

Abstract
Indolizinones are under-explored N-heterocycles that react with exquisite chemo- and stereoselectivity. An exploration of the fundamental reactivity of these azabicycles demonstrates the potential to relay stereochemical information from the ring-fusion to newly formed stereocenters on the bicyclic core. The indolizinone diene undergoes selective hydrogenation and readily participates in Diels–Alder cycloadditions as well as ene reactions. The vinylogous amide embedded in the five-membered ring is resistant to reaction when the diene is in place. However, removal of the diene allows for diastereoselective hydrogenation of, and 1,4-additions to, the vinylogous amide. These fundamental reactions with indolizinones have provided a structurally diverse array of products that hold promise in the context of natural product synthesis.
Introduction
Indolizinones are N-heterocycles that have only recently appeared in the chemical literature.1–5 These 6,5-N-fused bicycles contain eight contiguous sp2-hybridized atoms and a single sp3-carbon atom, which resides at the ring-fusion (see C9 of 1, Fig. 1). Both the dienamine contained within the six-membered ring and the vinylogous amide moiety in the five-membered ring could serve as functional handles, rendering indolizinones as useful precursors to indolizidine natural products, pharmaceutical agents and new materials.
Fig. 1.
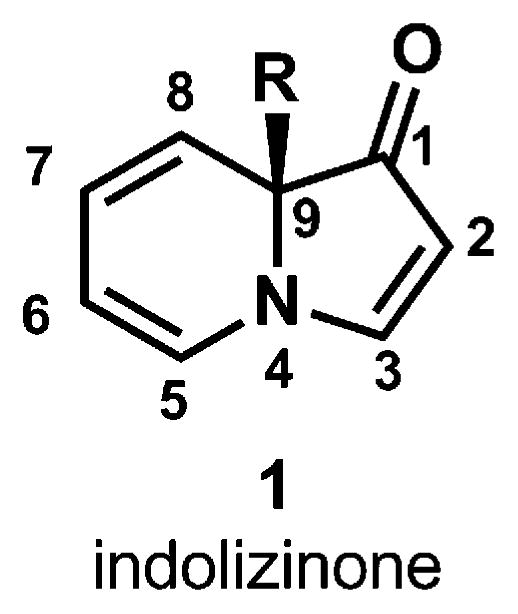
Indolizinone.
Methods for the synthesis of indolizinones were pioneered in our laboratories with platinum(II) and indium(III) salts1,3 and have been further developed by others with salts or complexes of copper,2 silver,6 gold6 and iodine.4 This transformation is proposed to proceed by cyclization of the pyridine moiety (see 2, Fig. 2A) onto a metal-activated alkyne to form an intermediate such as 3 followed by a stereoretentive Wagner–Meerwein shift of the propargylic substituent with concomitant proton transfer to afford the indolizinone product (4). Indolizinones are also accessible from propargylic alcohols using a greener protocol. Both our group7 and the Kim group5 discovered that heating tertiary alcohols such as 5 in a polar protic solvent (e.g., water or ethanol) affords indolizinone products in excellent yields (Fig. 2B).
Fig. 2.

Cycloisomerization of tertiary alcohols to form indolizinones (A) metal-catalyzed cyclization and (B) metal-free cyclization.
Although several protocols for the synthesis of indolizinones have recently emerged, little is known about their innate reactivity. Only two examples of reactions of indolizinones have appeared in the literature. In 2008, Paquette and co-workers described the dimerization of benzannulated indolizinones (see 7, Fig. 3A) via a light promoted [4+2] cyclization between the diene component of one indolizinone molecule and a double bond of the diene from another molecule of the indolizinone.8 The second disclosure of indolizinone chemistry came in 2010 from Kim and co-workers on the palladium-catalyzed cross-coupling of 2-iodoindolizinones (Fig. 3B).9 They demonstrated the competence of 2-iodoindolizinones as the halide component in Heck, Sonogoshira, and Suzuki–Miyaura reactions.
Fig. 3.
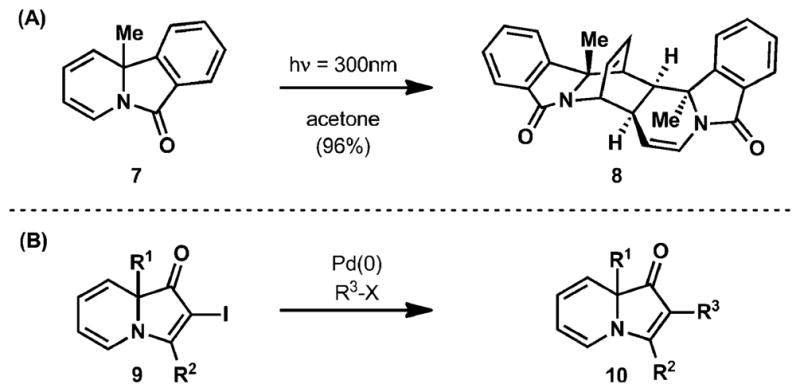
(A) Paquette’s photodimerization of pyrido[2,1-a]isoindol-6(4H)-one (7); (B) palladium-catalyzed cross couplings of 2-iodoin-dolizinone (9).
The relative lack of knowledge regarding indolizinone reactivity compared to other nitrogen-heterocycles (e.g., indoles10 and indolizines), coupled with our desire to use indolizinones as building blocks in the synthesis of complex molecules, spurred our investigation of these bicycles. In addition to understanding the types of reactions indolizinones would participate in, we were intrigued by the potential to relay the stereochemistry of the ring-fusion position (i.e., C9, Fig. 4) to the installation of additional stereocenters. Our studies have revealed the exquisite chemo- and stereoselectivity of several reactions that transform indolizinones into structurally diverse products with high diastereoselectivity in the formation of bonds at C3, C5, C6 and C8 (see asterisked positions in 11). The potential application of these products to the synthesis of indolizidine alkaloids is also discussed.
Fig. 4.
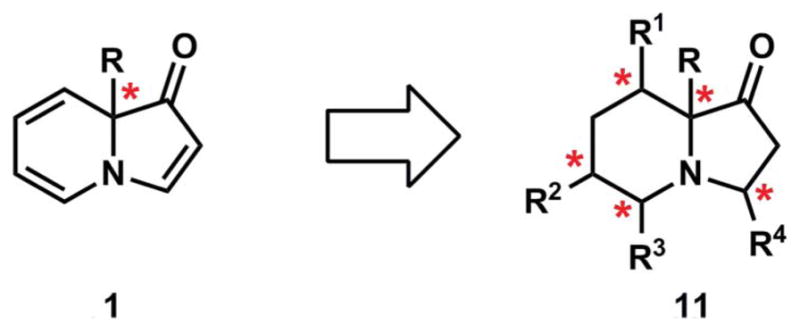
The relay of stereochemistry from the ring-fusion to positions on the indolizinone core.
Results and discussion
1. Hydrogenation
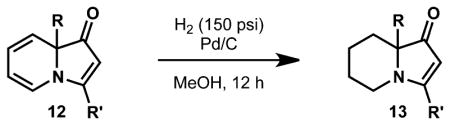
Indolizinones undergo regio- and diastereoselective hydrogenation under a variety of conditions. The electron-rich diene component is selectively reduced in the presence of the relatively electron-poor vinylogous amide double bond. Heterogeneous catalysts, such as Pd/C and RANEY® nickel, reduce only the indolizinone diene using pressures of hydrogen ranging from 1 to 15 atm.
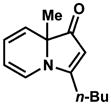
Exposing several indolizinones to Pd/C in methanol under hydrogen (150 psi) for 12 h led to high yields of the corresponding piperidine products (Table 1). Shorter reaction times and lower pressures of hydrogen led to incomplete conversion in some cases. Ethyl acetate and ethanol are also competent solvents for this transformation; however, the use of these solvents also led to incomplete conversion of starting material in some cases. The hydrogenation of indolizinones with alkyl or aryl substituents at C9 (entries 1–2) proceeded without event. Substitution on the vinylogous amide component is also tolerated and does not qualitatively affect the course of the reaction (entries 3–5).
Table 1.
Hydrogenation of the indolizinone diene
| |||
|---|---|---|---|
| Entry | Substrate | Product | Yield |
| 1 |
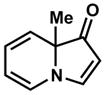 6 |
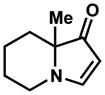 14 |
98% |
| 2 |
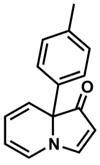 15 |
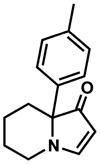 16 |
94% |
| 3 |
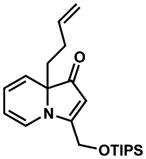 17 |
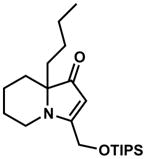 18 |
78% |
| 4 |
 19 |
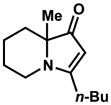 20 |
93% |
| 5 |
 21 |
 22 |
99% |
The hydrogenation of more substituted indolizinones provided insight into the facial selectivity of this transformation. We expected hydrogenation to occur from the less sterically hindered face of the bicycle, opposite the ring-fusion substituent. Tricyclic indolizinones 26 and 29 (Scheme 1) were synthesized and subjected to the hydrogenation conditions. The synthesis of indolizinones 26 and 29 began with a Boekelheide sequence on 2,3-cycloheptenopyridine (23) to regioselectively install the requisite hydroxyl group, affording alcohol 24 in 87% yield over three steps.11 Alcohol 24 was oxidized under Swern conditions to the corresponding ketone followed by 1,2-addition of the lithium acetylide of trimethylsilylacetylene. Subsequent removal of the silyl group provided cyclization substrate 25 in 52% yield over three steps. Upon heating in ethanol, 25 underwent ring-contractive cyclization to afford tricyclic indolizinone 26 in quantitative yield. In parallel, alcohol 24 was elaborated to alkylated indolizinone 29 in five steps (Scheme 1B). Several strategies for alkylation of pyridine 24 at C6 were explored, but ultimately, only the Chichibabin-type addition of n-butyllithium provided a single isomer of the desired product (27) in high yield. Alkylation product 27 was carried forward to indolizinone 29 in a manner analogous to the conversion of alcohol 24 to indolizinone 26.
Scheme 1.

Synthesis of (A) tricyclic indolizinone 26 and (B) alkylated indolizinone 29.
Exposure of 6,6,5-tricycle 26 to Pd/C under 150 psi of hydrogen provided a single diastereomer of the reduction product, which was assigned as 30 (Scheme 2A). The cis-ring fused product (30) constitutes the tricyclic core conserved throughout several members of the cylindricine family of marine alkaloids (e.g., 32, Scheme 2C).12–16 The alternative trans-ring fusion of the six-membered rings, which is not accessed by this method, is present in a related family of alkaloids, the lepadiformines (e.g., 33).17
Scheme 2.

(A) Hydrogenation of tricyclic indolizinone 26, (B) hydrogenation of alkylated indolizinone 29, and (C) select tricyclic marine natural products.
To probe the facial selectivity of the hydrogenation of the dienamine double bond most proximal to the indolizinone nitrogen (Δ5,6), tricyclic indolizinone 29 was exposed to the standard hydrogenation conditions. After 12 h, 29 was converted to 31, whereby the hydrogenation of both double bonds of the diene took place from the less hindered face, providing an entry to the 9-epi-lepadiformine core.
2. Diels–Alder cycloaddition
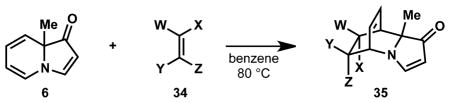
In addition to hydrogenation, the indolizinone dienamine participates in Diels–Alder cycloadditions with high regio- and diastereoselectivity. Using the Diels–Alder reaction, indolizinones are transformed into complex [2.2.2]azabicycles, which can be further processed into a structurally diverse set of products.
Cognizant of the possibility for dimerization of the indolizinone as observed by Paquette and co-workers under photo-irradiation (see Fig. 3A), reactive dienophiles were first investigated. Heating methylindolizinone (6) with maleic anhydride (36) in benzene at 80 °C afforded [2.2.2]azabicycle 37 as a single diastereomer in 96% yield (entry 1, Table 2). Under the same conditions, high yields as well as high diastereoselectivity (>20:1 d.r. by 1H NMR) were also observed in the reaction of methylindolizinone (6) with acrolein (38), methyl acrylate (40), t-butyl acrylate (42) and 2-chloroacrylonitrile (44). Methylindolizinone (6) reacted with 4-phenyl-1,2,4-triazole-3,5-dione (46) at 0 °C in dichloromethane to provide the hetero-Diels–Alder adduct 47 in 73% yield.
Table 2.
Diels–Alder cycloadditions of methylindolizinone (6) with various dienophiles
| |||
|---|---|---|---|
| Entry | Dienophile | Product | Yield |
| 1 |
 36 |
 37 |
96% |
| 2 |
 38 |
 39 |
84% |
| 3 |
 40 |
 41 |
94% |
| 4 |
 42 |
 43 |
91% |
| 6 |
 44 |
 45 |
93% |
| 6 |
 46 |
 47 |
73% |
In a further exploration of dienophiles for the Diels–Alder cycloaddition with methylindolizinone (6), dimethylacetylenedicarboxylate (DMAD, 48) was combined with 6 and heated to 80 °C in benzene for 12 h (Scheme 3). The sole product recovered from this reaction was a highly symmetrical aromatic compound that was identified as dimethyl phthalate (50), which could arise from the retro-Diels–Alder of the anticipated product, [2.2.2]azabicycle 49. Attempts to isolate Diels–Alder adduct 49 were not fruitful, but dimethyl phthalate (50) was consistently isolated as the major product. The generation of dimethyl phthalate (50) via a retro [4+2] of 49 would also produce azacyclopentadienone 51, which could not be isolated or trapped with either dienes or dienophiles.
Scheme 3.

Reaction of methylindolizinone (6) with DMAD (48).
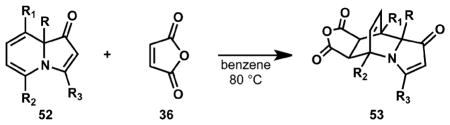
A variety of indolizinones bearing different substituents readily participate in Diels–Alder cycloadditions, which have been investigated with maleic anhydride (Table 3). In addition to varied substitution at the ring fusion of the indolizinone, alkyl and aryl groups on the vinylogous amide component (see entries 4 and 5) do not qualitatively affect the reaction. Tricyclic indolizinone 29 provided an extra challenge in the Diels–Alder reaction, where a productive cycloaddition generates a quaternary and tertiary center in quantitative yield (see 59).
Table 3.
Diels–Alder cycloadditions of indolizinones and maleic anhydride
| |||
|---|---|---|---|
| Entry | Indolizinone | Product | Yield |
| 1 |
 6 |
 37 |
96% |
| 2 |
 54 |
 55 |
67% |
| 3 |
 15 |
 56 |
89% |
| 4 |
 19 |
 57 |
93% |
| 5 |
 21 |
 58 |
91% |
| 6 |
 29 |
 59 |
88% |
As illustrated (Tables 2 and 3), the Diels–Alder reactions consistently afforded a single diastereomer of the [2.2.2]azabicyclic product (>20:1 d.r. by 1H NMR). To determine the relative stereochemistry of the Diels–Alder adducts, X-ray diffraction studies of 55 and 64 were undertaken. The crystal structures of 55 and 64 (Fig. 5) confirm the syn relationship between the unsaturated two-carbon bridge and the C9 substituent that results from the approach of the dienophile from the face of the indolizinone opposite the ring-fusion substituent. Furthermore, the crystal structures of 55 and 64 provide additional information about the Diels–Alder transition states. In 55, the syn relationship between the anhydride moiety and the two-carbon unsaturated bridge suggests an endo-transition state. The cyano group of 64 is anti to the unsaturated bridge providing evidence for a transition state in which the chlorine atom resides in the endo-position.
Fig. 5.

X-ray crystal structures of Diels–Alder adducts 55 and 64.
The rapid generation of complexity afforded by indolizinones through Diels–Alder cycloadditions presents an intriguing opportunity to assemble highly caged skeletons by employing an intramolecular variant. To this end, two indolizinone substrates were investigated which possessed a tethered dienophile: homoallylindolizinone 54 and ester 62. Heating homoallylindolizinone 54 at temperatures up to 120 °C resulted only in recovery of the starting material (Scheme 4). Presumably, the [4+2] cycloaddition of 54 does not proceed because of poorly matched electronics. To access a substrate with an electron deficient dienophile, homoallylindolizinone 54 was converted toα,β-unsaturated ester 62 via cross metathesis with methyl crotonate mediated by the Grubbs II catalyst.
Scheme 4.

Intramolecular Diels–Alder cycloadditions.
Despite the strain inherent in the desired product, heating 62 in m-xylenes at 150 °C for 12 h led to the desired [2.2.2]bicycle 63, which could be isolated in quantitative yield (Scheme 4). The intramolecular Diels–Alder provides a product with a different stereochemical relationship between the C9 ring-fusion substituent and the unsaturated two-carbon bridge as compared to the intermolecular reaction. The tethered dienophile can only approach the diene from the same face of the molecule, resulting in an anti relationship between the unsaturated two-carbon bridge and the ring-fusion substituent. The highly caged skeleton (see 63) generated in the intramolecular Diels–Alder reaction is unprecedented.
With rapid access to [2.2.2]azabicycles, the potential to convert these complex intermediates to indolizidine natural products and natural product-like structures was investigated. Efforts to synthesize tricyclic marine alkaloids (e.g., cylindricine and lepadiformine alkaloids, see 32 and 33, respectively, Fig. 2C) from Diels–Alder adducts led to several unanticipated discoveries.
For example, efforts to remove the chlorine and nitrile groups from [2.2.2]azabicycle 6418 provided an example of the anomalous reactivity of the Diels–Alder adducts (Scheme 5). Exposure of 64 to dissolving metal conditions afforded a single disubstituted aromatic compound, which was identified (by 1H NMR and mass spectral analysis) as 66. Arene 66 could arise from an initial reductive C–Cl bond cleavage to afford anion 65. Subsequent fragmentation to cleave the C–N bond of the [2.2.2]bicycle (see arrows, 65) would form a cyclohexadiene intermediate, which upon oxidation in air, would give arene 66. Although fragmentation of the [2.2.2]azabicycle was unproductive in the synthesis of indolizidine alkaloids, the potential exists for transforming products such as 66 into biologically relevant classes of molecules, such as protease inhibitors.19
Scheme 5.

Fragmentation of [2.2.2]azabicycle under dissolving metal conditions.
The carbon framework of the [2.2.2]azabicycle can be manipulated to afford a functionalized piperidine ring via oxidative cleavage of the unsaturated two-carbon bridge. The oxidative cleavage substrate 70 was accessed in two steps from indolizinone 29 via Diels–Alder cycloaddition with acryloyl chloride and a subsequent Barton decarboxylation (Scheme 6). Attempts to dihydroxylate the unsaturated two-carbon bridge of 70 were accompanied by oxidation of the vinylogous amide moiety. Over-oxidation was avoided by first reducing the double bond of the vinylogous amide, then subjecting the tertiary amine product to catalytic osmium tetroxide followed by sodium periodate. This protocol provided dialdehyde 71 as a mixture with its hydrate forms. Conversion of [2.2.2]azabicycle 70 to functionalized piperidine 71 could be useful in the context of natural product total synthesis. For example, stereoretentive decarbonylation of 71 would afford tricycle 72, which could lead to marine alkaloids such as lepadiformine C (73). Our efforts in this regard have thus far led only to complex mixtures from which 72 could not be isolated.
Scheme 6.

Oxidative cleavage of [2.2.2]azabicycle 70.
Alternatively, the unsaturated two-carbon bridge of [2.2.2]azabicycle 64 (Scheme 7) can be engaged in olefin metathesis reactions. Treatment of [2.2.2]azabicycle 64 with Grubbs second generation catalyst 81 in refluxing benzene effected a ring-opening/ring-closing metathesis to afford tricycle 79 in 73% yield. The syn relationship between the homoallyl group and the two-carbon unsaturated bridge of 64 enforces the stereochemistry in 79, which possesses appropriate functional handles for elaboration to cylindricine K (80).
Scheme 7.

Synthesis of the tricyclic cylindricine core via ring-opening/ring-closing metathesis of 64.
3. Ene reaction
As previously illustrated (Table 2, entry 6), indolizinone dienes can readily engage in Diels–Alder cycloadditions; however, with an appropriate substituent on the diene (e.g., 29, Scheme 8), the ene reaction can be a competing pathway. For example, although cycloaddition of methyl indolizinone 6 with PTAD (46) provided the Diels–Alder adduct 47 (entry 6, Table 2), treatment of tricyclic indolizinone 29 with PTAD (46) gave rise to the ene product (82) as a single diastereomer (Scheme 8).
Scheme 8.

Ene reaction of indolizinone 29 with PTAD.
4. Chemistry of the vinylogous amide
In our exploration of the reactivity of the dienamine moiety of indolizinones, the vinylogous amide was left untouched under the vast majority of the reaction conditions. We, therefore, were interested in learning (1) the identity of nucleophiles and conditions necessary for successful additions to the vinylogous amide and (2) to which position nucleophiles would add. To answer these questions, we first focused our efforts on two classes of nucleophiles: Grignard reagents and hydride sources.
A series of experiments wherein methylindolizinone (6) was treated with Grignard reagents in THF or diethyl ether at temperatures ranging from −78 to −42 °C resulted in the recovery of the indolizinone starting material. At temperatures above −42 °C, only complex mixtures of extremely polar products were obtained. Likewise, treatment of 6 with a variety of hydride sources including sodium borohydride, diisobutylaluminum hydride, and triethylsilane did not accomplish clean reduction and led only to recovery of starting material or the isolation of an unidentifiable mixture of products.
These unfruitful attempts to add nucleophiles to the indolizinone vinylogous amide led us to consider the potential outcomes of nucleophilic addition. In one scenario, the nucleophile could add in a 1,2-fashion to the vinylogous amide to generate an allylic alcohol such as 83 (Fig. 6). This product is a trienamine (83), which was expected to be highly reactive. The alternative reaction pathway, 1,4-addition to the vinylogous amide, would result in dienamine 84. Although dienamine 84 might be slightly more stable than trienamine 83, similar undesired reactions would likely ensue from this product as well.
Fig. 6.

Potential products of nucleophilic addition to the vinylogous amide.
With this insight into the challenges associated with nucleophilic additions to indolizinones, we sought to isolate the vinylogous amide in order to study its chemistry. Methylindolizinone (6) was hydrogenated to provide vinylogous amide 14. Upon treatment of 14 with vinylmagnesium bromide at temperatures ranging from −78 °C to ambient temperature, the starting material was recovered. However, when several equivalents of Grignard reagent were employed in conjunction with increased reaction temperatures, the 1,4-addition product was obtained as a 3:1 mixture of diastereomers 87:86 (Scheme 9). The major diastereomer possesses a trans relationship between the ring-fusion methyl group and the newly added vinyl substituent as established through nOe experiments.
Scheme 9.

Grignard addition to vinylogous amide 14.
It was expected that addition from the more sterically accessible face of the vinylogous amide, opposite the methyl group at C9, would provide a single diastereomer. However, the mixture of diastereomers implied one of the following scenarios: (1) the vinylmagnesium bromide was adding from both faces of the five-membered ring or (2) the enolate, formed as a direct result of 1,4-addition, could collapse to eliminate the nitrogen group (Scheme 10, see 88 → 89). In addition to isolating a mixture of diastereomers of the ketone product (86 and 87), a trace amount of product was obtained that did not possess a terminal vinyl group, yet contained two more carbon atoms than vinylogous amide 14. 1H NMR data for this minor product suggests a mixture of similar products, which were assigned as 6,7-bicycles 90 and 91 that differ only in the position of the double bond contained within the seven-membered ring. Mechanistically, the seven-membered ring could form from the ring-opened intermediate (89) by 1,6-addition of the amide to form the seven-membered ring.
Scheme 10.

Potential mechanism for erosion of diastereoselectivity in conjugate addition.
In order to gain insight into the poor diastereoselectivity observed in the addition of vinyl Grignard to the vinylogous amide, we sought to trap the enolate intermediate (88). In the event the enolate was leading to fragmentation of the five-membered ring, trapping the enolate could avoid racemization of the stereocenter created upon conjugate addition. If instead the lack of diastereoselectivity stemmed from a lack of facial selectivity in the addition, the same mixture of diastereomers should be obtained when the enolate is trapped. In practice, the addition of TBSOTf to the reaction mixture successfully intercepted the enolate and led to a single diastereomer of silyl enol ether 92, which hydrolyzed to afford ketone 87 (Scheme 11). The TBSOTf had two effects on this transformation: (1) a single diastereomer of the product could be obtained and (2) the activation of the vinylogous amide by the TBSOTf made it possible for the nucleophile to add at lower temperatures.
Scheme 11.

Activation and trapping with TBSOTf.
Similar to the addition of carbon nucleophiles, hydrides also add in a conjugate fashion to the vinylogous amide. To achieve synthetically useful yields of the reduced product, it was necessary to first activate the vinylogous amide to increase its electrophilicity. A variety of Lewis acids were screened for this purpose. In addition to silylating reagents such as TBSOTf and TMSOTf that provide silyl enol ether products, boron trifluoride diethyl etherate was also sufficient for amide activation. In parallel with the Lewis acid screen, a variety of hydride sources were evaluated for the conjugate reduction of the vinylogous amide. DIBAL-H, Superhydride, and L-Selectride were competent in this reduction in the presence of an exogenous Lewis acid. Superhydride provided the highest and most reproducible yields (Scheme 12).
Scheme 12.

Conjugate reduction of the vinylogous amide.
Further experimentation unveiled conditions for the selective reduction of the vinylogous amide in the presence of esters, anhydrides, and ketones. Under strongly acidic conditions, the vinylogous amide was reduced by NaCNBH3 (Fig. 7). This reagent, famous for its ability to reduce iminium ions formed in reductive amination over carbonyls, can competently deliver a hydride to the iminium ion (95) formed upon treatment of the vinylogous amide with 2 N HCl.
Fig. 7.

Reduction of the vinylogous amide with sodium cyanoborohydride.
Conclusions
The first in depth exploration of the innate reactivity of indolizinones has uncovered several regio- and stereoselective reactions. Specifically, the indolizinone diene can be hydrogenated, engaged in a Diels–Alder cycloaddition, or participate in an ene reaction. The vinylogous amide has proven to be significantly less reactive, requiring activation for the addition of nucleophiles to occur.
Beyond illuminating the reactivity of indolizinones, we anticipate that the reactions discussed herein, will set the stage for the synthesis of complex molecules from indolizinone building blocks. Our initial endeavors have provided access to the core of tricyclic marine alkaloids such as the cylindricines and lepadiformines. We intend to apply these new discoveries in indolizinone reactivity to the synthesis of indolizidine alkaloids and other complex natural product-like alkaloid structures. Overall, indolizinones are unique molecules that hold promise across a broad range of fields from materials to medicine.
Supplementary Material
Footnotes
Electronic supplementary information (ESI) available. CCDC reference numbers 834076 and 834077. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ob06423a
References
- 1.Smith CR, Bunnelle EM, Rhodes AJ, Sarpong R. Org Lett. 2007;9:1169–1171. doi: 10.1021/ol0701971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yan B, Zhou YB, Zhang H, Chen JJ, Liu YH. J Org Chem. 2007;72:7783–7786. doi: 10.1021/jo070983j. [DOI] [PubMed] [Google Scholar]
- 3.Bunnelle EM, Smith CR, Lee SK, Singaram SW, Rhodes AJ, Sarpong R. Tetrahedron. 2008;64:7008–7014. doi: 10.1016/j.tet.2008.02.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi J, Lee GH, Kim I. Synlett. 2008:1243–1249. [Google Scholar]
- 5.Kim I, Choi J, Lee S, Lee GH. Synlett. 2008:2334–2338. [Google Scholar]
- 6.Seregin IV, Schammel AW, Gevorgyan V. Org Lett. 2007;9:3433–3436. doi: 10.1021/ol701464j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The cyclization of tertiary alcohols such as 5 in water to obtain indolizinone products was developed in our laboratories concurrently with Kim and coworkers’ (see ref. 5) report of the EtOH version of this metal-free reaction.
- 8.Paquette LA, Dura RD, Gallucci JC. Heterocycles. 2008;76:129–132. [Google Scholar]
- 9.Kim K, Kim I. J Comb Chem. 2010;12:379–382. doi: 10.1021/cc100015k. [DOI] [PubMed] [Google Scholar]
- 10.Sundberg RJ. Indoles. Academic Press; London: 1996. [Google Scholar]
- 11.Gudmundsson K, Boggs SD. 11,574,583. US Patent. 2007
- 12.Blackman AJ, Li CP, Hockless DCR, Skelton BW, White AH. Tetrahedron. 1993;49:8645–8656. [Google Scholar]
- 13.Li CP, Blackman AJ. Aust J Chem. 1994;47:1355–1361. [Google Scholar]
- 14.Li CP, Blackman AJ. Aust J Chem. 1995;48:955–965. [Google Scholar]
- 15.Schar P, Cren S, Renaud P. Chimia. 2006;60:131–141. [Google Scholar]
- 16.Weinreb SM. Chem Rev. 2006;106:2531–2549. doi: 10.1021/cr050069v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biard JF, Guyot S, Roussakis C, Verbist JF, Vercauteren J, Weber JF, Boukef K. Tetrahedron Lett. 1994;35:2691–2694. [Google Scholar]
- 18.See Supporting Information for details on the preparation of 64†.
- 19.Peukert S, Sun YC, Zhang R, Hurley B, Sabio M, Shen XY, Gray C, Dzink-Fox J, Tao JS, Cebula R, Wattanasin S. Bioorg Med Chem Lett. 2008;18:1840–1844. doi: 10.1016/j.bmcl.2008.02.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.