doi: 10.1038/nature01859. [DOI] [PubMed] [Google Scholar]
- 20.Ghorbel MT, Becker KG, Henley JM. Profile of changes in gene expression in cultured hippocampal neurones evoked by the GABAB receptor agonist baclofen. Physiol Genomics. 2005;22:93–98. doi: 10.1152/physiolgenomics.00202.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Helm KA, Haberman RP, Dean SL, Hoyt EC, Melcher T, Lund PK, Gallagher M. GABAB receptor antagonist SGS742 improves spatial memory and reduces protein binding to the cAMP response element (CRE) in the hippocampus. Neuropharmacology. 2005;48:956–964. doi: 10.1016/j.neuropharm.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 22.Barthel F, Kienlen Campard P, Demeneix BA, Feltz P, Loeffler JP. GABAB receptors negatively regulate transcription in cerebellar granular neurons through cyclic AMP responsive element binding protein-dependent mechanisms. Neuroscience. 1996;70:417–427. doi: 10.1016/0306-4522(95)00380-0. [DOI] [PubMed] [Google Scholar]
- 23.Guyon A, Leresche N. Modulation by different GABAB receptor types of voltage-activated calcium currents in rat thalamocortical neurones. J Physiol. 1995;485(Pt 1):29–42. doi: 10.1113/jphysiol.1995.sp020710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mintz IM, Bean BP. GABAB receptor inhibition of P-type Ca2+ channels in central neurons. Neuron. 1993;10:889–898. doi: 10.1016/0896-6273(93)90204-5. [DOI] [PubMed] [Google Scholar]
- 25.Chen G, van den Pol AN. Presynaptic GABAB autoreceptor modulation of P/Q-type calcium channels and GABA release in rat suprachiasmatic nucleus neurons. J Neurosci. 1998;18:1913–1922. doi: 10.1523/JNEUROSCI.18-05-01913.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bussières N, El Manira A. GABA(B) receptor activation inhibits N- and P/Q-type calcium channels in cultured lamprey sensory neurons. Brain Res. 1999;847:175–185. doi: 10.1016/s0006-8993(99)02002-8. [DOI] [PubMed] [Google Scholar]
- 27.Lambert NA, Wilson WA. High-threshold Ca2+ currents in rat hippocampal interneurones and their selective inhibition by activation of GABA(B) receptors. J Physiol. 1996;492(Pt 1):115–127. doi: 10.1113/jphysiol.1996.sp021294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- 29.Lüscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron. 1997;19:687–695. doi: 10.1016/s0896-6273(00)80381-5. [DOI] [PubMed] [Google Scholar]
- 30.Andrade R, Malenka RC, Nicoll RA. A G protein couples serotonin and GABAB receptors to the same channels in hippocampus. Science. 1986;234:1261–1265. doi: 10.1126/science.2430334. [DOI] [PubMed] [Google Scholar]
- 31.Inanobe A, Morishige KI, Takahashi N, Ito H, Yamada M, Takumi T, Nishina H, Takahashi K, Kanaho Y, Katada T. G beta gamma directly binds to the carboxyl terminus of the G protein-gated muscarinic K+ channel, GIRK1. Biochem Biophys Res Commun. 1995;212:1022–1028. doi: 10.1006/bbrc.1995.2072. [DOI] [PubMed] [Google Scholar]
- 32.Huang CL, Slesinger PA, Casey PJ, Jan YN, Jan LY. Evidence that direct binding of G beta gamma to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron. 1995;15:1133–1143. doi: 10.1016/0896-6273(95)90101-9. [DOI] [PubMed] [Google Scholar]
- 33.Wickman KD, Iñiguez-Lluhl JA, Davenport PA, Taussig R, Krapivinsky GB, Linder ME, Gilman AG, Clapham DE. Recombinant G-protein beta gamma-subunits activate the muscarinic-gated atrial potassium channel. Nature. 1994;368:255–257. doi: 10.1038/368255a0. [DOI] [PubMed] [Google Scholar]
- 34.Reuveny E, Slesinger PA, Inglese J, Morales JM, Iñiguez-Lluhi JA, Lefkowitz RJ, Bourne HR, Jan YN, Jan LY. Activation of the cloned muscarinic potassium channel by G protein beta gamma subunits. Nature. 1994;370:143–146. doi: 10.1038/370143a0. [DOI] [PubMed] [Google Scholar]
- 35.Saint DA, Thomas T, Gage PW. GABAB agonists modulate a transient potassium current in cultured mammalian hippocampal neurons. Neurosci Lett. 1990;118:9–13. doi: 10.1016/0304-3940(90)90236-3. [DOI] [PubMed] [Google Scholar]
- 36.Fernández-Alacid L, Aguado C, Ciruela F, Martín R, Colón J, Cabañero MJ, Gassmann M, Watanabe M, Shigemoto R, Wickman K, et al. Subcellular compartment-specific molecular diversity of pre- and post-synaptic GABA-activated GIRK channels in Purkinje cells. J Neurochem. 2009;110:1363–1376. doi: 10.1111/j.1471-4159.2009.06229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ladera C, del Carmen Godino M, José Cabañero M, Torres M, Watanabe M, Luján R, Sánchez-Prieto J. Pre-synaptic GABA receptors inhibit glutamate release through GIRK channels in rat cerebral cortex. J Neurochem. 2008;107:1506–1517. doi: 10.1111/j.1471-4159.2008.05712.x. [DOI] [PubMed] [Google Scholar]
- 38.Pérez-Garci E, Gassmann M, Bettler B, Larkum ME. The GABAB1b isoform mediates long-lasting inhibition of dendritic Ca2+ spikes in layer 5 somatosensory pyramidal neurons. Neuron. 2006;50:603–616. doi: 10.1016/j.neuron.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 39.Kavalali ET, Zhuo M, Bito H, Tsien RW. Dendritic Ca2+ channels characterized by recordings from isolated hippocampal dendritic segments. Neuron. 1997;18:651–663. doi: 10.1016/s0896-6273(00)80305-0. [DOI] [PubMed] [Google Scholar]
- 40.Chalifoux JR, Carter AG. GABAB receptor modulation of synaptic function. Curr Opin Neurobiol. 2011;21:339–344. doi: 10.1016/j.conb.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sabatini BL, Svoboda K. Analysis of calcium channels in single spines using optical fluctuation analysis. Nature. 2000;408:589–593. doi: 10.1038/35046076. [DOI] [PubMed] [Google Scholar]
- 42.Kulik A, Vida I, Fukazawa Y, Guetg N, Kasugai Y, Marker CL, Rigato F, Bettler B, Wickman K, Frotscher M, et al. Compartment-dependent colocalization of Kir3.2-containing K+ channels and GABAB receptors in hippocampal pyramidal cells. J Neurosci. 2006;26:4289–4297. doi: 10.1523/JNEUROSCI.4178-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park HW, Jung H, Choi KH, Baik JH, Rhim H. Direct interaction and functional coupling between voltage-gated CaV1.3 Ca2+ channel and GABAB receptor subunit 2. FEBS Lett. 2010;584:3317–3322. doi: 10.1016/j.febslet.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 44.Bowery NG, Hill DR, Hudson AL, Doble A, Middlemiss DN, Shaw J, Turnbull M. (-)Baclofen decreases neurotransmitter release in the mammalian CNS by an action at a novel GABA receptor. Nature. 1980;283:92–94. doi: 10.1038/283092a0. [DOI] [PubMed] [Google Scholar]
- 45.Kaupmann K, Huggel K, Heid J, Flor PJ, Bischoff S, Mickel SJ, McMaster G, Angst C, Bittiger H, Froestl W, et al. Expression cloning of GABA(B) receptors uncovers similarity to metabotropic glutamate receptors. Nature. 1997;386:239–246. doi: 10.1038/386239a0. [DOI] [PubMed] [Google Scholar]
- 46.Steiger JL, Bandyopadhyay S, Farb DH, Russek SJ. cAMP response element-binding protein, activating transcription factor-4, and upstream stimulatory factor differentially control hippocampal GABABR1a and GABABR1b subunit gene expression through alternative promoters. J Neurosci. 2004;24:6115–6126. doi: 10.1523/JNEUROSCI.1200-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, Mosbacher J, Bischoff S, Kulik A, Shigemoto R, et al. GABA(B)-receptor subtypes assemble into functional heteromeric complexes. Nature. 1998;396:683–687. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- 48.Kuner R, Köhr G, Grünewald S, Eisenhardt G, Bach A, Kornau HC. Role of heteromer formation in GABAB receptor function. Science. 1999;283:74–77. doi: 10.1126/science.283.5398.74. [DOI] [PubMed] [Google Scholar]
- 49.Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, Yao WJ, Johnson M, Gunwaldsen C, Huang LY, et al. GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature. 1998;396:674–679. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]
- 50.White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, Barnes AA, Emson P, Foord SM, Marshall FH. Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature. 1998;396:679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- 51.Ng GY, Clark J, Coulombe N, Ethier N, Hebert TE, Sullivan R, Kargman S, Chateauneuf A, Tsukamoto N, McDonald T, et al. Identification of a GABAB receptor subunit, gb2, required for functional GABAB receptor activity. J Biol Chem. 1999;274:7607–7610. doi: 10.1074/jbc.274.12.7607. [DOI] [PubMed] [Google Scholar]
- 52.Galvez T, Parmentier ML, Joly C, Malitschek B, Kaupmann K, Kuhn R, Bittiger H, Froestl W, Bettler B, Pin JP. Mutagenesis and modeling of the GABAB receptor extracellular domain support a venus flytrap mechanism for ligand binding. J Biol Chem. 1999;274:13362–13369. doi: 10.1074/jbc.274.19.13362. [DOI] [PubMed] [Google Scholar]
- 53.Galvez T, Prezeau L, Milioti G, Franek M, Joly C, Froestl W, Bettler B, Bertrand HO, Blahos J, Pin JP. Mapping the agonist-binding site of GABAB type 1 subunit sheds light on the activation process of GABAB receptors. J Biol Chem. 2000;275:41166–41174. doi: 10.1074/jbc.M007848200. [DOI] [PubMed] [Google Scholar]
- 54.Bernard P, Guedin D, Hibert M. Molecular modeling of the GABA/GABA(B) receptor complex. J Med Chem. 2001;44:27–35. doi: 10.1021/jm000915o. [DOI] [PubMed] [Google Scholar]
- 55.Liu J, Maurel D, Etzol S, Brabet I, Ansanay H, Pin JP, Rondard P. Molecular determinants involved in the allosteric control of agonist affinity in the GABAB receptor by the GABAB2 subunit. J Biol Chem. 2004;279:15824–15830. doi: 10.1074/jbc.M313639200. [DOI] [PubMed] [Google Scholar]
- 56.Galvez T, Duthey B, Kniazeff J, Blahos J, Rovelli G, Bettler B, Prézeau L, Pin JP. Allosteric interactions between GB1 and GB2 subunits are required for optimal GABA(B) receptor function. EMBO J. 2001;20:2152–2159. doi: 10.1093/emboj/20.9.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Binet V, Brajon C, Le Corre L, Acher F, Pin JP, Prézeau L. The heptahelical domain of GABA(B2) is activated directly by CGP7930, a positive allosteric modulator of the GABA(B) receptor. J Biol Chem. 2004;279:29085–29091. doi: 10.1074/jbc.M400930200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Urwyler S, Gjoni T, Koljatić J, Dupuis DS. Mechanisms of allosteric modulation at GABAB receptors by CGP7930 and GS39783: effects on affinities and efficacies of orthosteric ligands with distinct intrinsic properties. Neuropharmacology. 2005;48:343–353. doi: 10.1016/j.neuropharm.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 59.Duthey B, Caudron S, Perroy J, Bettler B, Fagni L, Pin JP, Prézeau L. A single subunit (GB2) is required for G-protein activation by the heterodimeric GABA(B) receptor. J Biol Chem. 2002;277:3236–3241. doi: 10.1074/jbc.M108900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Havlickova M, Prezeau L, Duthey B, Bettler B, Pin JP, Blahos J. The intracellular loops of the GB2 subunit are crucial for G-protein coupling of the heteromeric gamma-aminobutyrate B receptor. Mol Pharmacol. 2002;62:343–350. doi: 10.1124/mol.62.2.343. [DOI] [PubMed] [Google Scholar]
- 61.Robbins MJ, Calver AR, Filippov AK, Hirst WD, Russell RB, Wood MD, Nasir S, Couve A, Brown DA, Moss SJ, et al. GABA(B2) is essential for g-protein coupling of the GABA(B) receptor heterodimer. J Neurosci. 2001;21:8043–8052. doi: 10.1523/JNEUROSCI.21-20-08043.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Margeta-Mitrovic M, Jan YN, Jan LY. Ligand-induced signal transduction within heterodimeric GABA(B) receptor. Proc Natl Acad Sci USA. 2001;98:14643–14648. doi: 10.1073/pnas.251554798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Margeta-Mitrovic M, Jan YN, Jan LY. Function of GB1 and GB2 subunits in G protein coupling of GABA(B) receptors. Proc Natl Acad Sci USA. 2001;98:14649–14654. doi: 10.1073/pnas.251554498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Margeta-Mitrovic M, Jan YN, Jan LY. A trafficking checkpoint controls GABA(B) receptor heterodimerization. Neuron. 2000;27:97–106. doi: 10.1016/s0896-6273(00)00012-x. [DOI] [PubMed] [Google Scholar]
- 65.Pagano A, Rovelli G, Mosbacher J, Lohmann T, Duthey B, Stauffer D, Ristig D, Schuler V, Meigel I, Lampert C, et al. C-terminal interaction is essential for surface trafficking but not for heteromeric assembly of GABA(b) receptors. J Neurosci. 2001;21:1189–1202. doi: 10.1523/JNEUROSCI.21-04-01189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Calver AR, Robbins MJ, Cosio C, Rice SQ, Babbs AJ, Hirst WD, Boyfield I, Wood MD, Russell RB, Price GW, et al. The C-terminal domains of the GABA(b) receptor subunits mediate intracellular trafficking but are not required for receptor signaling. J Neurosci. 2001;21:1203–1210. doi: 10.1523/JNEUROSCI.21-04-01203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Couve A, Filippov AK, Connolly CN, Bettler B, Brown DA, Moss SJ. Intracellular retention of recombinant GABAB receptors. J Biol Chem. 1998;273:26361–26367. doi: 10.1074/jbc.273.41.26361. [DOI] [PubMed] [Google Scholar]
- 68.Gassmann M, Haller C, Stoll Y, Abdel Aziz S, Biermann B, Mosbacher J, Kaupmann K, Bettler B. The RXR-type endoplasmic reticulum-retention/retrieval signal of GABAB1 requires distant spacing from the membrane to function. Mol Pharmacol. 2005;68:137–144. doi: 10.1124/mol.104.010256. [DOI] [PubMed] [Google Scholar]
- 69.Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- 70.Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- 71.Couve A, Thomas P, Calver AR, Hirst WD, Pangalos MN, Walsh FS, Smart TG, Moss SJ. Cyclic AMP-dependent protein kinase phosphorylation facilitates GABA(B) receptor-effector coupling. Nat Neurosci. 2002;5:415–424. doi: 10.1038/nn833. [DOI] [PubMed] [Google Scholar]
- 72.González-Maeso J, Wise A, Green A, Koenig JA. Agonist-induced desensitization and endocytosis of heterodimeric GABAB receptors in CHO-K1 cells. Eur J Pharmacol. 2003;481:15–23. doi: 10.1016/j.ejphar.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 73.Tosetti P, Bakels R, Colin-Le Brun I, Ferrand N, Gaiarsa JL, Caillard O. Acute desensitization of presynaptic GABAB-mediated inhibition and induction of epileptiform discharges in the neonatal rat hippocampus. Eur J Neurosci. 2004;19:3227–3234. doi: 10.1111/j.0953-816X.2004.03413.x. [DOI] [PubMed] [Google Scholar]
- 74.Pontier SM, Lahaie N, Ginham R, St-Gelais F, Bonin H, Bell DJ, Flynn H, Trudeau LE, McIlhinney J, White JH, et al. Coordinated action of NSF and PKC regulates GABAB receptor signaling efficacy. EMBO J. 2006;25:2698–2709. doi: 10.1038/sj.emboj.7601157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kanaide M, Uezono Y, Matsumoto M, Hojo M, Ando Y, Sudo Y, Sumikawa K, Taniyama K. Desensitization of GABA(B) receptor signaling by formation of protein complexes of GABA(B2) subunit with GRK4 or GRK5. J Cell Physiol. 2007;210:237–245. doi: 10.1002/jcp.20863. [DOI] [PubMed] [Google Scholar]
- 76.Virlon B, Firsov D, Cheval L, Reiter E, Troispoux C, Guillou F, Elalouf JM. Rat G protein-coupled receptor kinase GRK4: identification, functional expression, and differential tissue distribution of two splice variants. Endocrinology. 1998;139:2784–2795. doi: 10.1210/endo.139.6.6078. [DOI] [PubMed] [Google Scholar]
- 77.Sallese M, Salvatore L, D’Urbano E, Sala G, Storto M, Launey T, Nicoletti F, Knöpfel T, De Blasi A. The G-protein-coupled receptor kinase GRK4 mediates homologous desensitization of metabotropic glutamate receptor 1. FASEB J. 2000;14:2569–2580. doi: 10.1096/fj.00-0072com. [DOI] [PubMed] [Google Scholar]
- 78.Perroy J, Adam L, Qanbar R, Chénier S, Bouvier M. Phosphorylation-independent desensitization of GABA(B) receptor by GRK4. EMBO J. 2003;22:3816–3824. doi: 10.1093/emboj/cdg383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ando Y, Hojo M, Kanaide M, Takada M, Sudo Y, Shiraishi S, Sumikawa K, Uezono Y. S(+)-ketamine suppresses desensitization of γ-aminobutyric acid type B receptor-mediated signaling by inhibition of the interaction of γ-aminobutyric acid type B receptors with G protein-coupled receptor kinase 4 or 5. Anesthesiology. 2011;114:401–411. doi: 10.1097/ALN.0b013e318204e003. [DOI] [PubMed] [Google Scholar]
- 80.Schwenk J, Metz M, Zolles G, Turecek R, Fritzius T, Bildl W, Tarusawa E, Kulik A, Unger A, Ivankova K, et al. Native GABA(B) receptors are heteromultimers with a family of auxiliary subunits. Nature. 2010;465:231–235. doi: 10.1038/nature08964. [DOI] [PubMed] [Google Scholar]
- 81.Cruz HG, Ivanova T, Lunn ML, Stoffel M, Slesinger PA, Lüscher C. Bi-directional effects of GABA(B) receptor agonists on the mesolimbic dopamine system. Nat Neurosci. 2004;7:153–159. doi: 10.1038/nn1181. [DOI] [PubMed] [Google Scholar]
- 82.Fairfax BP, Pitcher JA, Scott MG, Calver AR, Pangalos MN, Moss SJ, Couve A. Phosphorylation and chronic agonist treatment atypically modulate GABAB receptor cell surface stability. J Biol Chem. 2004;279:12565–12573. doi: 10.1074/jbc.M311389200. [DOI] [PubMed] [Google Scholar]
- 83.Grampp T, Sauter K, Markovic B, Benke D. Gamma-aminobutyric acid type B receptors are constitutively internalized via the clathrin-dependent pathway and targeted to lysosomes for degradation. J Biol Chem. 2007;282:24157–24165. doi: 10.1074/jbc.M702626200. [DOI] [PubMed] [Google Scholar]
- 84.Grampp T, Notz V, Broll I, Fischer N, Benke D. Constitutive, agonist-accelerated, recycling and lysosomal degradation of GABA(B) receptors in cortical neurons. Mol Cell Neurosci. 2008;39:628–637. doi: 10.1016/j.mcn.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 85.Mutneja M, Berton F, Suen KF, Lüscher C, Slesinger PA. Endogenous RGS proteins enhance acute desensitization of GABA(B) receptor-activated GIRK currents in HEK-293T cells. Pflugers Arch. 2005;450:61–73. doi: 10.1007/s00424-004-1367-1. [DOI] [PubMed] [Google Scholar]
- 86.Kuramoto N, Wilkins ME, Fairfax BP, Revilla-Sanchez R, Terunuma M, Tamaki K, Iemata M, Warren N, Couve A, Calver A, et al. Phospho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron. 2007;53:233–247. doi: 10.1016/j.neuron.2006.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Carling D. AMP-activated protein kinase: balancing the scales. Biochimie. 2005;87:87–91. doi: 10.1016/j.biochi.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 88.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 89.Brock C, Boudier L, Maurel D, Blahos J, Pin JP. Assembly-dependent surface targeting of the heterodimeric GABAB Receptor is controlled by COPI but not 14-3-3. Mol Biol Cell. 2005;16:5572–5578. doi: 10.1091/mbc.E05-05-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Vargas KJ, Terunuma M, Tello JA, Pangalos MN, Moss SJ, Couve A. The availability of surface GABA B receptors is independent of gamma-aminobutyric acid but controlled by glutamate in central neurons. J Biol Chem. 2008;283:24641–24648. doi: 10.1074/jbc.M802419200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pooler AM, Gray AG, McIlhinney RA. Identification of a novel region of the GABA(B2) C-terminus that regulates surface expression and neuronal targeting of the GABA(B) receptor. Eur J Neurosci. 2009;29:869–878. doi: 10.1111/j.1460-9568.2009.06636.x. [DOI] [PubMed] [Google Scholar]
- 92.Hannan S, Wilkins ME, Dehghani-Tafti E, Thomas P, Baddeley SM, Smart TG. GABAB receptor internalisation is regulated by the R2 subunit. J Biol Chem. 2011:Epub ahead of print. doi: 10.1074/jbc.M110.220814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wilkins ME, Li X, Smart TG. Tracking cell surface GABAB receptors using an alpha-bungarotoxin tag. J Biol Chem. 2008;283:34745–34752. doi: 10.1074/jbc.M803197200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maier PJ, Marin I, Grampp T, Sommer A, Benke D. Sustained glutamate receptor activation down-regulates GABAB receptors by shifting the balance from recycling to lysosomal degradation. J Biol Chem. 2010;285:35606–35614. doi: 10.1074/jbc.M110.142406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Laffray S, Tan K, Dulluc J, Bouali-Benazzouz R, Calver AR, Nagy F, Landry M. Dissociation and trafficking of rat GABAB receptor heterodimer upon chronic capsaicin stimulation. Eur J Neurosci. 2007;25:1402–1416. doi: 10.1111/j.1460-9568.2007.05398.x. [DOI] [PubMed] [Google Scholar]
- 96.Ungewickell EJ, Hinrichsen L. Endocytosis: clathrin-mediated membrane budding. Curr Opin Cell Biol. 2007;19:417–425. doi: 10.1016/j.ceb.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 97.Terunuma M, Vargas KJ, Wilkins ME, Ramírez OA, Jaureguiberry-Bravo M, Pangalos MN, Smart TG, Moss SJ, Couve A. Prolonged activation of NMDA receptors promotes dephosphorylation and alters postendocytic sorting of GABAB receptors. Proc Natl Acad Sci USA. 2010;107:13918–13923. doi: 10.1073/pnas.1000853107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guetg N, Abdel Aziz S, Holbro N, Turecek R, Rose T, Seddik R, Gassmann M, Moes S, Jenoe P, Oertner TG, et al. NMDA receptor-dependent GABAB receptor internalization via CaMKII phosphorylation of serine 867 in GABAB1. Proc Natl Acad Sci USA. 2010;107:13924–13929. doi: 10.1073/pnas.1000909107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- 100.Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol. 2009;10:623–635. doi: 10.1038/nrm2745. [DOI] [PubMed] [Google Scholar]
- 101.Kantamneni S, Holman D, Wilkinson KA, Corrêa SA, Feligioni M, Ogden S, Fraser W, Nishimune A, Henley JM. GISP binding to TSG101 increases GABA receptor stability by down-regulating ESCRT-mediated lysosomal degradation. J Neurochem. 2008;107:86–95. doi: 10.1111/j.1471-4159.2008.05580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458:445–452. doi: 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- 103.Vigot R, Barbieri S, Bräuner-Osborne H, Turecek R, Shigemoto R, Zhang YP, Luján R, Jacobson LH, Biermann B, Fritschy JM, et al. Differential compartmentalization and distinct functions of GABAB receptor variants. Neuron. 2006;50:589–601. doi: 10.1016/j.neuron.2006.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vogt KE, Nicoll RA. Glutamate and gamma-aminobutyric acid mediate a heterosynaptic depression at mossy fiber synapses in the hippocampus. Proc Natl Acad Sci USA. 1999;96:1118–1122. doi: 10.1073/pnas.96.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chalifoux JR, Carter AG. GABAB receptors modulate NMDA receptor calcium signals in dendritic spines. Neuron. 2010;66:101–113. doi: 10.1016/j.neuron.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pan BX, Dong Y, Ito W, Yanagawa Y, Shigemoto R, Morozov A. Selective gating of glutamatergic inputs to excitatory neurons of amygdala by presynaptic GABAb receptor. Neuron. 2009;61:917–929. doi: 10.1016/j.neuron.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cimarosti H, Kantamneni S, Henley JM. Ischaemia differentially regulates GABA(B) receptor subunits in organotypic hippocampal slice cultures. Neuropharmacology. 2009;56:1088–1096. doi: 10.1016/j.neuropharm.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Anju TR, Abraham PM, Antony S, Paulose CS. Alterations in cortical GABAB receptors in neonatal rats exposed to hypoxic stress: role of glucose, oxygen, and epinephrine resuscitation. Mol Cell Biochem. 2010;343:1–11. doi: 10.1007/s11010-010-0491-9. [DOI] [PubMed] [Google Scholar]
- 109.Blundon JA, Zakharenko SS. Dissecting the components of long-term potentiation. Neuroscientist. 2008;14:598–608. doi: 10.1177/1073858408320643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ramírez OA, Vidal RL, Tello JA, Vargas KJ, Kindler S, Härtel S, Couve A. Dendritic assembly of heteromeric gamma-aminobutyric acid type B receptor subunits in hippocampal neurons. J Biol Chem. 2009;284:13077–13085. doi: 10.1074/jbc.M900575200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Scita G, Di Fiore PP. The endocytic matrix. Nature. 2010;463:464–473. doi: 10.1038/nature08910. [DOI] [PubMed] [Google Scholar]