

Abstract
The tumor suppressor Brca1 plays an important role in protecting mammalian cells against genomic instability, but little is known about its modes of action. In this work we demonstrate that recombinant human Brca1 protein binds strongly to DNA, an activity conferred by a domain in the center of the Brca1 polypeptide. As a result of this binding, Brca1 inhibits the nucleolytic activities of the Mre11/Rad50/Nbs1 complex, an enzyme implicated in numerous aspects of double-strand break repair. Brca1 displays a preference for branched DNA structures and forms protein–DNA complexes cooperatively between multiple DNA strands, but without DNA sequence specificity. This fundamental property of Brca1 may be an important part of its role in DNA repair and transcription.
The tumor suppressor gene BRCA1 was cloned several years ago through its link to inherited breast cancer (1). Since then, hundreds of mutations in the BRCA1 gene have been found in affected families. Approximately 50% of inherited breast cancer cases are estimated to result from BRCA1 mutations, and nearly all families with a history of both ovarian and breast cancer carry mutations in the gene (2). Studies of mammalian cells deficient in Brca1 have suggested that it is involved in DNA double-strand break repair, transcription-coupled repair, and cell cycle control, all of which are important for maintaining genomic stability (for a review, see ref. 3).
One of the early clues linking Brca1 to DNA repair was its association with Rad51, the primary RecA homolog in eukaryotic cells (4). The Brca1 protein colocalizes with Rad51 in nuclear dots during S phase and in response to DNA damage, suggesting that it may also be involved in homologous recombination and recombinational repair. The proliferation defects and embryonic lethality observed in mice with targeted disruptions of the BRCA1 gene (5–9) are very similar to the phenotypes of mice lacking Rad51 or Brca2, another factor associated with familial breast cancer (10, 11). All of these embryos are sensitive to ionizing radiation, exhibit high levels of chromosomal abnormalities, and can be partially rescued by p53 mutations.
Recently, Brca1 was also found to associate with Rad50, part of the Mre11/Rad50/Nbs1(nibrin) complex (M/R/N) (12, 13), which is known to be involved in both nonhomologous end-joining and homologous recombination in yeast and vertebrate cells (14–18). The Nbs1 component of the complex is phosphorylated in response to DNA damage by ATM (19–22), a kinase that also phosphorylates Brca1 after the introduction of double-strand breaks (23, 24). The Brca1 foci, which appear after ionizing radiation, colocalize, in a subset of the cell population, with nuclear foci formed by M/R/N (12, 13), again suggesting a link between Brca1 and the cellular response to DNA double-strand breaks. How these foci form and what draws Brca1 to these foci are unknown.
Another consequence of ionizing radiation is the accumulation of oxidized bases, which are removed preferentially from transcriptionally active genes in a process known as transcription-coupled repair. Brca1-deficient cells exhibit defects in transcription-coupled repair, suggesting a link between Brca1 and base excision repair (25). This link may be manifested through Brca1 association with mismatch repair enzymes which are required for transcription-coupled repair (12). Alternatively, the link to transcription-coupled repair may be through transcription, as Brca1 has been reported to associate with components of the RNA polymerase II holoenzyme (26) and the chromatin remodeling complex SWI/SNF (27).
In addition to direct repair of DNA damage, the cellular response to DNA-damaging agents relies on checkpoint mechanisms to prevent cells with damage from traversing the cell cycle. Brca1 also plays a role in these systems, as evidenced by the defective G2-M checkpoint in mouse cells lacking exon 11 of Brca1 (28) and by its influence on the expression of several genes involved in checkpoint functions, including p53, p21, and GADD45 (29–32). ATM phosphorylation of the CtIP protein was recently found to regulate the association between Brca1 and CtIP, which in turn affects GADD45 expression, thus identifying another link between ATM, Brca1, and cell cycle control (32).
Brca1 is clearly an important component of the mammalian response to DNA damage; however, very little is known about the mechanisms of its action in DNA repair. In this work we demonstrate that Brca1 inhibits the exonuclease activities of the M/R/N complex. This inhibition is a result of very strong, sequence-nonspecific DNA binding by Brca1 protein, mediated by a domain in the center of the Brca1 polypeptide. Both the full-length protein and the isolated DNA-binding domain exhibit a preference for branched DNA structures; this property may underlie the observed correlation of this protein with double-strand break repair.
Materials and Methods
Protein Expression and Purification from Insect Cells.
Full-length human Brca1 was expressed with the use of the transfer vector pDC218, which was made by cloning full-length BRCA1 into pUNI15 and then transferred to a baculovirus vector by plasmid fusion (33). Baculovirus made from pDC218 was used to infect 600 ml of Sf21 cells, and the culture was harvested after 48 h and frozen in liquid nitrogen. The cell pellet was thawed and resuspended in buffer A (50 mM Tris⋅HCl, pH 7.4/500 mM NaCl/10% glycerol/5 mM DTT). Cells were lysed by homogenization and brief sonication in the presence of 0.5% Tween-20 and protease inhibitors (Roche Molecular Biochemicals), then centrifuged at 100,000 × g for 2 h at 4°C. The supernatant was loaded onto FLAG antibody resin (Sigma) and washed extensively with buffer A and then with buffer B (50 mM Tris⋅HCl, pH 7.4/10 mM NaCl/10% glycerol/5 mM DTT). Brca1 was eluted with 5 mg/ml FLAG peptide (Sigma) in buffer C (25 mM Tris⋅HCl, pH 7.4/100 mM NaCl/10% glycerol/5 mM DTT/0.1% Tween-20). Fractions containing Brca1 were dialyzed against buffer C for 2 h; aliquots were frozen in liquid nitrogen and stored at −80°C.
Protein Expression and Purification from Escherichia coli.
pDC78, -79, -80, -81, -99, and -208 were transformed into the BL21 DE3 E. coli strain (Novagen). Cells were induced with 0.2 mM isopropyl β-d-thiogalactoside for 2 h at 37°C, harvested, and frozen at −80°C. After thawing, each pellet was resuspended in 50 mM Tris⋅HCl (pH 8.0) containing 10% sucrose. The mixture was adjusted to contain 10 mM DTT, 10 mM EDTA, 0.1 mg/ml lysozyme, and 1 mM PMSF and incubated on ice for 15 min. The lysed cells were adjusted again to contain 0.5 M KCl and 1% Tween-20 and incubated on ice for 15 min before centrifugation at 100,000 × g for 1 h. Each supernatant was loaded onto ≈1 ml glutathione S-transferase (GST)-Sepharose (Amersham Pharmacia) and washed with buffer D (25 mM Tris⋅HCl, pH 8.0/10 mM KCl/10% glycerol/2 mM DTT/1% Tween-20). The fusion proteins were eluted with 10 mM reduced glutathione in buffer D containing 0.1% Tween-20 and dialyzed for 2 h against the same buffer before freezing in liquid nitrogen.
Antibodies.
The Western blots shown in Fig. 1A used antibodies directed against the FLAG epitope (Sigma) or amino acids 1005–1313 of human Brca1 (Ab-4; Oncogene Science). The gel mobility shift assays shown in Fig. 2A included Ab-4 as well as an antibody directed against the N-terminal region of Brca1 (Ab-1, Oncogene Science).
Figure 1.

Brca1 inhibits M/R/N nuclease activity. (A) Recombinant human Brca1 containing a FLAG affinity tag, purified from insect cells, was separated by SDS/PAGE and stained with Coomassie blue. The full-length protein, as well as the truncated product (see text), is shown in the lane marked Brca1. Sizes of molecular weight markers in lane M.W. are as indicated. Western blots of the Brca1 preparation with antibodies specific for the N-terminal FLAG tag or amino acids 1005–1313 of Brca1 are shown at the right. (B) Nuclease assays were performed with ≈25 ng of M/R/N or 25 ng of Mre11 per reaction, as indicated, with varying amounts of Brca1: 20 ng, lanes 3 and 7; 40 ng, lanes 4 and 8, and 80 ng, lanes 5 and 9. The double-stranded DNA substrate was labeled with 32P at the 5′ end of the strand with a recessed 3′ end. The substrate was incubated for 15 min at 37°C with Brca1, and then the Mre11 (or M/R/N, as indicated) was added, and the reaction continued for another 25 min before separation on a denaturing polyacrylamide gel.
Figure 2.

Brca1 binds DNA. (A) Gel mobility shift assays were performed with full-length Brca1 (lanes 2–6) or the isolated truncated product (lanes 7–9). Approximately 10 ng of protein was added in lanes 2 and 7, 20 ng in lanes 3 and 8, and 40 ng in lanes 4–6 and lane 9. The proteins were mixed with a 32P-labeled double-stranded DNA substrate and incubated for 10 min at room temperature before electrophoresis in a 0.7% 1/2× TBE (90 mM Tris/64.6 mM boric acid/2.5 mM EDTA, pH 8.3) agarose gel. In lanes 5 and 6, 100 ng of two different antibodies directed against Brca1 was also included in the binding reaction. (B) Gel mobility shift assays were performed as in A, except with fragments of Brca1 expressed in E. coli as GST fusion proteins. Approximately 300 ng of each fragment was incubated with the DNA substrate per reaction. The amino acid endpoints of each Brca1 fragment are shown above the lanes. (C) Schematic representation of the Brca1 polypeptide and the regions covered by the various fragments expressed in E. coli. The DNA-binding activity of the fragments is indicated in the right column, with ++ denoting the highest level of binding, and + indicating partial levels of binding. The Rad50 binding domain shown for comparison at the bottom is from yeast two-hybrid data by Zhong et al. (13).
DNA Substrates.
In Figs. 1B, 2A and B, 3A, and 4A, lanes 1–3, the double-stranded oligonucleotide substrate was composed of TP74 and TP124 (34); TP74 was labeled with 32P at the 5′ end. The gel shift substrate used in Fig. 3 was composed of DAR40 (labeled with 32P at the 5′ end) annealed with DG113 (35). The branched oligonucleotide substrate used in Fig. 3B, lanes 7 to 9 in Fig. 4A, and Fig. 4B was composed of DG26 (GGCTTAGACACTGTGCACAGTGCTACAGACTGGAACAAAAACCCTGCAG), DG81 (CTGCAGGGTTTTTGTTCCAGTCTGTAGCACTGTGGAAGACAGGCCAGATC), and DG27 (CACAGTGTCTAAGCC); DG26 was labeled with 32P at the 5′ end. The Y-structure substrate used in lanes 4–6 in Fig. 4A was composed of DG26 annealed to DG81; DG26 was labeled with 32P at the 5′ end. The 4-way junction used in Fig. 4, A and B, was composed of TP9 (36) annealed to TP 303 (TGCATGCTGAGACTTCTCATTACACAGTGCTACAGACTGGAACAAAAACCCTGCAG), TP305 (GACCTGGCACGTAGGACAGCATGGGATCTGGCCTGTCTTACAGTACAATGCATTGTACATGAACGTAGCATC), and TP306 (GATGCTACGTTCATGTACAATGCATTGTACTGTAATGAGAAGTCTCAGCATGCA); TP9 was labeled with 32P at the 5′ end in Fig. 4A (unlabeled in Fig. 4B). The linear duplex used as a competitor in Fig. 4B was composed of DG113 annealed with DAR40. The plasmid used in Fig. 4B–D was pUC19, which was linearized in Fig. 4C and Fig. 5 with AflIII and cut into fragments in Fig. 4D with ApaLI, PvuI, and HindIII.
Figure 3.

The isolated DNA-binding domain of Brca1 exhibits activities similar to those of the full-length protein. (A) Nuclease assays were performed as in Fig. 1B, except with the Brca1 fragments added to the reactions with M/R/N protein. Approximately 80 ng of each of the fragments was added to the DNA before incubation with M/R/N. (B) Gel mobility shift assays were performed as in Fig. 2A, except here comparing titrations of full-length Brca1 with the 452-1079 Brca1 fragment. Amounts of full-length protein added were 6.3 ng (lane 2), 12.5 ng (lane 3), 25 ng (lane 4), and 50 ng (lane 5). Amounts of 452-1079 fragment added were 30 ng (lane 6), 62.5 ng (lane 7), 125 ng (lane 8), 250 ng (lane 9), and 500 ng (lane 10). The DNA substrate in this experiment was a 32P-labeled branched DNA with a 3′ flap, and all reactions contained 2 mM MgCl2.
Figure 4.

Brca1 binds preferentially to branched structures and long DNAs. (A) Gel mobility shift assays were performed with 25 ng of full-length Brca1, added to reactions as indicated, on 32P-labeled linear (lanes 1–3), Y-structure (lanes 4–6), 3′ flap (lanes 7–9), and four-way junction (lanes 10–12) substrates, shown diagrammatically at the top of the figure. The flaps on the branched substrates were 16 nt in length; for other details on the substrates see Materials and Methods. Reactions contained either 2 mM MgCl2 (+) or 0.5 mM EDTA (−) and were separated in 0.7% agarose gels. (B) Gel mobility shift assays were performed as in A with 25 ng of full-length Brca1 on the 32P-labeled flap substrate. Varying amounts of unlabeled competitor DNAs were also present from the start of the reactions. Linear duplex DNA was added in lanes 2–5, four-way junctions in lanes 7–10, and supercoiled plasmid DNA in lanes 12–15. The amounts of competitor were varied from 1.56 ng to 100 ng, as indicated. The arrow identifies an additional band formed when unlabeled plasmid is present in the binding reaction, suggesting that both the labeled DNA substrate and unlabeled plasmid DNA are present in the complex. (C) Quantitative comparison of Brca1 DNA binding to the branched substrate in the presence of various DNA competitors. The amounts of protein–DNA complex II formed in the presence of linear, branched, four-way junction, supercoiled plasmid, and linearized plasmid competitors are shown in relation to the total amount of labeled substrate present in each reaction. (D) Gel mobility shift assays were performed as in A on three different lengths of 32P-labeled linear DNA fragments, as indicated at the top of the figure. The amounts of full-length Brca1 present in the reactions were 0.9 ng (lanes 2, 8, 14), 1.8 ng (lanes 3, 9, 15), 3.75 ng (lanes 4, 10, 16), 7.5 ng (5, 11, 17), and 15 ng (lanes 6, 12, 18).
Figure 5.
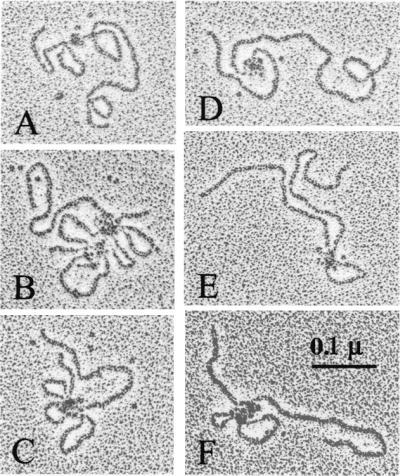
Brca1 DNA binding visualized by electron microscopy. The 452-1079 aa fragment of Brca1 was incubated with linearized pUC19 DNA, sprayed onto carbon grids, rotary shadowed, and visualized in an electron microscope. The complexes shown in A–F were representative of the total population of protein-bound DNA molecules observed.
Reaction Conditions.
Nuclease reactions contained 25 mM 4-morpholinepropanesulfonic acid (pH 7.0), 40–60 mM NaCl, 4 mM DTT, 1 mM MnCl2, 0.1 mg/ml BSA, and 0.05 pmol DNA substrate in a volume of 10 μl. Reactions were incubated at 37°C for 30 min before stopping with 0.5% SDS and 10 mM EDTA, separation on a 15% denaturing polyacrylamide gel, and PhosphorImager analysis (Molecular Dynamics).
Gel mobility shift assays contained 20 mM Tris⋅HCl (pH 7.4), 40–60 mM NaCl, 4 mM DTT, 0.1 mg/ml BSA, 0.1% Triton X-100, and 0.01–0.05 pmol DNA substrate in a volume of 10 μl. Assays shown in Fig. 3B, Fig. 4A (as indicated), and Fig. 4D also contained 2 mM MgCl2. Reactions were incubated at room temperature before loading on a 0.7% agarose gel and run at 5 V/cm for 2–4 h. The gels were dried onto DE81 paper (Whatman) and analyzed by PhosphorImager. Kd values were obtained by estimating protein concentrations (by Bradford assay and intensity on SDS/PAGE gels) at which 50% of the probe was present in a bound complex.
Electron Microscopy.
Binding reactions (20 μl) contained 40 ng 452-1079 GST-fusion Brca1 fragment and 50 ng pUC19 DNA linearized with AflIII, as well as 25 mM 4-morpholinepropanesulfonic acid buffer (pH 7), 4 mM DTT, 50 mM NaCl, and 5 mM MgCl2. A reaction containing equivalent amounts of the 1021–1552 aa fragment of Brca1 and linearized pUC19 was done in parallel. The samples were prepared for rotary shadowing as previously described (37). The shadowing was performed in a Balzers 301 freeze fracture apparatus at room temperature, with a platinum evaporation angle of 7°.
Results
To investigate the biochemical properties of Brca1, the full-length protein with an N-terminal affinity tag (FLAG) was expressed in insect cells. The protein was expressed poorly, but a small amount of the full-length polypeptide was recovered (Fig. 1A). In addition to full-length Brca1 (208 kDa), a smaller product of ≈70 kDa was recovered, which coeluted with the full-length protein in ion exchange and gel filtration separations (“truncated”). Both the 70-kDa and the 208-kDa products reacted with antibodies directed against the N terminus of Brca1, whereas only the full-length product reacted with an antibody directed against residues in the C-terminal half of Brca1 (Fig. 1A). Thus the smaller product is most likely a C-terminal truncation of the full-length protein. Because the N-terminal domain of Brca1 can mediate homodimerization (38), it is likely that the full-length and truncated proteins are present in a multimeric complex. A small amount of the truncated protein was isolated (data not shown), but preparations of the full-length protein always contained some of the truncated product.
Brca1 Inhibits M/R/N Nuclease Activity.
The association of Brca1 with the M/R/N complex (12) led us to consider the possibility that Brca1 might regulate the nuclease activity of M/R/N. As shown in Fig. 1B, the presence of Brca1 inhibited exonuclease activity by recombinant human M/R/N complex on a 5′ end-labeled linear substrate (lanes 3–5). However, the full-length preparation also inhibited exonuclease activity by Mre11 alone (lanes 7–9), indicating that interactions with Rad50 are not responsible for the inhibition. Brca1 itself has no detectable nuclease activity (data not shown).
Brca1 Binds DNA.
Because the inhibitory activity of Brca1 appeared to be independent of protein–protein interactions, we attempted to determine whether our Brca1 preparation interacted with DNA directly. In a gel mobility shift assay, full-length Brca1 retarded the mobility of a 50-bp double-stranded DNA fragment (Fig. 2A, lanes 2–4). This complex could be supershifted with two different antibodies directed against Brca1, confirming that the protein is indeed present in the shifted species (lanes 5 and 6). The truncated product by itself did not form a complex with the labeled DNA (Fig. 2A, lanes 7–9). All gel mobility shift assays were performed in low-percentage agarose gels, as the complexes were too large to enter polyacrylamide matrices (data not shown).
To determine which domain of Brca1 might be responsible for DNA binding, we tested GST fusions of different fragments of Brca1 expressed in E. coli (23). A polypeptide containing amino acids 452-1079 caused the same mobility shift as the full-length protein (Fig. 2B, lane 3). A smaller fragment within that region, amino acids 504–802, also bound to the probe, but less efficiently (lane 6). A diagram of the fragments in relation to the full-length Brca1 ORF is shown in Fig. 2C. Removal of the GST affinity tag from the 452-1079 fragment increased the mobility of the protein–DNA complex but had no effect on the overall level of DNA binding exhibited by the protein (data not shown). Therefore, the GST domain was not artificially promoting cooperative interactions.
When we tested the various fragments of Brca1 expressed in E. coli, we found that the fragments that exhibited DNA binding activity (452–1079 and 504–802) were also the only ones that inhibited the M/R/N exonuclease (Fig. 3A, lanes 4 and 7). The inhibition of nuclease activity thus fully correlates with the DNA-binding activity of Brca1.
Both the full-length Brca1 preparation and the 452-1079 fragment yield two types of complexes with DNA, designated I and II (Fig. 3B). Complex I migrates close to the position of the unbound probe but is progressively retarded in mobility with increasing protein concentrations. Complex II, in contrast, has very low mobility in the agarose gel and appears to form cooperatively from complex I. Complex II also becomes further retarded in the gel as more and more protein is added (lanes 9 and 10). Data from gel mobility shift assays suggest that the Kd for complex II is ≈250 nM for the 452-1079 fragment (assuming a molecular mass of 96 kDa). Although the low concentration of our full-length Brca1 protein preparation prevents us from obtaining 50% binding under these conditions, the Kd for complex II is estimated to be ≈100 nM (assuming a molecular mass of 208 kDa). The affinity of the Brca1 DNA-binding domain for DNA in complex I is difficult to measure in gel shift assays because of the small separation between the bound and unbound species at low protein concentrations, but we have estimated that its Kd is at least 100- to 1000-fold lower than that of complex II, less than or equal to 0.1–0.3 nM.
Brca1 Exhibits Higher Affinity for Branched DNA Structures.
We observed that Brca1 formed the larger type II complex with 3- to 4-fold higher affinity on DNA containing single-stranded flaps or double-stranded branches, as shown in Fig. 4A. The full-length Brca1 preparation exhibited the highest affinity for four-way junctions, but there were no significant differences among various types of flap structures. The difference in affinity was as much as 6- to 7-fold between linear and junction DNA in reactions containing physiological levels of magnesium.
The higher affinity for branched structures was confirmed in competition assays (Fig. 4B), which showed that a four-way junction competed for binding with a labeled branched substrate 3- to 5-fold better than a linear duplex. The quantitation of these data (Fig. 4C) shows that the affinity increases from linear to increasingly branched structures. Brca1 does not bind strongly to short single-stranded DNA (approximately the same binding characteristics as short linear duplexes; data not shown).
Interestingly, plasmid DNA was found to be at least 2- to 3-fold more effective as a competitor than the best oligonucleotide structures (moles of nucleotides held constant). Quantitation of these data (Fig. 4C) shows that linearizing the plasmid made no difference in its ability to compete with a labeled substrate for Brca1 binding (Fig. 4C). When plasmid DNA was used as an unlabeled competitor in gel shift assays, a much more retarded complex was observed at low concentrations of the competitor (Fig. 4B, lane 12, indicated by the arrow), suggesting that this complex contains both labeled oligonucleotide and unlabeled plasmid DNA species. The appearance of this complex with the addition of the unlabeled plasmid shows that Brca1 proteins can bind multiple DNA strands simultaneously (in this case, a large plasmid molecule and a short labeled oligonucleotide).
Because Brca1 exhibits low affinity for short linear duplexes and high affinity for long plasmid DNA, we looked for a length-dependent transition between the two. Gel shift assays on different sized DNA fragments indeed showed that long fragments of several hundred nucleotides formed complex II much more efficiently than smaller DNAs (Fig. 4D). The transition occurred between 300 and approximately 500 bp. We have not observed any effect of short, single-stranded overhangs on the relative affinity of Brca1 for DNA molecules (data not shown).
The Brca1 DNA-Binding Domain Forms DNA Loops.
To determine what types of structures were being formed by Brca1 on DNA, we rotary-shadowed plasmid DNA bound by the 452-1079 aa fragment and visualized the complexes with an electron microscope. As shown in Fig. 5, the linearized 2.7-kb plasmids can be seen in contact with globular protein molecules. In many cases the DNA formed loops in the presence of the Brca1 fragment, with a large number of proteins bound together at the base of each loop (Fig. 5, B–E). Counts of DNA molecules from randomly acquired micrographs showed that 73% (n = 96) were clearly associated with protein. In contrast, none of the DNA molecules (n = 223) were bound by a fragment containing amino acids 1021–1552 of Brca1 (data not shown), a fragment that does not exhibit DNA binding in gel shift assays. Both polypeptides contained a GST affinity tag and were purified in tandem. Thus the large protein–DNA complexes are seen only with the DNA-binding domain of Brca1.
Consistent with our competition analysis in gel shift assays, the electron microscope data show that the Brca1 DNA-binding domain does not bind to DNA ends and also does not appear to prefer any specific binding location on the plasmid DNA. The size of the loops varied between complexes, as did the distance of the complexes from the DNA ends and the number of loops per complex.
The electron microscope images demonstrate that the 452-1079 fragment multimerizes on DNA binding. It is not possible at this resolution to determine the specific number of fragment monomers present in each complex, but the largest multimers often contain 8–12 protein molecules bound to DNA. In contrast, the free protein visible on the grids appears to be uniform in size and is not aggregated, suggesting that multimerization is specific to the DNA-bound state.
Discussion
The product of the tumor suppressor gene BRCA1 is clearly involved in maintaining genomic stability during cell proliferation, as evidenced by the chromosomal abnormalities in cell lines deficient in Brca1 and the developmental defects and embryonic lethality of Brca1 null mice (for a review see ref. 3). The large, 208-kDa protein has been reported to associate (either directly or indirectly) with a score of different factors involved in DNA repair, cell cycle regulation, and transcription (12, 26, 27), but a coherent mechanistic model for Brca1 action has not yet emerged. In this study we show that Brca1 binds strongly to DNA; this property may play a role in targeting it to sites of DNA replication and damage repair.
Brca1 DNA Binding.
We observed two types of Brca1–DNA complexes in gel mobility shift assays. Type I complexes migrated close to the unbound DNA probe and did not show significant structure specificity. The Kd of this complex was estimated to be less than 0.1–0.3 nM, which represents extremely tight DNA binding for a nonspecific DNA-binding protein.
Type II complexes, on the other hand, exhibit characteristics of cooperative binding by Brca1 and are greatly affected by the structure and length of the DNA molecule. Brca1 forms complex II with a 6- to 7-fold preference for flaps, branched DNA, or four-way junctions over short DNA fragments. On linear DNA, Brca1 forms complex II much more efficiently when the molecules are longer than 300–500 bp. In the electron microscope, these complexes appeared to contain many molecules of Brca1 in a single, large group that bridged several different sites on the plasmid DNA. The large number of Brca1 molecules bound together in the complex is consistent with the cooperativity of complex II formation in the gel assay. In addition, the DNA loops provide an explanation for both the length dependence of complex formation as well as for the large reduction in mobility of the complex through gel matrices.
Brca1 Binding of Multiple DNA Sites.
The electron microscope images also document a few instances where more than one DNA molecule is part of the Brca1 protein–DNA complex (as in Fig. 5B). This situation was anticipated because the Brca1–DNA complex could be supershifted in gel mobility shift assays by the addition of unlabeled plasmid DNA, indicating that both the probe and plasmid were present in the shifted complex. The images of type II complexes suggest that there may be a correlation between the number of Brca1 molecules and the number of DNA strands bound. The largest protein complexes connect six or more DNA sites, whereas smaller protein multimers are associated with only two or three sites. This observed accumulation of proteins into large DNA-bound complexes is also consistent with the gel mobility shifts (Fig. 3B), indicating that complex II can be further retarded by the addition of more protein.
Possible Functions of Brca1 DNA Binding.
Many recent observations link the Brca1 protein to events that take place during S phase. Expression and phosphorylation of Brca1 fluctuate during the cell cycle and peak during S phase (39–41), and Brca1 can be visualized in punctate nuclear dots that largely coincide with Rad51 nuclear dots during DNA replication (4). These foci disappear in response to DNA damage and then reappear at sites that coincide with foci of Rad51, PCNA, and Brca2 and with areas of localized single-strand DNA (42–44). Recently it was demonstrated that the locations of Brca1 foci originate as sites of histone H2AX phosphorylation, which occurs at sites of double-strand breaks (45, 46). The accumulation of all of these factors at sites of DNA damage suggest that Brca1 is targeting areas of the genome that are undergoing damage-induced replication and recombinational repair. Our observation of DNA binding by Brca1 suggests that it may bind directly to DNA intermediates in the form of collapsed replication forks and DNA surrounding double-strand breaks or sites of oxidative damage.
It is also possible that Brca1 may have a protective role in facilitating DNA repair, by preventing attack on naked DNA intermediates. Brca1-deficient mouse cells exhibit a deficiency in homologous repair of double-strand breaks and an increase in nonhomologous mechanisms of repair (47). The DNA repair phenotype of these cells suggests that Brca1 plays an inhibitory role in NHEJ, a scenario consistent with the inhibitory effects of Brca1 DNA binding on M/R/N nuclease activity. The experiments shown here demonstrate the ability of Brca1 to bind to DNA intermediates; how this activity is integrated into the pathways of DNA repair in vivo has yet to be determined.
Acknowledgments
We thank Jenny Hinshaw at the National Institutes of Health for facilitating our electron microscopy experiments. T.T.P. was supported by a Helen Hay Whitney postdoctoral fellowship. S.J.E. is a member of the Howard Hughes Medical Institute.
Abbreviations
- M/R/N
Mre11/Rad50/Nbs1
- GST
glutathione S-transferase
Footnotes
See commentary on page 5952.
References
- 1.Miki Y, Swensen J, Shattuck-Eidens D, Futreal P A, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett L M, Ding W, et al. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- 2.Gayther S A, Pharoah P D, Ponder B A. J Mammary Gland Biol Neoplasia. 1998;3:365–376. doi: 10.1023/a:1018779830743. [DOI] [PubMed] [Google Scholar]
- 3.Deng C X, Scott F. Oncogene. 2000;19:1059–1064. doi: 10.1038/sj.onc.1203269. [DOI] [PubMed] [Google Scholar]
- 4.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston D M. Cell. 1997;88:265–275. doi: 10.1016/s0092-8674(00)81847-4. [DOI] [PubMed] [Google Scholar]
- 5.Shen S X, Weaver Z, Xu X, Li C, Weinstein M, Chen L, Guan X Y, Ried T, Deng C X. Oncogene. 1998;17:3115–3124. doi: 10.1038/sj.onc.1202243. [DOI] [PubMed] [Google Scholar]
- 6.Ludwig T, Chapman D L, Papaioannou V E, Efstratiadis A. Genes Dev. 1997;11:1226–1241. doi: 10.1101/gad.11.10.1226. [DOI] [PubMed] [Google Scholar]
- 7.Liu C Y, Flesken-Nikitin A, Li S, Zeng Y, Lee W H. Genes Dev. 1996;10:1835–1843. doi: 10.1101/gad.10.14.1835. [DOI] [PubMed] [Google Scholar]
- 8.Hakem R, de la Pompa J L, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, et al. Cell. 1996;85:1009–1023. doi: 10.1016/s0092-8674(00)81302-1. [DOI] [PubMed] [Google Scholar]
- 9.Gowen L C, Johnson B L, Latour A M, Sulik K K, Koller B H. Nat Genet. 1996;12:191–194. doi: 10.1038/ng0296-191. [DOI] [PubMed] [Google Scholar]
- 10.Sharan S K, Morimatsu M, Albrecht U, Lim D S, Regel E, Dinh C, Sands A, Eichele G, Hasty P, Bradley A. Nature (London) 1997;386:804–810. doi: 10.1038/386804a0. [DOI] [PubMed] [Google Scholar]
- 11.Lim D S, Hasty P. Mol Cell Biol. 1996;16:7133–7143. doi: 10.1128/mcb.16.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Cortez D, Yazdi P, Neff N, Elledge S J, Qin J. Genes Dev. 2000;14:927–939. [PMC free article] [PubMed] [Google Scholar]
- 13.Zhong Q, Chen C F, Li S, Chen Y, Wang C C, Xiao J, Chen P L, Sharp Z D, Lee W H. Science. 1999;285:747–750. doi: 10.1126/science.285.5428.747. [DOI] [PubMed] [Google Scholar]
- 14.Yamaguchi-Iwai Y, Sonoda E, Sasaki M S, Morrison C, Haraguchi T, Hiraoka Y, Yamashita Y M, Yagi T, Takata M, Price C, et al. EMBO J. 1999;18:6619–6629. doi: 10.1093/emboj/18.23.6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haber J E. Cell. 1998;95:583–586. doi: 10.1016/s0092-8674(00)81626-8. [DOI] [PubMed] [Google Scholar]
- 16.Chen C, Kolodner R D. Nat Genet. 1999;23:81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 17.Bressan D A, Baxter B K, Petrini J H. Mol Cell Biol. 1999;19:7681–7687. doi: 10.1128/mcb.19.11.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boulton S J, Jackson S P. EMBO J. 1998;17:1819–1828. doi: 10.1093/emboj/17.6.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu X, Ranganathan V, Weisman D S, Heine W F, Ciccone D N, O'Neill T B, Crick K E, Pierce K A, Lane W S, Rathbun G, et al. Nature (London) 2000;405:477–482. doi: 10.1038/35013089. [DOI] [PubMed] [Google Scholar]
- 20.Zhao S, Weng Y C, Yuan S S, Lin Y T, Hsu H C, Lin S C, Gerbino E, Song M H, Zdzienicka M Z, Gatti R A, et al. Nature (London) 2000;405:473–477. doi: 10.1038/35013083. [DOI] [PubMed] [Google Scholar]
- 21.Lim D S, Kim S T, Xu B, Maser R S, Lin J, Petrini J H, Kastan M B. Nature (London) 2000;404:613–617. doi: 10.1038/35007091. [DOI] [PubMed] [Google Scholar]
- 22.Gatei M, Young D, Cerosaletti K M, Desai-Mehta A, Spring K, Kozlov S, Lavin M F, Gatti R A, Concannon P, Khanna K. Nat Genet. 2000;25:115–119. doi: 10.1038/75508. [DOI] [PubMed] [Google Scholar]
- 23.Cortez D, Wang Y, Qin J, Elledge S J. Science. 1999;286:1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- 24.Gatei M, Scott S P, Filippovitch I, Soronika N, Lavin M F, Weber B, Khanna K K. Cancer Res. 2000;60:3299–3304. [PubMed] [Google Scholar]
- 25.Gowen L C, Avrutskaya A V, Latour A M, Koller B H, Leadon S A. Science. 1998;281:1009–1012. doi: 10.1126/science.281.5379.1009. [DOI] [PubMed] [Google Scholar]
- 26.Scully R, Anderson S F, Chao D M, Wei W, Ye L, Young R A, Livingston D M, Parvin J D. Proc Natl Acad Sci USA. 1997;94:5605–5610. doi: 10.1073/pnas.94.11.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bochar D A, Wang L, Beniya H, Kinev A, Xue Y, Lane W S, Wang W, Kashanchi F, Shiekhattar R. Cell. 2000;102:257–265. doi: 10.1016/s0092-8674(00)00030-1. [DOI] [PubMed] [Google Scholar]
- 28.Xu X, Weaver Z, Linke S P, Li C, Gotay J, Wang X W, Harris C C, Ried T, Deng C X. Mol Cell. 1999;3:389–395. doi: 10.1016/s1097-2765(00)80466-9. [DOI] [PubMed] [Google Scholar]
- 29.Harkin D P, Bean J M, Miklos D, Song Y H, Truong V B, Englert C, Christians F C, Ellisen L W, Maheswaran S, Oliner J D, et al. Cell. 1999;97:575–586. doi: 10.1016/s0092-8674(00)80769-2. [DOI] [PubMed] [Google Scholar]
- 30.Somasundaram K, Zhang H, Zeng Y X, Houvras Y, Peng Y, Wu G S, Licht J D, Weber B L, El-Deiry W S. Nature (London) 1997;389:187–190. doi: 10.1038/38291. [DOI] [PubMed] [Google Scholar]
- 31.Somasundaram K, MacLachlan T K, Burns T F, Sgagias M, Cowan K H, Weber B L, el-Deiry W S. Oncogene. 1999;18:6605–6614. doi: 10.1038/sj.onc.1203284. [DOI] [PubMed] [Google Scholar]
- 32.Li S, Ting N S, Zheng L, Chen P L, Ziv Y, Shiloh Y, Lee E Y, Lee W H. Nature (London) 2000;406:210–215. doi: 10.1038/35018134. [DOI] [PubMed] [Google Scholar]
- 33.Liu Q, Li M Z, Leibham D, Cortez D, Elledge S J. Curr Biol. 1998;8:1300–1309. doi: 10.1016/s0960-9822(07)00560-x. [DOI] [PubMed] [Google Scholar]
- 34.Paull T T, Gellert M. Proc Natl Acad Sci USA. 2000;97:6409–6414. doi: 10.1073/pnas.110144297. . (First published May 23, 2000; 10.1073/pnas.110144297) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paull T T, Gellert M. Genes Dev. 1999;13:1276–1288. doi: 10.1101/gad.13.10.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paull T T, Gellert M. Mol Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- 37.Tyler J M, Branton D. J Ultrastruct Res. 1980;71:95–102. doi: 10.1016/s0022-5320(80)90098-2. [DOI] [PubMed] [Google Scholar]
- 38.Meza J E, Brzovic P S, King M C, Klevit R E. J Biol Chem. 1999;274:5659–5665. doi: 10.1074/jbc.274.9.5659. [DOI] [PubMed] [Google Scholar]
- 39.Vaughn J P, Davis P L, Jarboe M D, Huper G, Evans A C, Wiseman R W, Berchuck A, Iglehart J D, Futreal P A, Marks J R. Cell Growth Differ. 1996;7:711–715. [PubMed] [Google Scholar]
- 40.Ruffner H, Verma I M. Proc Natl Acad Sci USA. 1997;94:7138–7143. doi: 10.1073/pnas.94.14.7138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen Y, Farmer A A, Chen C F, Jones D C, Chen P L, Lee W H. Cancer Res. 1996;56:3168–3172. [PubMed] [Google Scholar]
- 42.Chen J, Silver D P, Walpita D, Cantor S B, Gazdar A F, Tomlinson G, Couch F J, Weber B L, Ashley T, Livingston D M, Scully R. Mol Cell. 1998;2:317–328. doi: 10.1016/s1097-2765(00)80276-2. [DOI] [PubMed] [Google Scholar]
- 43.Raderschall E, Golub E I, Haaf T. Proc Natl Acad Sci USA. 1999;96:1921–1926. doi: 10.1073/pnas.96.5.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scully R, Chen J, Ochs R L, Keegan K, Hoekstra M, Feunteun J, Livingston D M. Cell. 1997;90:425–435. doi: 10.1016/s0092-8674(00)80503-6. [DOI] [PubMed] [Google Scholar]
- 45.Paull T T, Rogakou E P, Yamazaki V, Kirchgessner C U, Gellert M, Bonner W M. Curr Biol. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 46.Rogakou E P, Boon C, Redon C, Bonner W M. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moynahan M E, Chiu J W, Koller B H, Jasin M. Mol Cell. 1999;4:511–518. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]