

Abstract
Hypoxia-induced pulmonary vasoconstriction in patients with a medical history of high-altitude pulmonary edema (HAPE) may involve activation of the endothelin-1 (ET-1) pathway. We, therefore, compared the effect of the ETA/ETB receptor antagonist, bosentan, on pulmonary artery systolic pressure (PASP) in healthy subjects with (HS: HAPE subjects, n=5) or without a HAPE-history (CS: Control subjects, n=10). A double-blind, placebo-controlled, randomized, crossover design was performed in order to study the effects on PASP of a single oral dose of bosentan (250 mg) after 90 min exposure to normobaric hypoxia (FiO2 =0.12). In normoxia, PASP, evaluated by echocardiography, was 23.4±2.7 mmHg in CS and 28±5.8 mmHg in HS (NS). During the placebo period, hypoxia induced a significant decrease in SaO2, PaO2 and PCO2 and increase in pH in both CS and HS. Pulmonary arterial systolic pressure was also significantly increased (+8.5±5.0 mmHg in CS; +13.4±3.1 mmHg in HS) and reached significantly higher levels in HS than in CS (P=0.02). Bosentan significantly but similarly blunted the hypoxia-induced increase in PASP in both CS (Bosentan: 27.0±3.3 mmHg; placebo: 32.1±3.5 mmHg; P<0.01) and HS (Bosentan: 35.0±2.9 mmHg; placebo: 41.4±7.6 mmHg; P<0.05), (CS 5.2±5.3 vs. HS -6.4±5.2 mmHg, NS). Bosentan did not have a major effect on the hypoxia-induced changes in blood gas, or on cardiac output (CO) and systemic blood pressure (SBP), which were not modified by hypoxia. Plasma ET-1 in hypoxia during the bosentan period was 2.8 times higher than during for both CS and HS. A single oral dose of bosentan similarly blunted the hypoxia-induced increase in PASP both in healthy and HAPE-susceptible subjects, without altering CO or SBP.
Keywords: endothelin-1, hypoxia, pulmonary hypertension
INTRODUCTION
Endothelin, which is a potent pulmonary vasoconstrictor, has been involved in the pathophysiological pathway of high-altitude pulmonary edema (HAPE). Indeed, the main mechanism for edema constitution appears to be hypoxic pulmonary vasoconstriction that leads to a larger increase in pulmonary artery pressure (PAP) in subjects with HAPE susceptibility than in subjects without HAPE history. In mountaineers, the increase in plasma ET-1 concentration at high altitude is correlated with the increase in PAP, and is larger in subjects susceptible to HAPE than in subjects with no HAPE history.[1,2] Nonselective ETA/ETB endothelin receptor blockade in animal models prevent hypoxic pulmonary vasoconstriction and reduces acute and chronic hypoxia-induced pulmonary hypertension.[3–5] Thus, we and other investigators have shown that pulmonary arterial systolic pressure (PASP) in healthy subjects exposed to high altitude was significantly lower after endothelin-1 (ET-1) receptor blockade by bosentan than after placebo.[6,7] However, at the present time no data are available for subjects with a previous history of HAPE.
The aim of this study was therefore to investigate, in healthy mountaineers with a past history of HAPE in comparison with control subjects, the effects of a single oral dose of bosentan on PASP and cardiac hemodynamics during an acute normobaric-hypoxia test at rest and during submaximal exercise.
MATERIALS AND METHODS
Fifteen men (age: 37±8 years) with experience in mountain hiking were recruited for the study. Five of them had previously experienced at least 2 HAPE episodes at high altitude, defined as acute dyspnea with cyanosis at an altitude >3,000 m. with a need to descend to an altitude <2,000 m (HAPE subjects group, HS). The 10 remaining healthy subjects had not previously experienced HAPE, despite regular mountain hiking at or above the same altitude (control subjects group, CS). The results for the latter group have been published in detail elsewhere.[7] None of the subjects was acclimatized to altitude or was on any medication. They were nonsmokers and free of disease at the time of the study. All subjects gave written informed consent to participate to the study. The study was approved by the “Comité des Personnes se Prêtant à la Recherche Biomédicale, Paris-Cochin” and was carried out in accordance with the Declaration of Helsinki principles.
Study design
Briefly, the two groups were compared in a two-period, randomized, double-blind, crossover study in order to investigate the hemodynamic and biochemical effects of a single oral 250 mg dose of bosentan (Actelion, Ltd, Basel, Switzerland) or placebo during hypoxia on 2 distinct days separated by at least 2 days washout interval. The dose of bosentan used is the recommended dose for patients with primary or secondary pulmonary hypertension. Subjects were instructed to avoid strenuous physical activity 1 week before the investigation. All investigations were performed at the Georges Pompidou Hospital Clinical Investigation Center in Paris (40-50 m. above sea level) at normal barometric pressure.
On each study day at 8 a.m., after a light caffeine- and fat-free breakfast, baseline echocardiography (with PASP measurement) was carried out and blood samples for plasma ET-1 concentration and blood gas measurements were taken at rest in normoxia and during a submaximal exercise test at 8:30 a.m. At 9:30 a.m., 250 mg bosentan or a matching placebo was given orally with 250 ml tap water, according to the randomization procedure. At 11:30 a.m., the subject was exposed to normobaric hypoxia for 90 minutes. During exposure to the normobaric hypoxic gas mixture, a second set of echocardiography and blood sample analyses were carried out, at rest at 12:30 p.m. (3 hours. after bosentan or placebo ingestion) and during a second submaximal exercise test at 1 p.m.
Oxygen saturation (SaO2) by ear oximetry (Oximax N-595, Nellcor, Boulder, Colo., USA), systolic and diastolic blood pressure (SBP and DBP, respectively) by a semiautomatic validated oscillometric device (Nippon Collins, Komaki City, Japan) and heart rate (HR) were continuously monitored.
Normobaric hypoxia
Normobaric hypoxia was achieved by inhalation of a gas mixture of O2(FiO2 =0.12) and N2 at normal barometric pressure through a tightly secure facial mask; this pressure was equivalent to the oxygen partial pressure found at an altitude of 4,300 m (Altitrainer, SM-TEC, Lausanne, Switzerland).
Submaximal exercise test
The submaximal exercise tests were performed in the semisupine position on a cycle ergometer. The test started with a workload of 40 W. This was then increased by 10 W every minute until reaching 30% of the maximal heart rate during exercise in normoxia, which was calculated as follows: Resting HR + (maximal predicted HR – resting HR) × 0.3. Maximal predicted heart rate was calculated as 220 – age (years). For each subject, the same target HR during exercise was used in normobaric hypoxic conditions.
Echocardiographic measurements
Echocardiography was performed with a 3.5 MHz probe (Vivid 7, GE Healthcare, Milwaukee, Wis., USA), and the recordings were analyzed off-line. Left heart disease and patent foramen ovale were excluded on the first study day in all subjects by left heart standard measurements and a contrast test.[8] The PASP, defined as equal to the RV systolic pressure, was calculated using the modified Bernoulli equation (PASP = 4VTR2 + RAP), where VTR is the tricuspid regurgitation peak velocity, measured by continuous-wave Doppler and RAP the right atrial pressure. The right atrial pressure was estimated as a function of the inferior vena cava diameter and its variation during breathing. Pulmonary flow was recorded by pulsed-wave Doppler and acceleration time was measured from onset to the peak velocity of pulmonary flow. Pulmonary vascular resistance (PVR) was determined using the formula PVR (Wood Unit, WU) = VTR/VTIRVOT + 0.16, where VTIRVOT is the velocity time-integral of the right ventricle outflow tract.[9] Cardiac output (CO) was calculated as the product of the velocity-time integral and the area of the LV outflow tract and HR. Early tricuspid inflow wave, E, and annular tricuspid velocity, Et, were measured by Doppler imaging for the tricuspid ratio: E/Et calculation. During the exercise tests, only VTR, pulmonary flow, tricuspid flow, and tricuspid annular velocities were recorded.
Laboratory methods
Plasma ET-1 concentrations were determined with a luminescent immunoassay kit “QET00” (R & D Systems Europe Ltd, Abingdon, UK). The quantification limit was 0.25 pg/ml PaO2, PCO2, and pHa were measured in arterialized blood samples taken from an ear lobe prewarmed with capsaicin cream.
Statistics
Baseline characteristics and echocardiographic measurements between subjects with or without HAPE history were compared using the Wilcoxon rank sum test.
Experimental and treatment effects were assessed using analysis of variance (ANOVA) for a cross-over design in the CS group.[7] Results were expressed as mean differences with their 95% confidence intervals (CI). P values less than 0.05 were considered significant.
Given the small sample size of the HS group (n=5), we used the Wilcoxon signed-rank test to analyze the differences between bosentan and placebo in the different experimental conditions as listed above. Results were expressed as mean differences with their range. Because of the small sample size of the HS group and the general lack of power of nonparametric tests (Wilcoxon test), the lowest two-tailed P value could not be lower than 0.063, whatever the difference between paired observations. However, P=0.063 suggests that all five paired differences were in the same direction and thus had the same sign.
Statistical analyses were performed with SAS statistical software (version 9.1, Cary, NC, USA).
RESULTS
Differences between control and HAPE-subjects during the placebo period: Baseline measurements and effects of exercise or normobaric hypoxia
At screening, clinical characteristics of CS and HS were similar, with the exception of SBP and DBP, which were significantly higher in HS than in CS (Table 1).
Table 1.
Control and HAPE subject characteristics at screening and baseline
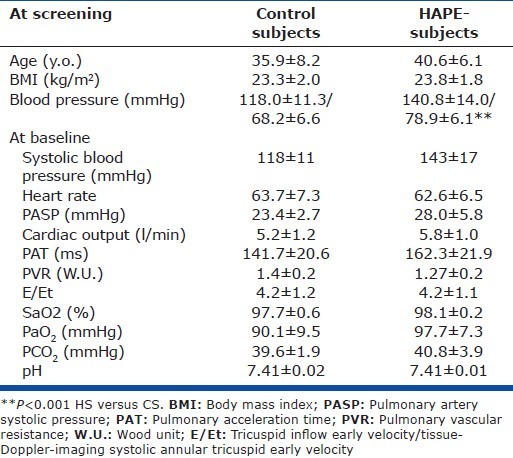
Baseline PASP in normoxia was not significantly different between CS and HS (Table 1 and Fig. 1). Three HS had a baseline PASP between 30 and 35 mmHg, whereas the greatest PASP measured in CS was 28 mmHg. Pulmonary acceleration time, PVR, Et/Ea ratio, and CO at rest did not significantly differ between the two groups (Table 1). Baseline HR, SaO2, and blood gases were similar between HS and CS (Table 1).
Figure 1.
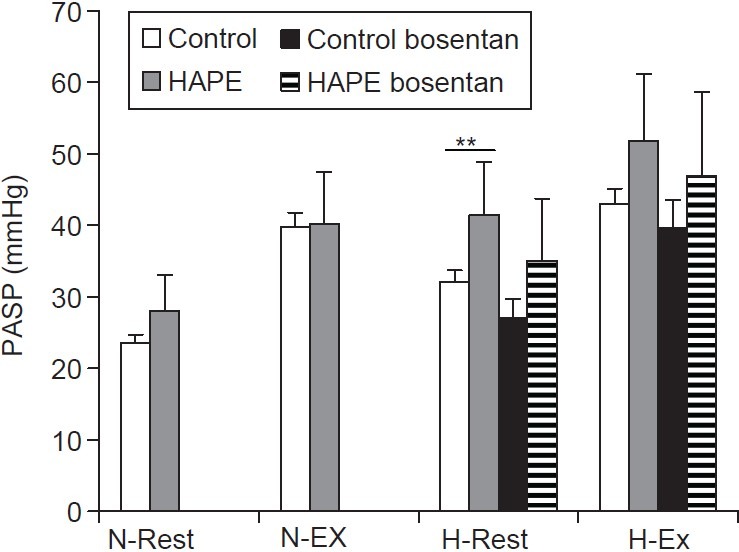
Systolic pulmonary arterial pressure (PASP) in HAPE subjects (HS, gray bars) and control subjects (CS, open bars) during normoxia at rest (N-Rest) and exercise (N-Ex), and during hypoxia exposure at rest (H-Rest) and exercise (H-Ex). *P=0.02 HS vs. CS).
During exercise in normoxia, the workload necessary to reach 30% of the maximal predicted HR was similar in HS (116±11 W achieved in 8.6±1.14 min. on Day 1) and in CS (106±22 W, 8.5±3.6 min. on Day 1). Pulmonary arterial systolic pressure increased similarly in both groups; it reached 38±9 mmHg on Day 1 and 40±7 mmHg on Day 2 in HS, and 39±11 mmHg on Day 1 and 39±9 mmHg on Day 2 in CS (NS between groups; Fig. 1). Five CS and 2 HS reached a PASP > 45 mmHg during exercise. As expected, SBP increased during exercise (CS 163±23 and HS 173±11 mmHg), with no significant difference between HS and CS at the maximal workload. Heart rate, SaO2, and blood gases and echocardiographic measurements (pulmonary acceleration time, PVR, CO) were similar between HS and CS during exercise (not shown).
In both CS and HS, exposure to normobaric hypoxia induced, a large and similar decrease in SaO2 (CS: 82.6±7.8%, HS: 78.9±5.7%), PaO2 (CS: 42.1±6.8 mmHg, HS: 41.1±6.6 mmHg), and PaCO2 (CS: 33.4±4.4 mmHg, HS: 36.9±3.2 mmHg) and a similar increase in pH (CS: 7.48±0.03, HS: 7.46±0.01) at rest. Pulmonary arterial systolic pressure at rest increased significantly by +8.5±5.0 mmHg in CS and by +13.4±3.1 mmHg in HS after 60- to 90-minute exposure to normobaric hypoxia (P= 0.075 CS vs. HS). It reached significantly higher levels in HS (41.4±7.6 mmHg) than in CS (32.1±3.5 mmHg, P=0.02; Fig. 1). The hypoxia-induced increase in PASP was accompanied by a similar parallel increase in PVR in the CS: (1.7±0.2 WU, P<0.05) and the HS (1.8±0.4 WU, P=0.063) and a decrease in pulmonary acceleration time, which was significant only in HS (132.8±14.8 ms, P=0.063). Systemic blood pressure and CO did not significantly change with exposure to normobaric hypoxia, in either group (not shown). Plasma ET-1 concentrations at rest slightly increased with hypoxia in CS (+35%) but not in HS (Fig. 2).
Figure 2.
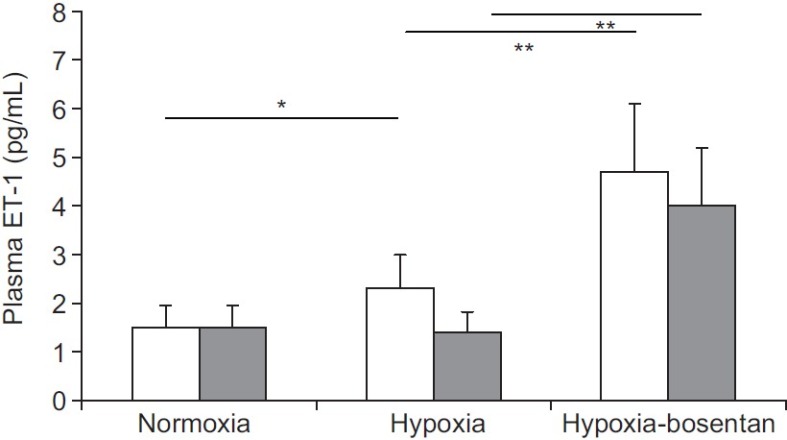
Plasma endothelin-1 (ET-1) concentrations in control (CS, open bar) and HAPE (HS, gray bar) subjects. *P=0.04; **P<0.01.
In both CS and HS, exercise in normobaric hypoxic conditions induced a further decrease in SaO2 (CS: 72.3±3.0%, HS 69.5±8.1 %), PaO2 (CS: 34.3±2.9 mmHg, HS 32.7±4.2 mmHg) and PaCO2(CS: 29.7±4.3 mmHg, HS 31.4±3.8 mmHg) with no significant difference between the two groups. Maximal workload, and the time to reach the expected HR, decreased substantially during normobaric hypoxia in both CS and HS (workload: 69±14.5 W vs. 72±14.8 W, respectively, NS; HR: 4.20±1.75 min vs. 4.20±1.48 min, respectively, NS). A further increase in PASP, by +11±7.9 mmHg in CS and by +10.4±13 mmHg in HS, was observed (NS between groups; Fig. 1). The PASP reached at maximal workload in hypoxic conditions was higher in HS (51.8±12 mmHg) than in CS (43±8.5 mmHg) but the difference between groups was not significant (Fig. 1; P=0.21). Four HS patients had PAPS > 45 mmHg during exercise in normobaric hypoxia.
Differences between control and HAPE-subjects: Effects of bosentan at rest and exercise during normobaric hypoxia
Bosentan was well tolerated in all subjects and induced significantly 3- to 4-fold higher plasma ET-1 concentrations in hypoxic conditions at rest compared to placebo (Fig. 2).
During normobaric hypoxia at rest, PASP after bosentan was not significantly different in CS and HS (CS: 27.0±3.3 mmHg and HS: 35.0±2.9 mmHg, P=NS), although bosentan significantly blunted the hypoxia-induced increase in PASP for both CS and HS compared to placebo (CS: 5.2 mmHg, 95% CI [2.4; 9.9], P=0.002 bosentan vs. placebo; HS: 6.4 mmHg, 95%CI [3.4; 13.1], P=0.063 bosentan vs. placebo Fig. 1). All HS had PASP <40 mmHg at rest during the bosentan period. The increase in PVR did not significantly change between bosentan and placebo periods for either group (CS: 1.5±0.2 WU, HS: 1.5±0.3 WU).
Bosentan had no significant effect on the magnitude of the exercise-induced increase in PASP during hypoxia (exercise-rest) in either CS (bosentan: +13.4±11.3 mmHg; placebo: +11.0±7.9 mmHg; P=0.68) or HS (bosentan: +11.9±11.9 mmHg; placebo: +10.4±13.0 mmHg; P=0.81). However, after bosentan intake, PASP during exercise in hypoxia remained higher for HS than for CS (not shown) and lower than after placebo, for both CS (39.6±12.6 mmHg) and HS (46.85±11.3 mmHg), although these differences were not statistically significant (Fig. 1).
There was no significant difference between bosentan and placebo for either SBP or echocardiographic parameters in hypoxic conditions at rest or exercise (not shown). Bosentan had no significant effect on SaO2, PaCO2, PaO2, and pH at rest or during exercise in hypoxic conditions in either group (not shown).
DISCUSSION
High-altitude pulmonary edema occurs in <1% of otherwise healthy subjects during ascent to high altitude. Level of altitude, prior acclimatization, rate of ascent, exertion, cold, individual genetic susceptibility, and presence of patent foramen ovale are the main clinical determinants for HAPE.[10] However, searching for other potential pathophysiological pathways in order to detect the subjects susceptible to develop HAPE and to find new therapeutic strategies to prevent HAPE occurrence is still a topical subject. Our results show that, in subjects with or without medical history of HAPE, blockade of ETA and ETB receptors by a single oral dose of bosentan 250 mg similarly reduces the hypoxia-induced pulmonary hypertension at rest. Although bosentan did not significantly reduce the magnitude of the increase in PASP due to exercise in hypoxic conditions, PASP remained 3-5 mmHg lower after bosentan intake than after placebo intake in both groups. The vasodilatory effect of bosentan was restricted to the pulmonary circulation and bosentan had no effect on right ventricular systolic or on systemic hemodynamics (SBP, CO and HR). Despite its favorable pulmonary hemodynamic effect, acute administration of bosentan had no significant effect on gas exchange in hypoxic conditions in control or HAPE subjects.
Differences between CS and HS during the placebo period
Several attempts have been made to identify HAPE-susceptible individuals and to apply preventive measures before an ascent to high altitude (reviewed in Reference 10). These subjects appear to have an increase pulmonary vasoreactivity which may be revealed by hypoxia and/or exercise. As previously reported, we first confirmed that HS had normal resting PASP at sea level in normoxia.[11] However, in our study, resting PASP was slightly higher, although nonsignificantly, in HS than in CS. Part of the difference in resting PASP between the two groups may be due to the higher SBP in HS than CS.[12] Second, we confirmed that for a similar hypoxia-induced decrease in SaO2 and PaO2 at rest, HS had more pronounced hypoxic pulmonary vasoconstriction (increase in PVR) than CS, thus achieving a significantly higher PASP. This was consistent with the results of previous studies.[11,13–15] However, there was an overlap of PASP values between HS and CS that did not allow HS to be distinguished from CS. Further, larger studies are needed in order to establish if the hypoxia test at rest could serve as a screening test to detect HAPE-susceptible subjects before altitude exposure. Third, in contrast to previous findings,[11] PASP values recorded during exercise at a similar workload in normoxic conditions (≈110 W for a HR ≈120 bpm in both groups) did not significantly differ between HS and CS. Indeed, Grunig et al.[11] suggested that a PASP > 45 mmHg during submaximal exercise in normoxia may be a useful screening test to identify HAPE susceptible subjects. We observed that PASP at exercise in normoxia was >45 mmHg only in 2 HS and that 2 CS reached PASP above this threshold during exercise. However, these data should be interpreted with caution because of the small sample size of the two studies. Finally, the workload achieved under normoxic (≈110 W, 8–9 minutes) and hypoxic (≈70 W, 4-5 minutes) conditions were similar between HS and CS.
The larger increase in PASP in HS than in CS during hypoxia at rest and exercise suggests an increased intrinsic vasoreactivity of their pulmonary circulation in response to hypoxic stress, potentially involving ET-1 transduction pathways (reviews in[16] and[17] ). We found that a 90-minute exposure to normobaric hypoxia induced a slight increase in plasma ET-1 concentrations in CS but not in HS, probably because of its small sample size. Previous studies have found plasma ET-1 concentration to be higher in HAPE susceptible subjects than in nonsusceptible subjects, with the increase in plasma ET-1 concentration correlated to the increase in PASP after exposure to high altitude.[1,2] Berger et al. also found that at high altitude there is a transpulmonary gain in ET-1 plasma concentration while at sea level there is a transpulmonary loss.[18] This may be due to increased ET-1 release from endothelial cells and/or increased conversion of big ET-1.[19,20] However, measurement of endothelins in plasma is not a sensitive method for investigating the effects of endothelins within the vascular wall, whereas the magnitude of the response to a pharmacological of ETA/ETB receptors represents at best an integrated evaluation of the degree of the endothelin system involvement.
Bosentan effects at rest and during exercise during normobaric hypoxia in HS and CS
The selected dose of bosentan (250 mg) effectively blocked ETA/ETB receptors in both CS and HS, with a similar 3-fold increase in plasma ET-1 concentrations.[21–23] It also blunted the hypoxia-induced increase in PASP at rest in both CS and HS subjects, with a similar decrease (15-20%) in PASP. This is consistent with the results of the ACME-1 study, in which PASP was 30% lower after 3-day bosentan treatment (62.5–125 mg o.d.) than after placebo in healthy subjects after an ascent to high altitude.[6] In our study, bosentan did not significantly reduce the magnitude of the exercise/hypoxia-induced increase in PASP in both groups. However, PASP after bosentan treatment remained 3–5 mmHg lower than after placebo in both groups (NS). Although HS reached a higher PASP than CS during both rest and exercise under hypoxic conditions, the magnitude of the blunting effect of bosentan on PASP increase did not differ between HS and CS groups; however, PASP after bosentan treatment remained higher in HS than in CS.
The absence of a significant difference between bosentan and placebo during exercise can be explained by increased between-subject variability in PASP and difficulty in obtaining adequate Doppler signal during exercise. Furthermore, the small sample size of the study also contributes to the absence of significant response. The effect of bosentan on PASP was probably due to its selective pulmonary vasodilatory effect at the selected dose.[24] Indeed, the blunting effect of bosentan on the PASP increase was not associated with any change in CO, SBP or HR.
Finally, a single oral dose of bosentan had no major effect on SaO2 or blood gases at rest or during exercise under hypoxic conditions, suggesting that in contrast with Faoro et al.[25] a single oral dose of bosentan does not have an acute effect on oxygen transfer capacity in these experimental conditions in normal or HAPE-subjects.
CONCLUSION
Our preliminary findings demonstrate that (1) the ET1 system is indeed activated by hypoxia independently of individual susceptibility to HAPE, and (2) this activation can be effectively and partly reversed by an ETA/ETB receptor antagonist. However, the fact that the magnitude of the reduction in PASP after bosentan treatment was similar between HS and CS suggests that hypoxia-induced activation of the ET-1 system is not any greater in HS than in CS in our experimental conditions. Therefore, the PASP response during hypoxia to a single oral dose of 250 mg of bosentan cannot serve as a useful screening test to identify HAPE susceptible subjects. However, these results should be confirmed by further studies involving more subjects.
ACKNOWLEDGMENTS
The authors thank the nursing staff of the Clinical Investigation Centre at the Georges Pompidou Hospital who ran the protocol, Dr. Laure Marelle who recruited the subjects and Dr. Jean-Louis Paul who performed the ET1 dosages. They also thank Dr. Virgine Gressin (Actélion, France) and Dr. Martine Clozel (Actélion, Basle) to have kindly provided bosentan and its matching placebo.
Footnotes
Source of Support: This work was supported by a grant from Assistance Publique des Hôpitaux de Paris (Contrat d’Initiative à la Recherche Clinique: AOR05053). Clinical trial registration information: www.clinicaltrials.govNCT00260819
Conflict of Interest: None declared.
REFERENCES
- 1.Sartori C, Vollenweider L, Loffler BM, Delabays A, Nicod P, Bärtsch P, et al. Exaggerated endothelin release in high-altitude pulmonary edema. Circulation. 1999;99:2665–8. doi: 10.1161/01.cir.99.20.2665. [DOI] [PubMed] [Google Scholar]
- 2.Goerre S, Wenk M, Bartsch P, Lüscher TF, Niroomand F, Hohenhaus E, et al. Endothelin-1 in pulmonary hypertension associated with high-altitude exposure. Circulation. 1995;91:359–64. doi: 10.1161/01.cir.91.2.359. [DOI] [PubMed] [Google Scholar]
- 3.Eddahibi S, Raffestin B, Clozel M, Levame M, Adnot S. Protection from pulmonary hypertension with an orally active endothelin receptor antagonist in hypoxic rats. Am J Physiol. 1995;268(2 Pt 2):H828–35. doi: 10.1152/ajpheart.1995.268.2.H828. [DOI] [PubMed] [Google Scholar]
- 4.Willette RN, Ohlstein EH, Mitchell MP, Sauermelch CF, Beck GR, Luttmann MA, et al. Nonpeptide endothelin receptor antagonists.VIII: Attentuation of acute hypoxia-induced pulmonary hypertension in the dog. J Pharmacol Exp Ther. 1997;280:695–701. [PubMed] [Google Scholar]
- 5.Underwood DC, Bochnowicz S, Osborn RR, Luttmann MA, Hay DW. Nonpeptide endothelin receptor antagonists. X. Inhibition of endothelin-1- and hypoxia-induced pulmonary pressor responses in the guinea pig by the endothelin receptor antagonist, SB 217242. J Pharmacol Exp Ther. 1997;283:1130–7. [PubMed] [Google Scholar]
- 6.Modesti PA, Vanni S, Morabito M, Modesti A, Marchetta M, Gamberi T, et al. Role of endothelin-1 in exposure to high altitude: Acute Mountain Sickness and Endothelin-1 (ACME-1) study. Circulation. 2006;114:1410–6. doi: 10.1161/CIRCULATIONAHA.105.605527. [DOI] [PubMed] [Google Scholar]
- 7.Pham I, Wuerzner G, Richalet JP, Peyrard S, Azizi M. Endothelin receptors blockade blunts hypoxia-induced increase in PAP in humans. Eur J Clin Invest. 2010;40:195–202. doi: 10.1111/j.1365-2362.2010.02254.x. [DOI] [PubMed] [Google Scholar]
- 8.Allemann Y, Hutter D, Lipp E, Sartori C, Duplain H, Egli M, et al. Patent foramen ovale and high-altitude pulmonary edema. JAMA. 2006;296:2954–8. doi: 10.1001/jama.296.24.2954. [DOI] [PubMed] [Google Scholar]
- 9.Abbas AE, Fortuin FD, Schiller NB, Appleton CP, Moreno CA, Lester SJ. A simple method for noninvasive estimation of pulmonary vascular resistance. J Am Coll Cardiol. 2003;41:1021–7. doi: 10.1016/s0735-1097(02)02973-x. [DOI] [PubMed] [Google Scholar]
- 10.Hackett PH, Roach RC. High-altitude illness. N Engl J Med. 2001;345:107–14. doi: 10.1056/NEJM200107123450206. [DOI] [PubMed] [Google Scholar]
- 11.Grunig E, Mereles D, Hildebrandt W, Swenson ER, Kübler W, Kuecherer H, et al. Stress Doppler echocardiography for identification of susceptibility to high altitude pulmonary edema. J Am Coll Cardiol. 2000;35:980–7. doi: 10.1016/s0735-1097(99)00633-6. [DOI] [PubMed] [Google Scholar]
- 12.Abergel E, Chatellier G, Toussaint P, Dib JC, Menard J, Diebold B. Doppler-derived pulmonary arterial systolic pressure in patients with known systemic arterial pressures. Am J Cardiol. 1996;77:767–9. doi: 10.1016/s0002-9149(97)89216-8. [DOI] [PubMed] [Google Scholar]
- 13.Hultgren HN, Grover RF, Hartley LH. Abnormal circulatory responses to high altitude in subjects with a previous history of high-altitude pulmonary edema. Circulation. 1971;44:759–70. doi: 10.1161/01.cir.44.5.759. [DOI] [PubMed] [Google Scholar]
- 14.Kawashima A, Kubo K, Kobayashi T, Sekiguchi M. Hemodynamic responses to acute hypoxia, hypobaria, and exercise in subjects susceptible to high-altitude pulmonary edema. J Appl Physiol. 1989;67:1982–9. doi: 10.1152/jappl.1989.67.5.1982. [DOI] [PubMed] [Google Scholar]
- 15.Yagi H, Yamada H, Kobayashi T, Sekiguchi M. Doppler assessment of pulmonary hypertension induced by hypoxic breathing in subjects susceptible to high altitude pulmonary edema. Am Rev Respir Dis. 1990;142:796–801. doi: 10.1164/ajrccm/142.4.796. [DOI] [PubMed] [Google Scholar]
- 16.Weir EK, Lopez-Barneo J, Buckler KJ, Archer SL. Acute oxygen-sensing mechanisms. N Engl J Med. 2005;353:2042–55. doi: 10.1056/NEJMra050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartsch P, Mairbaurl H, Maggiorini M, Swenson ER. Physiological aspects of high-altitude pulmonary edema. J Appl Physiol. 2005;98:1101–10. doi: 10.1152/japplphysiol.01167.2004. [DOI] [PubMed] [Google Scholar]
- 18.Berger MM, Dehnert C, Bailey DM, Luks AM, Menold E, Castell C, et al. Transpulmonary plasma ET-1 and nitrite differences in high altitude pulmonary hypertension. High Alt Med Biol. 2009;10:17–24. doi: 10.1089/ham.2008.1053. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi H, Soma S, Muramatsu M, Oka M, Fukuchi Y. Upregulation of ET-1 and its receptors and remodeling in small pulmonary veins under hypoxic conditions. Am J Physiol Lung Cell Mol Physiol. 2001;280:L1104–14. doi: 10.1152/ajplung.2001.280.6.L1104. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi H, Soma S, Muramatsu M, Oka M, Ienaga H, Fukuchi Y. Discrepant distribution of big endothelin (ET)-1 and ET receptors in the pulmonary artery. Eur Respir J. 2001;18:5–14. doi: 10.1183/09031936.01.00075501. [DOI] [PubMed] [Google Scholar]
- 21.Bohm F, Pernow J, Lindstrom J, Ahlborg G. ETA receptors mediate vasoconstriction, whereas ETB receptors clear endothelin-1 in the splanchnic and renal circulation of healthy men. Clin Sci (Lond) 2003;104:143–51. doi: 10.1042/CS20020192. [DOI] [PubMed] [Google Scholar]
- 22.Fukuroda T, Fujikawa T, Ozaki S, Ishikawa K, Yano M, Nishikibe M. Clearance of circulating endothelin-1 by ETB receptors in rats. Biochem Biophys Res Commun. 1994;199:1461–5. doi: 10.1006/bbrc.1994.1395. [DOI] [PubMed] [Google Scholar]
- 23.Weber C, Schmitt R, Birnboeck H, Hopfgartner G, van Marle SP, Peeters PA, et al. Pharmacokinetics and pharmacodynamics of the endothelin-receptor antagonist bosentan in healthy human subjects. Clin Pharmacol Ther. 1996;60:124–37. doi: 10.1016/S0009-9236(96)90127-7. [DOI] [PubMed] [Google Scholar]
- 24.Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]
- 25.Faoro V, Boldingh S, Moreels M, Martinez S, Lamotte M, Unger P, et al. Bosentan Decreases Pulmonary Vascular Resistance and Improves Exercise Capacity in Acute Hypoxia. Chest. 2009;135:1215–22. doi: 10.1378/chest.08-2222. [DOI] [PubMed] [Google Scholar]