

Abstract
Pulmonary hypertension (PH) is a common complication of interstitial lung disease (ILD), particularly in idiopathic pulmonary fibrosis (IPF) and ILD associated with connective tissue disease, where the underlying pathology is often a nonspecific interstitial pneumonia (NSIP) pattern. The degree of PH in ILD is typically mild to moderate and radiographic changes of ILD are usually prominent. We describe four patients with idiopathic NSIP and severe PH (mPAP > 40 mmHg). The average mean pulmonary artery pressure was 51±7 mmHg and pulmonary vascular resistance was 13±4 Wood's units. Pulmonary function was characterized by mild restriction (total lung capacity 63–94% predicted) and profound reductions in DLCO (19–53% predicted). Computed tomographic imaging revealed minimal to moderate interstitial thickening without honeycombing. In two of the cases, an initial clinical diagnosis of idiopathic pulmonary arterial hypertension was made. Both were treated with intravenous epoprostenol, which was associated with worsening of hypoxemia. All four patients died or underwent lung transplant within 4 years of PH diagnonsis. Lung pathology in all four demonstrated fibrotic NSIP with medial thickening of the small and medium pulmonary arteries, and proliferative intimal lesions that stained negative for endothelial markers (CD31 and CD34) and positive for smooth muscle actin. There were no plexiform lesions. Severe pulmonary hypertension can therefore occur in idiopathic NSIP, even in the absence of advanced radiographic changes. Clinicians should suspect underlying ILD as the basis for PH when DLCO is severely reduced or gas exchange deteriorates with pulmonary vasodilator therapy.
Keywords: interstitial lung disease, pulmonary artery pressure, vasculopathy
INTRODUCTION
Pulmonary hypertension (PH) is a common complication of interstitial lung disease (ILD). It is associated with reduced exercise capacity, derangements in gas exchange,[1–3] and increased mortality.[1,2,4–6] Estimates of the prevalence of PH in ILD have varied from 28% to 85%, with the highest rates observed in idiopathic pulmonary fibrosis (IPF).[1,2,5,7–11] PH is particularly common in ILD associated with connective tissue diseases, where the underlying pathology is frequently a nonspecific interstitial pneumonia (NSIP) pattern. There are sparse data regarding the prevalence and clinical features of PH in idiopathic NSIP. In the absence of connective tissue disease, ILD-associated PH is typically not severe, with mean pulmonary artery pressures (mPAP) usually under 40 mmHg.[1,6,12] We report the clinical and pathological features of four patients with idiopathic NSIP and severe PH (mPAP > 40 mmHg).
Methods
We identified four patients referred to a specialized pulmonary hypertension program with pathologically proven NSIP and severe pulmonary hypertension (defined by a mPAP > 40 mmHg). While two patients (#2 and #3) had low-level antinuclear antibody (ANA) titers (1:40, 1:80), there was no other clinical evidence of a connective tissue disorder. HIV antibody was undetectable in all four patients, and thromboembolic disease was excluded through perfusion lung scanning. None of the patients demonstrated significant left heart abnormalities on echocardiography.
The pathologic specimens were reexamined by an experienced pulmonary pathologist to confirm the NSIP pattern and to characterize the pulmonary vascular morphology. Five-micron thick tissue sections were stained with routine H and E stains and Movat pentachrome histochemical stain. Immunohistochemical staining for CD31, CD34, smooth muscle actin (SMA), and factor VIII was performed by an automated stainer (BOND-MAX, Leica Microsystems, Inc.) All immunohistochemical staining was performed with corresponding positive and negative controls.
The presence of NSIP was confirmed based upon the presence of uniform interstitial fibrosis and chronic mononuclear interstitial fibrosis with temporal homogeneity and lack of granulomas, hyaline membranes, or fibroblastic foci. The histologic sections were also evaluated for features of pulmonary hypertension, including arterial medial thickening, subintimal proliferation, muscularization of the venous walls, plexiform lesions, a chronic perivascular inflammatory infiltrate and, a perivascular deposition of hemosiderin and capillary angiomatosis. The number of sections available for evaluation ranged between 4 and 12 sections.
CASE REPORTS
Patient 1 was seen in-clinic following 3 years of progressive dyspnea with exertion and an episode of syncope. Past medical history was notable for remote intravenous cocaine and heroin use resulting in chronic hepatitis C infection. He had been treated with interferon and ribavirin to achieve an undetectable viral load. Liver function tests and prothrombin time were normal and there was no evidence of portal hypertension. A computed tomography (CT) scan of the chest showed minimal interstitial thickening and bronchiectasis in the lung bases (Fig. 1). Pulmonary function tests (PFTs) and right heart catheterization (RHC) findings are shown in (Table 1). A clinical diagnosis of pulmonary arterial hypertension (PAH) was made and he was treated initially with bosentan, followed by subcutaneous treprostinil, with a subsequent switch to intravenous epoprostenol and sildenafil. There was no clinical benefit appreciated with any of these regimens. Over the course of 3 years, his WHO functional class worsened from class III-IV and his 6-minute walk distance fell from 401 to 253 m. Oxygenation deteriorated progressively, eventually requiring 6 l/minute of supplemental oxygen. A bilateral lung transplant was performed 4 years after initial presentation. Pathologic examination of the explanted lungs revealed a diffuse cellular and fibrotic non-specific interstitial pneumonia pattern (Fig. 2).
Figure 1.
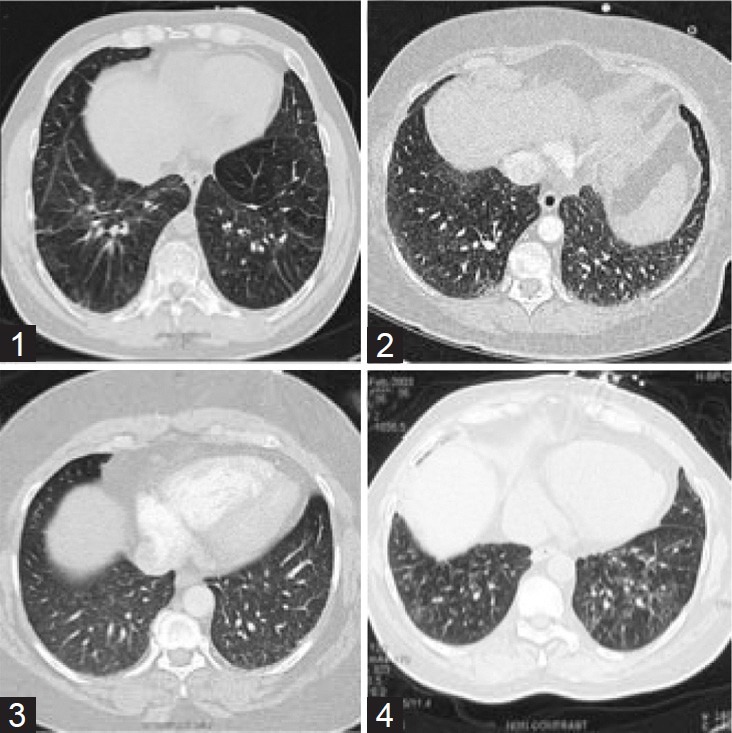
Representative CT cuts from patients 1-4 demonstrating interstitial changes with reticular ground-glass opacities predominantly in the lower lobes.
Table 1.
Patient characteristics
Figure 2.
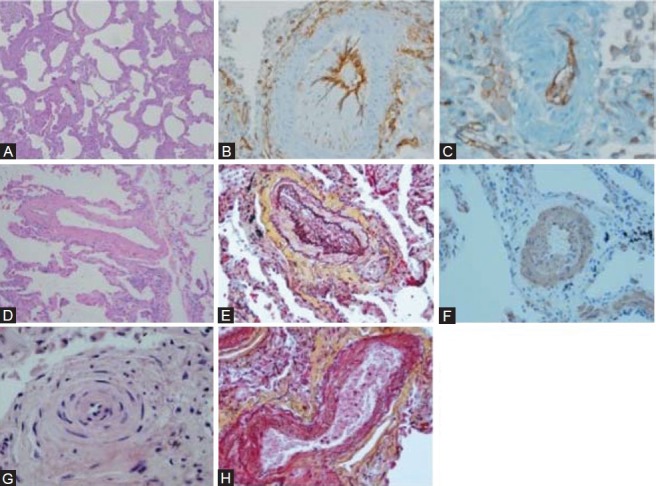
Representative pathology. (A) Uniform interstitial fibrosis and mild chronic inflammation characteristic of fibrotic NSIP (H and E, ×4). (B) CD34 (×20) and (C) CD31 stains demonstrate subintimal proliferation by non-endothelial cells (×20). (D) H and E staining (×10) and (E) Movat staining (×20) demonstrate arterial medial hypertrophy with eccentric intimal proliferation. (F) Medial thickening and subintimal proliferation consisting of smooth muscle cells (smooth muscle actin stain, ×20). (G) Concentric intimal proliferation (H and E, ×20). (H) Venous muscularization (Movat statin, ×20).
Patient 2 presented with progressive exertional dyspnea over the preceding 9 years. She had been given a diagnosis of chronic obstructive pulmonary disease (COPD) and treated with bronchodilators and intermittent oral corticosteroids. The latter were often associated with temporary symptomatic improvement. Her past medical history was notable for longstanding diabetes and hypertension. She had a remote 20 pack-year smoking history. Four months prior to her initial assessment, RHC demonstrated severe pre-capillary pulmonary hypertension. PFTs revealed normal lung volumes with a profound reduction in diffusing capacity (DLCO; Table 1). Chest CT imaging was free of parenchymal lung disease, but demonstrated mediastinal and hilar lymphadenopathy measuring up to 25 mm in maximal nodal diameter. Intravenous epoprostenol was started for a clinical diagnosis of idiopathic PAH. After transient symptomatic improvement, the patient experienced progressive worsening of dyspnea and an increase in oxygen requirement from 2 to 10 l/minute on an epoprostenol dose of 14 ng/kg/min. At presentation to our clinic, she reported WHO functional class IV limitations and demonstrated decompensated right heart failure on physical examination. A thin-section (1-mm) CT of the chest exhibited a subtle pattern of diffuse reticulation with patchy ground-glass opacities in the lung bases (Fig. 1). Given these findings and the poor clinical response to PAH therapy, a diagnosis of pulmonary veno-occlusive disease was suspected. The epoprostenol dose was tapered and sildenafil introduced. Two months later, sudden death occurred. Autopsy demonstrated an extensive fibrotic NSIP pattern. There was also severe 3-vessel coronary artery disease (CAD) without evidence of myocardial infarction.
Patient 3 presented to the pulmonary clinic with chronic cough and mild dyspnea. Her past medical history was significant for hypertension, obesity (BMI of 38), and active smoking (40 pack-years). Initial pulmonary function testing demonstrated normal lung volumes (TLC: 86%) with a mild reduction in DLCO (73%). Chest CT imaging revealed subtle, diffuse reticular opacities without traction bronchiectasis or honeycombing (Fig. 1). A surgical lung biopsy demonstrated a fibrotic NSIP pattern. Baseline physical exam and echocardiogram done at presentation were not suggestive of PH. Prednisone (0.5 mg/kg/day) was initiated for declining PFTs 3 months later with subsequent improvement in symptoms and pulmonary function, after which steroid therapy was tapered off. Five years later she experienced a non-ST elevation myocardial infarction. A right heart catheterization in this setting showed a pulmonary artery pressure of 90/36 (mean 56 mmHg) with a pulmonary artery wedge pressure of 16 mmHg. Echocardiography at that time showed a dilated right heart with normal left sided function. The interstitial process was more prominent on imaging, and hypoxemia was now present (Table 1). Mild obstructive sleep apnea (Apnea Hypopnea Index: 11.1) was also diagnosed. Continuous positive airway pressure therapy and supplemental oxygen were initiated. Her cardiopulmonary status continued to decline, with recurrent hospitalizations for right heart failure. A repeat RHC 7 years after her initial presentation with ILD demonstrated severe PAH with RV failure. Progressive restriction and diffusion defects were noted on PFT's (Table 1). The patient subsequently expired due to refractory hypoxemic respiratory failure.
Patient 4 was referred to the clinic with a concomitant diagnosis of ILD and PH. His symptoms had begun 4 years prior with progressive exertional dyspnea (WHO class IV), cough and development of lower extremity edema. His past medical history was significant for diabetes mellitus, single-vessel CAD, and poorly controlled systemic hypertension. Physical examination demonstrated signs of PH and right heart failure, as well as crackles on lung auscultation. Chest CT scan demonstrated mild patchy subpleural and peribronchovascular reticular opacities with minimal honeycombing and traction bronchiectasis. There was also moderate mediastinal adenopathy up to 22 mm. in maximal diameter (Fig. 1). PFT and RHC findings are shown in Table 1. An open-lung biopsy showed extensive fibrotic nonspecific interstitial pneumonia. He was started on prednisone and azathioprine with a good initial response. Fifteen months later, right heart failure associated with atrial fibrillation occurred. He subsequently died suddenly at home.
Pulmonary vascular morphology
Lung pathology of all 4 patients demonstrated interstitial fibrosis and mild chronic inflammation characteristic of fibrotic NSIP (Fig. 2). An extensive pulmonary arterial vasculopathy was also present in all cases, and was characterized by varying degrees of medial thickening. In addition, there were widespread intimal lesions in medium and small pulmonary arteries resulting in considerable luminal narrowing and obliteration. The majority of these lesions consisted of eccentric and concentric, nonlaminar intimal fibrosis. In three of the four cases, there were clear concentric “onion-skin” intimal proliferation lesions focally. The intimal lesions stained negative for CD31 and CD34 (endothelial cell markers), but positive for smooth muscle actin. Movat staining also demonstrated muscularization of the pulmonary veins, without evidence of pulmonary veno-occlusive disease. Plexiform or angiomatoid lesions were absent.
DISCUSSION
We report four cases of severe pulmonary hypertension associated with pathologically proven NSIP and no evidence of connective tissue disease (CTD). In each case, the vascular proliferation consisted of smooth muscle cells, which is in contrast to the intimal proliferation of endothelial cells observed in idiopathic PAH.[13]
Studies evaluating the prevalence and severity of PH in idiopathic interstitial pneumonia (IIP) have been small, and generally have not focused on NSIP.[14,15] One study included 13 patients with NSIP and reported a 46% incidence of pulmonary hypertension. The average mean pulmonary arterial pressure (mPAP) in that study was 27.6 mmHg.[16] An echocardiographic study reported the incidence of PH to be 28% among 70 patients with IIP, 11 of whom had NSIP. The average sPAP in this group was 30.2 mmHg, though the individual sPAP of the NSIP patients was not reported.[7] In a study by Dorfmüller et al., three patients with severe pulmonary hypertension (mPAP range 51-62 mmHg) were found to have NSIP. However, all three patients carried a known diagnosis of either limited scleroderma or mixed CTD.[17] To our knowledge, this is the first clinicopathologic description of severe pulmonary hypertension, defined as an mPAP >40 mmHg, in a group of patients with idiopathic NSIP. Patient 1 had a history of hepatic cirrhosis and treatment with interferon-alpha, both of which have been associated with an increased incidence of PH.[18,19] However, there was no evidence of portal hypertension or liver dysfunction, and his symptoms of dyspnea preceded his treatment with interferon. Therefore, these are unlikely to be significant contributors to his PH. Patient 3 had a history of mild obstructive sleep apnea (OSA). While OSA can be associated with mild pulmonary hypertension, severe PH, as seen in this patient is rare.[3,20,21]
Pathogenesis of PH in ILD
PH in patients with ILD is thought to result largely from vascular destruction and remodeling with vessel fibrosis.[22] Remodeled vessels lack an elastin layer and have decreased vascular compliance.[23] Chronic hypoxemia likely also plays a limited role.[3] All four patients in our series had a prominent vasculopathy with medial thickening and intimal proliferation. Concentric intimal proliferation, as is often seen in cases of severe pulmonary arterial hypertension (PAH)[13,17] was present in three patients.
In contrast to the intimal proliferation of endothelial cells observed in idiopathic PAH,[13] the intimal proliferative process observed in these cases seems to be one involving smooth muscle cells. This is evidenced by the absence of staining with CD34 and CD31, and positive smooth muscle actin staining. Other lesions often associated with pulmonary hypertension of this severity, such as plexiform and angiomatoid lesions,[13] were notably absent in these patients. Severe pulmonary hypertension[24–27] in the absence of plexiform lesions[24,26,28] has been described in pulmonary veno-occlusive disease. In addition, arteriolar medial hypertrophy[25,26] and intimal proliferation of pulmonary arterioles,[26] as was seen in our patients, have also been reported in patients with pulmonary veno-occlusive disease (PVOD). However, despite the fact that three patients demonstrated venous muscularization, there was no evidence of occlusion of pulmonary venules or preseptal pulmonary veins.
There is evidence that PH in ILD is caused in part by endothelial injury and dysfunctional endothelial cells with a resultant imbalance of vasoconstricting and vasodilating agents.[29] Endothelin-1 is known to cause vasoconstriction and smooth muscle cell proliferation of the pulmonary vasculature,[29] and increased serum,[30] endothelial cell,[29] and alveolar epithelial cell[31] expression of endothelin-1 has been associated with pulmonary hypertension.[32] Likewise, serotonin may also contribute to the development of idiopathic PAH[33,34] through a similar vasoconstricting effect on pulmonary vasculature.[35] Moreover, dysfunctional endothelial cells from lungs of patients with PH also demonstrate decreased levels of vasodilators, including prostacyclin,[29,36] nitric oxide,[29,37] and vasoactive intestinal peptide.[38] Finally, there is also evidence that factors such as platelet-derived growth factor (PDGF)[39] and TGF-β[40–42] may play a role in the development of PH. Whether or not similar mechanisms are responsible for PH in NSIP remains to be seen.
Pulmonary function tests
Despite severely elevated mean pulmonary pressures, the degree of restriction on pulmonary function testing was relatively modest. The patients in our series demonstrated a mild restrictive defect (total lung capacity 63-94% predicted) with a severe reduction in DLCO (19%-53% predicted). Several studies show PH to correlate poorly with the degree of restriction, and only modestly with DLCO in patients with IPF[5,10] and other forms of ILD.[7] Other reports regarding PFT abnormalities in patients with both idiopathic and nonidiopathic forms of NSIP have also demonstrated similar reductions in TLC and DLCO, though no correlation with mPAP was made.[43–45] The degree of pulmonary hypertension in our patients was not predicted by the degree of restriction on their PFTs, though a severe reduction in DLCO may have served as a clue to the presence of pulmonary vascular disease.
Radiographic data
CT imaging demonstrated interstitial changes with reticular groundglass opacities, predominantly in the lower lobes, and two patients had evidence of mild traction bronchiectasis (Fig. 1). The severity of pulmonary hypertension in our patients was relatively disproportionate to the radiographic findings, which is consistent with previous studies in ILD that also demonstrated a lack of correlation between the amount of fibrosis/interstitial thickening and the degree of pulmonary hypertension.[46,47] In Patients 1 and 2 the radiographic changes were quite subtle and led to an erroneous diagnosis of Idiopathic PAH.
Response to targeted PAH therapy
Patients 1 and 2 demonstrated worsening oxygenation in the setting of intravenous epoprostenol, likely due to increased perfusion of poorly ventilated areas of diseased lung. While similar clinical worsening with epoprostenol has been reported in patients with pulmonary veno-occlusive disease, such cases were associated with concomitant pulmonary edema,[25,27] which was absent in our patients.
Patients 1 and 4 demonstrated no clinical improvement with bosentan therapy. In patients with scleroderma-related ILD, bosentan did not significantly improve 6-minute walk distance,[48] hemodynamics, or survival.[49] A similar lack of benefit in 6-minute walk,[50] time to clinical worsening, mortality or quality of life[51] was also seen in trials of bosentan in IPF. In a recent randomized, placebo-controlled trial of sildenafil in IPF, 6-minute walk distance was not improved after 12 weeks of therapy. However, there was a small but significant benefit in arterial oxygenation, carbon monoxide diffusion capacity, degree of dyspnea and quality of life.[52] The pulmonary arteries of our patients demonstrated intimal lesions consisting of smooth muscle cells, as opposed to the endothelial cells observed in idiopathic PAH,[13] and this may explain the lack of clinical response to therapy traditionally used in the treatment of PAH.
CONCLUSIONS
Severe pulmonary hypertension with prominent pulmonary vascular remodeling can occur with NSIP in the absence of known CTD. Pulmonary function and radiographic abnormalities in these patients can be subtle. In contrast to the endothelial cell proliferation observed in IPAH, the vascular pathology observed in these cases involved abnormal proliferation of vascular smooth muscle cells. Recognition of NSIP or other parenchymal lung disease as the basis for severe PH is critical, as PAH therapy in such cases could result in worsening of gas exchange. Clinical outcomes are poor as illustrated here. Carefully designed clinical trials are required to identify effective treatment.
Footnotes
Source of Support: Nil,
Conflict of Interest: None declared.
REFERENCES
- 1.Lettieri CJ, Nathan SD, Barnett SD, Ahmad S, Shorr AF. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2006;129:746–52. doi: 10.1378/chest.129.3.746. [DOI] [PubMed] [Google Scholar]
- 2.Leuchte HH, Baumgartner RA, Nounou ME, Vogeser M, Neurohr C, Trautnitz M, et al. Brain natriuretic peptide is a prognostic parameter in chronic lung disease. Am J Respir Crit Care Med. 2006;173:744–50. doi: 10.1164/rccm.200510-1545OC. [DOI] [PubMed] [Google Scholar]
- 3.Girgis RE, Mathai SC. Pulmonary hypertension associated with chronic respiratory disease. Clin Chest Med. 2007;28:219–32. doi: 10.1016/j.ccm.2006.11.006. x. [DOI] [PubMed] [Google Scholar]
- 4.Nadrous HF, Pellikka PA, Krowka MJ, Swanson KL, Chaowalit N, Decker PA, et al. The impact of pulmonary hypertension on survival in patients with idiopathic pulmonary fibrosis. Chest. 2005;128:616S–7S. doi: 10.1378/chest.128.6_suppl.616S-a. [DOI] [PubMed] [Google Scholar]
- 5.Nadrous HF, Pellikka PA, Krowka MJ, Swanson KL, Chaowalit N, Decker PA, et al. Pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Chest. 2005;128:2393–9. doi: 10.1378/chest.128.4.2393. [DOI] [PubMed] [Google Scholar]
- 6.Hamada K, Nagai S, Tanaka S, Handa T, Shigematsu M, Nagao T, et al. Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest. 2007;131:650–6. doi: 10.1378/chest.06-1466. [DOI] [PubMed] [Google Scholar]
- 7.Handa T, Nagai S, Miki S, Ueda S, Yukawa N, Fushimi Y, et al. Incidence of pulmonary hypertension and its clinical relevance in patients with interstitial pneumonias: Comparison between idiopathic and collagen vascular disease associated interstitial pneumonias. Intern Med. 2007;46:831–7. doi: 10.2169/internalmedicine.46.6342. [DOI] [PubMed] [Google Scholar]
- 8.Nathan SD, Noble PW, Tuder RM. Idiopathic pulmonary fibrosis and pulmonary hypertension: Connecting the dots. Am J Respir Crit Care Med. 2007;175:875–80. doi: 10.1164/rccm.200608-1153CC. [DOI] [PubMed] [Google Scholar]
- 9.Colombat M, Mal H, Groussard O, Capron F, Thabut G, Jebrak G, et al. Pulmonary vascular lesions in end-stage idiopathic pulmonary fibrosis: Histopathologic study on lung explant specimens and correlations with pulmonary hemodynamics. Hum Pathol. 2007;38:60–5. doi: 10.1016/j.humpath.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 10.Nathan SD, Shlobin OA, Ahmad S, Urbanek S, Barnett SD. Pulmonary hypertension and pulmonary function testing in idiopathic pulmonary fibrosis. Chest. 2007;131:657–63. doi: 10.1378/chest.06-2485. [DOI] [PubMed] [Google Scholar]
- 11.Nathan SD, Shlobin OA, Ahmad S, Koch J, Barnett SD, Ad N, et al. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration. 2008;76:288–94. doi: 10.1159/000114246. [DOI] [PubMed] [Google Scholar]
- 12.Shorr AF, Wainright JL, Cors CS, Lettieri CJ, Nathan SD. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J. 2007;30:715–21. doi: 10.1183/09031936.00107206. [DOI] [PubMed] [Google Scholar]
- 13.Tuder RM, Marecki JC, Richter A, Fijalkowska I, Flores S. Pathology of pulmonary hypertension. Clin Chest Med. 2007;28:23–42. doi: 10.1016/j.ccm.2006.11.010. vii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan RT, Kuzo R, Goodman LR, Siegel R, Haasler GB, Presberg KW Medical College of Wisconsin Lung Transplant Group. Utility of CT scan evaluation for predicting pulmonary hypertension in patients with parenchymal lung disease. Chest. 1998;113:1250–6. doi: 10.1378/chest.113.5.1250. [DOI] [PubMed] [Google Scholar]
- 15.Weitzenblum E, Ehrhart M, Rasaholinjanahary J, Hirth C. Pulmonary hemodynamics in idiopathic pulmonary fibrosis and other interstitial pulmonary diseases. Respiration. 1983;44:118–27. doi: 10.1159/000194537. [DOI] [PubMed] [Google Scholar]
- 16.Ahmad S, Barnett SD, Shlobin OA, Nathan SD. Comparison of the Prevalence of Pulmonary Arterial Hypertension (PAH) in Patients with Idiopathic Pulmonary Fibrosis (IPF) and Non-Specific Interstitial Pneumonia (NSIP) Am J Respir Crit Care Med. 2006;3:S242. [Google Scholar]
- 17.Dorfmuller P, Humbert M, Perros F, Sanchez O, Simonneau G, Müller KM, et al. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum Pathol. 2007;38:893–902. doi: 10.1016/j.humpath.2006.11.022. [DOI] [PubMed] [Google Scholar]
- 18.Naschitz JE, Slobodin G, Lewis RJ, Zuckerman E, Yeshurun D. Heart diseases affecting the liver and liver diseases affecting the heart. Am Heart J. 2000;140:111–20. doi: 10.1067/mhj.2000.107177. [DOI] [PubMed] [Google Scholar]
- 19.Dhillon S, Kaker A, Dosanjh A, Japra D, Vanthiel DH. Irreversible pulmonary hypertension associated with the use of interferon alpha for chronic hepatitis C. Dig Dis Sci. 2010;55:1785–90. doi: 10.1007/s10620-010-1220-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arias MA, Garcia-Rio F, Alonso-Fernandez A, Martinez I, Villamor J. Pulmonary hypertension in obstructive sleep apnoea: Effects of continuous positive airway pressure: A randomized, controlled cross-over study. Eur Heart J. 2006;27:1106–13. doi: 10.1093/eurheartj/ehi807. [DOI] [PubMed] [Google Scholar]
- 21.Alchanatis M, Tourkohoriti G, Kakouros S, Kosmas E, Podaras S, Jordanoglou JB. Daytime pulmonary hypertension in patients with obstructive sleep apnea: The effect of continuous positive airway pressure on pulmonary hemodynamics. Respiration. 2001;68:566–72. doi: 10.1159/000050574. [DOI] [PubMed] [Google Scholar]
- 22.Nathan SD. Pulmonary hypertension in interstitial lung disease. Int J Clin Pract. 2008 doi: 10.1111/j.1742-1241.2008.01624.x. [DOI] [PubMed] [Google Scholar]
- 23.Renzoni EA, Walsh DA, Salmon M, Wells AU, Sestini P, Nicholson AG, et al. Interstitial vascularity in fibrosing alveolitis. Am J Respir Crit Care Med. 2003;167:438–43. doi: 10.1164/rccm.200202-135OC. [DOI] [PubMed] [Google Scholar]
- 24.Thomas de Montpreville V, Dulmet E, Fadel E, Dartevelle P. Lymph node pathology in pulmonary veno-occlusive disease and pulmonary capillary heamangiomatosis. Virchows Arch. 2008;453:171–6. doi: 10.1007/s00428-008-0636-3. [DOI] [PubMed] [Google Scholar]
- 25.Montani D, Achouh L, Dorfmuller P, Le Pavec J, Sztrymf B, Tchérakian C, et al. Pulmonary veno-occlusive disease: Clinical, functional, radiologic, and hemodynamic characteristics and outcome of 24 cases confirmed by histology. Medicine (Baltimore) 2008;87:220–33. doi: 10.1097/MD.0b013e31818193bb. [DOI] [PubMed] [Google Scholar]
- 26.Holcomb BW, Jr, Loyd JE, Ely EW, Johnson J, Robbins IM. Pulmonary veno-occlusive disease: A case series and new observations. Chest. 2000;118:1671–9. [PubMed] [Google Scholar]
- 27.Palmer SM, Robinson LJ, Wang A, Gossage JR, Bashore T, Tapson VF. Massive pulmonary edema and death after prostacyclin infusion in a patient with pulmonary veno-occlusive disease. Chest. 1998;113:237–40. doi: 10.1378/chest.113.1.237. [DOI] [PubMed] [Google Scholar]
- 28.Montani D, O’Callaghan DS, Savale L, Jaïs X, Yaïci A, Maitre S, et al. Pulmonary veno-occlusive disease: Recent progress and current challenges. Respir Med. 2010;104(Suppl 1):S23–32. doi: 10.1016/j.rmed.2010.03.014. [DOI] [PubMed] [Google Scholar]
- 29.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 30.Stewart DJ, Levy RD, Cernacek P, Langleben D. Increased plasma endothelin-1 in pulmonary hypertension: Marker or mediator of disease? Ann Intern Med. 1991;114:464–9. doi: 10.7326/0003-4819-114-6-464. [DOI] [PubMed] [Google Scholar]
- 31.Giaid A, Michel RP, Stewart DJ, Sheppard M, Corrin B, Hamid Q. Expression of endothelin-1 in lungs of patients with cryptogenic fibrosing alveolitis. Lancet. 1993;341:1550–4. doi: 10.1016/0140-6736(93)90694-c. [DOI] [PubMed] [Google Scholar]
- 32.Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328:1732–9. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- 33.Herve P, Launay JM, Scrobohaci ML, Brenot F, Simonneau G, Petitpretz P, et al. Increased plasma serotonin in primary pulmonary hypertension. Am J Med. 1995;99:249–54. doi: 10.1016/s0002-9343(99)80156-9. [DOI] [PubMed] [Google Scholar]
- 34.Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, et al. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest. 2001;108:1141–50. doi: 10.1172/JCI12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morecroft I, Heeley RP, Prentice HM, Kirk A, MacLean MR. 5-hydroxytryptamine receptors mediating contraction in human small muscular pulmonary arteries: Importance of the 5-HT1B receptor. Br J Pharmacol. 1999;128:730–4. doi: 10.1038/sj.bjp.0702841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:1925–32. doi: 10.1164/ajrccm.159.6.9804054. [DOI] [PubMed] [Google Scholar]
- 37.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333:214–21. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- 38.Petkov V, Mosgoeller W, Ziesche R, Raderer M, Stiebellehner L, Vonbank K, et al. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest. 2003;111:1339–46. doi: 10.1172/JCI17500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Antoniades HN, Bravo MA, Avila RE, Galanopoulos T, Neville-Golden J, Maxwell M, et al. Platelet-derived growth factor in idiopathic pulmonary fibrosis. J Clin Invest. 1990;86:1055–64. doi: 10.1172/JCI114808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coker RK, Laurent GJ, Jeffery PK, du Bois RM, Black CM, McAnulty RJ. Localisation of transforming growth factor beta1 and beta3 mRNA transcripts in normal and fibrotic human lung. Thorax. 2001;56:549–56. doi: 10.1136/thorax.56.7.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Corrin B, Butcher D, McAnulty BJ, Dubois RM, Black CM, Laurent GJ, et al. Immunohistochemical localization of transforming growth factor-beta 1 in the lungs of patients with systemic sclerosis, cryptogenic fibrosing alveolitis and other lung disorders. Histopathology. 1994;24:145–50. doi: 10.1111/j.1365-2559.1994.tb01293.x. [DOI] [PubMed] [Google Scholar]
- 42.Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, et al. BMPR2 haploinsufficiency as the inherited molecular mechanism for primary pulmonary hypertension. Am J Hum Genet. 2001;68:92–102. doi: 10.1086/316947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Carvalho EF, Parra ER, de Souza R, A’B Saber AM, Machado Jde C, Capelozzi VL. Arterial and interstitial remodelling processes in non-specific interstitial pneumonia: Systemic sclerosis versus idiopathic. Histopathology. 2008;53:195–204. doi: 10.1111/j.1365-2559.2008.03072.x. [DOI] [PubMed] [Google Scholar]
- 44.Park IN, Jegal Y, Kim DS, Do KH, Yoo B, Shim TS, et al. Clinical course and lung function change of idiopathic nonspecific interstitial pneumonia. Eur Respir J. 2009;33:68–76. doi: 10.1183/09031936.00158507. [DOI] [PubMed] [Google Scholar]
- 45.Daniil ZD, Gilchrist FC, Nicholson AG, Hansell DM, Harris J, Colby TV, et al. A histologic pattern of nonspecific interstitial pneumonia is associated with a better prognosis than usual interstitial pneumonia in patients with cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med. 1999;160:899–905. doi: 10.1164/ajrccm.160.3.9903021. [DOI] [PubMed] [Google Scholar]
- 46.Zisman DA, Karlamangla AS, Ross DJ, Keane MP, Belperio JA, Saggar R, et al. High-resolution chest CT findings do not predict the presence of pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2007;132:773–9. doi: 10.1378/chest.07-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Devaraj A, Wells AU, Meister MG, Corte TJ, Hansell DM. The effect of diffuse pulmonary fibrosis on the reliability of CT signs of pulmonary hypertension. Radiology. 2008;249:1042–9. doi: 10.1148/radiol.2492080269. [DOI] [PubMed] [Google Scholar]
- 48.Seibold JR, Denton CP, Furst DE, Guillevin L, Rubin LJ, Wells A, et al. Randomized, prospective, placebo-controlled trial of bosentan in interstitial lung disease secondary to systemic sclerosis. Arthritis Rheum. 2010;62:2101–8. doi: 10.1002/art.27466. [DOI] [PubMed] [Google Scholar]
- 49.Launay D, Sitbon O, Le Pavec J, Savale L, Tchérakian C, Yaïci A, et al. Long-term outcome of systemic sclerosis-associated pulmonary arterial hypertension treated with bosentan as first-line monotherapy followed or not by the addition of prostanoids or sildenafil. Rheumatology (Oxford) 2010;49:490–500. doi: 10.1093/rheumatology/kep398. [DOI] [PubMed] [Google Scholar]
- 50.King TE, Jr, Behr J, Brown KK, du Bois RM, Lancaster L, de Andrade JA, et al. BUILD-1: A randomized placebo-controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;177:75–81. doi: 10.1164/rccm.200705-732OC. [DOI] [PubMed] [Google Scholar]
- 51.King TE, Jr, Brown KK, Raghu G, du Bois RM, Lynch DA, Martinez F, et al. BUILD-3: A randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;184:92–9. doi: 10.1164/rccm.201011-1874OC. [DOI] [PubMed] [Google Scholar]
- 52.Zisman DA, Schwarz M, Anstrom KJ, Collard HR, Flaherty KR, Hunninghake GW. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N Engl J Med. 2010;363:620–8. doi: 10.1056/NEJMoa1002110. [DOI] [PMC free article] [PubMed] [Google Scholar]