

Abstract

The initial 5-exo versus 6-endo cyclization of the acyl group onto the activated alkyne of propargylic esters has been found to be dependent on electronic effects of the acyl, alkyne, and propargylic carbon substituents. These electronic effects control the ratio of 2,3-disubstituted versus 1,3-disubstituted indolizine products formed when substrates bearing pyridines at the alkyne terminus are used.
Cycloisomerization transformations involving the activation of alkynes using late transition metals (e.g., Ru, Pt, Ag, and Au) are a very active area of current research in synthetic organic chemistry.1 A subset of these reactions involving propargylic esters2 have undergone a renaissance in the past decade, building on the pioneering studies of Ohloff and Rautenstrauch using Zn(II) and Pd(II) salts, respectively.3 The proposed mechanisms by which these cycloisomerizations proceed are mostly supported by empirical observations and may involve zwitterion, metallocarbenoid, or allene type intermediates as outlined in Scheme 1. However, a universally predictive model that delineates the role of catalysts and the electronic effects the alkyne substituents have in funneling a substrate through a specific reactive intermediate to product has yet to emerge.4 This shortcoming highlights the importance of initiating investigations that employ different metal complexes and propargylic ester substrates in an effort to understand the mechanisms of these processes.
Scheme 1.

Potential Cycloisomerization Mechanisms
A particularly successful approach to control initial 5-exo versus 6-endo nucleophilic attack of the ester carbonyl onto the alkyne (see 1 → 2 versus 1 → 6) is the introduction of substituents to electronically modulate the alkyne reactivity.4b,5 While propargylic esters bearing terminal alkynes typically undergo 5-exo-dig cyclization using Ru, Pt, or Au catalysts,6 internal alkynes bearing alkyl or aryl7 substituents usually undergo 6-endo-dig cyclization. Malacria was the first to report that ester substituents at the alkyne terminus lead exclusively to initial 5-exo-dig cyclization using PtCl2 as the catalyst.5 We have recently shown that this strategy is effective for the synthesis of indenes (e.g., 9, Scheme 2) using propargylic ester substrates such as 7.8
Scheme 2.

Pt-Catalyzed Indene Formation
Herein, we report a novel mode of reactivity of propargylic ester substrates bearing pyridine substituents at the alkyne terminus that undergo cycloisomerization to produce highly functionalized indolizine products. During the course of these studies, we have discovered remote electronic effects of substituents at the propargylic position to be important in biasing initial 5-exo versus 6-endo cyclization.
For example, two possible cycloisomerization pathways are open to propargylic ester substrate 10 (Scheme 3). Path A is predicated on the basis of our previous cycloisomerization studies, whereby 10 could undergo initial 5-exo-dig cyclization due to the electron-withdrawing capability of the 2-substituted pyridine at the alkyne terminus. Backbonding from the metal center (see 11) was expected to yield metallocarbenoid 12, which upon cyclization (via the intermediacy of zwitterion 13) would yield 2,3-disubstituted indolizine 14 following proton transfer. Alternatively, in Path B, initial 6-endo-dig cyclization (should the pyridine substituent exert electronic effects similar to a phenyl group) would furnish allene 15, which would react further (via 16) to form 1,3-disubstituted indolizine 17.
Scheme 3.

Proposed 1,3- and 2,3-Disubstituted Indolizine Formation
Our group has recently reported a cycloisomerization transformation for the synthesis of 1,3-disubstituted indolizines such as 17.9 However, general methods to synthesize 2,3-disubstituted indolizines (e.g., 14) are limited. Given the existing challenge of regioselective functionalization of indolizines,10 the transformation of 10 → 14 would be especially significant.
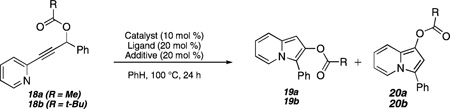
The propargylic ester substrate 18a (Table 1), which was prepared in a one-pot sequence in high overall yield from the corresponding pyridine acetylene, was used in our initial optimization studies.11 Using previous Pt(II)-catalyzed cycloisomerization studies from our laboratories as a starting point, we investigated the effect of combinations of platinum salts, solvents, and reaction conditions on the efficiency of the indolizine-forming transformation.
Table 1.
Optimization Studies
 | |||||
|---|---|---|---|---|---|
| entry | substrate | catalyst | ligand | additive | yield (%) (ratio 19:20)a |
| 1 | 18a | PtCl2 | – | – | 10 |
| 2 | 18a | PtCl2 | PPh3 | – | – |
| 3 | 18a | PtCl2 | P(C6F5)3 | – | 42 (3:1) |
| 4 | 18b | PtCl2 | P(C6F5)3 | – | 64 (9:1) |
| 5b | 18b | PtCl2 | P(C6F5)3 | Lil | 61 (15:1) |
| 6b | 18b | PtCl2 | P(C6F5)3 | LiCl | 42 (9:1) |
| 7b | 18b | PtI2 | P(C6F5)3 | – | 82 (18:1) |
Ratio determinations were made by comparison of 1H NMR resonances.
Reaction conducted at 80 °C.
The best initial results were obtained with PtCl2 as a catalyst and benzene as the solvent, which gave a 10% conversion with the majority of the mass balance accounted for by starting material (Table 1, entry 1). With the goal of using phosphine additives to modulate the reactivity of the Pt catalysts, we surveyed a range of phosphines including JohnPhos, 2-dicyclohexylphosphino biphenyl,12 and PPh3 (entry 2) as additives. Each of these cases returned the starting material. However, with the electron-deficient tris-(pentafluorophenyl)phosphine as an additive,13 a significant increase in yield (42%, entry 3) of indolizine products was observed, albeit as a 3:1 mixture comprising regioisomeric indolizines 19a and 20a.14 The acyl substituent also emerged as an important parameter with pivalates (see 18b, entry 4) giving significantly better selectivities and yields as compared to acetates (18a, entry 3).
Our continuing studies have sought to minimize the regioisomeric mixture of products (i.e., 19 versus 20). By analogy to the course of reaction of ester-terminated alkyne substrates (see Scheme 2), we hypothesized that a more electron-deficient pyridine fragment would favor an initial 5-exo-dig cyclization, which would lead predominantly to the desired 2,3-disubstituted indolizines via an overall 1,2-acyl shift. With this goal in mind, we surveyed a range of Lewis acids as additives, which could in theory accept electron density from the pyridine by coordination to the nitrogen atom. A variety of Lewis acid additives including InCl3, Yb(OTf)3, TMSCl, and AlCl3 as well as protic acids such as HCl led predominantly to decomposition. By contrast, we were gratified to find that LiI gave an improved 15:1 ratio of 19b/20b at 80 °C (entry 5). Encouraged by this result, other lithium salts such as LiCl (entry 6) were investigated as additives, but these did not show any added improvement. These results indicated that the iodide counterion was playing a crucial role. Thus, we attempted the reaction with PtI2, which gave an excellent yield and ratio favoring 19b (entry 7).
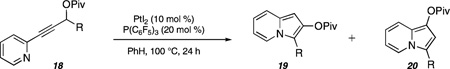
The cycloisomerization reaction proved to be general for a range of substrates employing the optimized conditions of 10 mol % of PtI2, 20 mol % of P(C6F5)3, 0.10 M in PhH at 100 °C for 24 h, as illustrated in Table 2. In general, substrates bearing alkyl substituents at the propargylic position (e.g., R = i-Bu or cyclohexyl) were unproductive and led predominantly to decomposition. However, with t-butyl or cyclopropyl substituents, a mixture of indolizine products was obtained in low yield (entries 1 and 2, respectively). Alkenyl substituents provided good yields and a high ratio favoring the desired indolizine (entries 3–5). Aromatic substituents at the propargylic position generally served as the best substrates for these reactions as evidenced by the exceptional product ratios and yields (e.g., entry 11).
Table 2.
Effect of Substitution at the Propargylic Position
 | |||
|---|---|---|---|
| entry | R | yield (%) | ratio (19:20)a |
| 1 | t-Bu | 11 | 4:1 |
| 2 | 49 | 3:1 | |
| 3 | 74 | 14:1 | |
| 4 | 54 | 14:1 | |
| 5 |  |
56 | >20:1 |
| 6 | 34 | >20:1 | |
| 7 | 73 | 11:1 | |
| 8 | 67 | 13:1 | |
| 9 |  |
76 | 7:1 |
| 10 | 72 | 10:1 | |
| 11 | 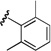 |
91 | >20:1 |
| 12 | |||
| (a) R = H | 75 | 13:1 | |
| (b) R = OMe | 74 | 13:1 | |
| (c) R = CF3 | 58 | 2:1 | |
| (d) R = CO2Me | 78 | 6:1 | |
| (e) R = CN | 31 | 3:1 | |
Ratio determinations were made by comparison of 1H NMR resonances.
A qualitative examination of our observations up to this stage suggests that substituents at the propargylic position that stabilize developing cationic character promote the formation of the 2,3-disubstituted indolizine products (presumably by favoring the 5-exo-dig cyclization pathway). To more closely probe this observation, several substrates possessing electronically differentiated aryl groups were prepared (entries 12a–e, Table 2). While electron-neutral (entry 12a) and electron-donating substituents (entry 12b) gave good yields and high ratios favoring the desired indolizine, electron-withdrawing groups led to diminished yields and lower product ratios (entries 12c–e). To the best of our knowledge, this study serves as the first investigation of electronic effects at the propargylic position on the course of cycloisomerizations of propargylic esters.
In addition to the dramatic effect of electronics at the propargylic position on observed product ratios, we have also found that the introduction of an electron-rich acyl group (p-methoxybenzoyl) in place of the pivalate has a significant effect. For example, ratios of >20:1 (67% yield) and 13:1 (56% yield) were obtained using 21b and 21d as substrates, respectively (Scheme 4; compare to 21a and 21c, which possess pivalate groups).
Scheme 4.

Effect of Acyl Substituent
With regard to the mechanism of these indolizine-forming reactions, several preparative experiments have provided additional insight. Using deuterated propargylic ester 24 (Scheme 5) as the substrate, indolizine 25-d was obtained with 60% deuterium incorporation at C-1.15 This observation supports the propargylic position as the source of the C-1 proton.
Scheme 5.

Crossover Experiment with Cycloisomerization Substrates
Furthermore, a crossover experiment employing 24-d and 18b led to all four possible deutero and protio products (as identified by 1H NMR), giving support to an intermolecular mechanism for proton transfer.16,17
In conclusion, we have shown for the first time a marked dependence of the initial metal-catalyzed mode of cyclization of propargylic esters on the electronic effects of the substituent at the propargylic position. Electron-donating groups favor 5-exo-dig cyclization, whereas more of the product resulting from initial 6-endo-dig cyclization is observed with electron-withdrawing groups. Additionally, the acyl group electronics influence the mode of cyclization. Using pyridine-terminated propargylic ester substrates, a novel cycloisomerization reaction for the synthesis of 2,3-disubstituted indolizines has been developed. Our current efforts are focused on probing the mechanisms of these transformations further and applying our methods to the synthesis of other heterocycles.
Supplementary Material
Acknowledgment
The authors are grateful to UC Berkeley, GlaxoSmithKline, Johnson Matthey, and Boehringer Ingelheim for support of this research.
Footnotes
Note Added after ASAP Publication. In Table 1, entry 7 of column 3 (catalyst) was incorrect in the version published ASAP September 11, 2007; the corrected version was published ASAP September 13, 2007.
Supporting Information Available: Experimental details and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For a recent review, see: Fürstner A, Davies PW. Angew. Chem. Int. Ed. 2007;46:3099. doi: 10.1002/anie.200604335.
- 2.For related reviews, see: Marion N, Nolan SP. Angew. Chem. Int. Ed. 2007;46:2750. doi: 10.1002/anie.200604773. Miki K, Uemura S, Ohe K. Chem. Lett. 2005;34:1068. Mendez M, Mamane V, Fürstner A. Chemtracts Org. Chem. 2003;16:397. For examples of Au- and Pt-catalyzed acyloxy migrations of propargylic esters, see: Wang S, Zhang L. J. Am. Chem. Soc. 2006;128:8414. doi: 10.1021/ja062777j. Mamane V, Gress T, Krause H, Fürstner A. J. Am. Chem. Soc. 2004;126:8654. doi: 10.1021/ja048094q. Fehr C, Galindo J. Angew. Chem. Int. Ed. 2006;45:2901. doi: 10.1002/anie.200504543. Cho EJ, Kim M, Lee D. Eur. J. Org. Chem. 2006:3074.
- 3.(a) Strickler H, Davis JB, Ohloff G. Helv. Chim. Acta. 1976;59:1328. [Google Scholar]; (b) Rautenstrauch V. J. Org. Chem. 1984;49:950. [Google Scholar]
- 4.Important advances have been made in identifying metal salts and ligand combinations and alkyne substituents that may exert influence on the course of the reaction; see for example: Marion N, de Frémont P, Lemiére G, Stevens ED, Fensterbank L, Malacria M, Nolan SP. Chem. Commun. 2006:2048. doi: 10.1039/b602839j. Barluenga J, Riesgo L, Vicene R, López LA, Tomás M. J. Am. Chem. Soc. 2007;129:7772. doi: 10.1021/ja072864r.
- 5.Cariou K, Mainetti E, Fensterbank L, Malacria M. Tetrahedron. 2004;60:9745. [Google Scholar]
- 6.(a) Miki K, Ohe K, Uemura S. Tetrahedron Lett. 2003;44:2019. [Google Scholar]; (b) Miki K, Ohe K, Uemura S. J. Org. Chem. 2003;68:8503. doi: 10.1021/jo034841a. [DOI] [PubMed] [Google Scholar]
- 7.For earlier discussions, see: Cho EJ, Kim M, Lee D. Org. Lett. 2006;8:5413. doi: 10.1021/ol062335c. Marion N, Diez-González S, de Frémont P, Noble AR, Nolan SP. Angew. Chem. Int. Ed. 2006;45:3647. doi: 10.1002/anie.200600571.
- 8.Prasad BAB, Yoshimoto FK, Sarpong R. J. Am. Chem. Soc. 2005;127:12468. doi: 10.1021/ja053192c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith CR, Bunnelle EM, Rhodes AR, Sarpong R. Org. Lett. 2007;9:1169. doi: 10.1021/ol0701971. (b) A related report recently appeared: Seregin IV, Schammel AW, Gevorgyan V. Org. Lett. 2007;9:3433. doi: 10.1021/ol701464j.
- 10.(a) Park CH, Ryabova V, Seregin IV, Sromek AW, Gevorgyan V. Org. Lett. 2004;6:1159. doi: 10.1021/ol049866q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sawada K, Okada S, Kuroda A, Watanabe S, Sawada Y, Tanaka H. Chem. Pharm. Bull. 2001;49:799. doi: 10.1248/cpb.49.799. [DOI] [PubMed] [Google Scholar]; (c) Ostby OB, Dalhus B, Gundersen L-L, Rise F, Bast A, Haenen GRMM. Eur. J. Org. Chem. 2000;22:3763. [Google Scholar]
- 11.For complete synthesis details, see Supporting Information.
- 12.(a) Tomori H, Fox JM, Buchwald SL. J. Org. Chem. 2000;65:5334. doi: 10.1021/jo000691h. [DOI] [PubMed] [Google Scholar]; (b) Fox JM, Huang X, Chieffi A, Buchwald SL. J. Am. Chem. Soc. 2000;122:1360. [Google Scholar]
- 13.For an example of the use of P(C6F5)3 as a ligand in Pt(II)-catalyzed cycloisomerizations, see: Hours AE, Snyder JK. Tetrahedron Lett. 2006;47:675.
- 14.The regioisomers 19 and 20 were inseparable by column chromatography. The major isomer (19) was identified using nOe studies.
- 15.Subsequent studies have shown that the proton at C-1 is readily exchangeable. Complete loss of deuterium from this position occurs upon purification of 25-d on SiO2.
- 16.The rates of reaction of 24-d and 18b were judged to be comparable.
-
17.Although further empirical evidence points to Path A (see Scheme 1) as being operative (ref 18), an alternate pathway involving conversion of, e.g., 18b to 19b via allene 26 and zwitterion 27 as intermediates cannot be unambiguously discounted.
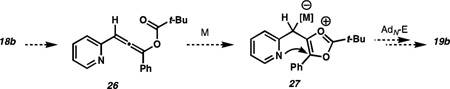 In our hands, catalysts such as CuI and CuCl, which have been reported to promote isomerizations related to 18b → 26 → 27 (refs 19 and 20), led only to decomposition. Furthermore, 1H NMR and preparative monitoring of these reactions did not identify a discrete intermediate such as 26.
In our hands, catalysts such as CuI and CuCl, which have been reported to promote isomerizations related to 18b → 26 → 27 (refs 19 and 20), led only to decomposition. Furthermore, 1H NMR and preparative monitoring of these reactions did not identify a discrete intermediate such as 26.
-
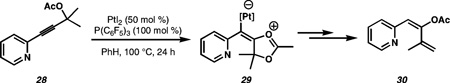
18.Our proposed mechanism is supported by the reaction of tertiary propargylic ester 28, which yields 30 under our reaction conditions.
- 19.Sromek AW, Kel’in AV, Gevorgyan V. Angew. Chem. Int. Ed. 2004;43:2280. doi: 10.1002/anie.200353535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Kel’in AV, Sromek AW, Gevorgyan V. J. Am. Chem. Soc. 2001;123:2074. doi: 10.1021/ja0058684. [DOI] [PubMed] [Google Scholar]; (b) Schwier T, Sromek AW, Yap DML, Chernyak D, Gevorgyan V. J. Am. Chem. Soc. 2007;129:9868. doi: 10.1021/ja072446m. [DOI] [PMC free article] [PubMed] [Google Scholar]


Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.