

Abstract
Endothelial dysfunction plays an important role in the pathogenesis of pulmonary arterial hypertension (PAH) in sickle cell disease (SCD). A variety of evidence suggests that circulating endothelial progenitor cells (EPCs) play an integral role in vascular repair. We hypothesized that SCD patients with PAH are deficient in EPCs, potentially contributing to endothelial dysfunction and disease progression. The number of circulating CD34+/CD14−/CD106+ EPCs was significantly lower in SCD patients with PAH than without PAH (P=0.025). CD34+/CD14−/CD106+ numbers significantly correlated with tricuspid regurgitation velocity (TRV, r=−0.44, P=0.033) 6-minute walk distance (6MWD, r= 0.72, P=0.001), mean pulmonary artery pressure (mPAP, r= −0.43, P=0.05), and pulmonary vascular resistance (PVR, r=−0.45, P=0.05). Other EPC subsets including CD31+/CD133+/CD146+ were similar between both groups. Numbers of EPCs did not correlate with age, sex, hemoglobin, WBC count, reticulocyte count, lactate dehydrogenase (LDH), iron/ferritin levels, and serum creatinine. These data indicate that subsets of EPC are lower in SCD patients with PAH than in those without PAH. Fewer EPCs in PAH patients may contribute to the pulmonary vascular pathology. Reduced number of EPCs in SCD patients with PAH might not only give potential insight into the pathophysiological mechanisms but also might be useful for identifying suitable therapeutic targets in these patients.
Keywords: associated pulmonary arterial hypertension, sickle cell disease, stem cell, pathogenic mechanism
INTRODUCTION
PAH is a major complication of SCD with the prevalence ranging from 20% to 30% based on Doppler echocardiographic studies.[1] Patients with SCD have a higher risk of death with even mild elevations in PAP compared to primary PH. Each 10 mmHg rise in mean PAP was found to be associated with a 1.7-fold increase in the rate of death.[2] Recent autopsy studies suggest that up to 75% of SCD patients have histological evidence of PAH at the time of death.[3]
Pathological changes seen in these patients are similar to those seen in other forms of PAH.[3] Specific mechanisms by which PAH develops in SCD remain poorly defined. Histological features of SCD-related PAH include intimal hyperplasia; smooth muscle proliferation; and formation of plexiform lesions causing vascular lumen obliteration.[4,5] These findings suggest abnormal endothelial homeostasis caused by impairment of endothelial repair.
Multiple lines of evidence suggest that endothelial progenitor cells (EPCs) play an important role in endothelial repair process.[6] EPCs are precursor cells that are typically considered to arise from mesodermal stem cells or hemangioblasts in the bone marrow.[7] Upon stimulation by various angiogenic factors including VEGF-A and SDF-1, these cells circulate to the site of ischemia or endothelial injury, where they proliferate and differentiate into mature endothelial cells and contribute to postnatal neovascularization and re-endothelialization.[6,8] EPCs lack mature endothelial markers, but coexpress markers of bone marrow origin such as CD34 or AC133 in addition to endothelial markers (VE-Cadherin or VEGFR-2).[7,9–11]
Since the discovery of EPCs by Asahara et al. in 1997,[12] numerous studies have shown that the number and function of progenitor cells correlate with cardiovascular risk factors, reflect endothelial impairment and are predictive of clinical outcome.[13–15] Numbers of circulating EPCs are also altered in pulmonary disease states.[16,17] These findings have fostered a growing interest in EPCs as a potential therapeutic target or predictive biomarker in PAH.[18,19]
The exact role of progenitor cells in preventing pulmonary vascular alterations in patients with SCD remains undetermined. In this study we sought to compare numbers of various EPC subsets in patients with SCD with and without PAH.
MATERIALS AND METHODS
Study population
Patients with known SCD were recruited from our hospital's SCD clinic. Patients with PH related to left heart disease, pulmonary disease, chronic thromboembolic disease, autoimmune or collagen vascular disease, sleep-associated disorders, HIV infection or liver disease were excluded. Patients with chronic renal insufficiency (serum creatinine ≥ 1.5 mg/dl), pregnancy, smoking or substance abuse, active sickle crisis or acute chest syndrome at the time of their echo were also excluded (Table 1). All participants were free of wounds, ulcers, retinopathy, recent surgery, inflammatory or malignant disease as these conditions might influence EPC number. Venous blood was collected at the time of echo study from all participants and processed within 24 hours of collection for evaluation of progenitor cells. This study was approved by the Downstate-Kings County Review Board (Chairperson Eli Freidman: IRB # 050409-KCH-S0096) and written informed consent was obtained from all individuals.
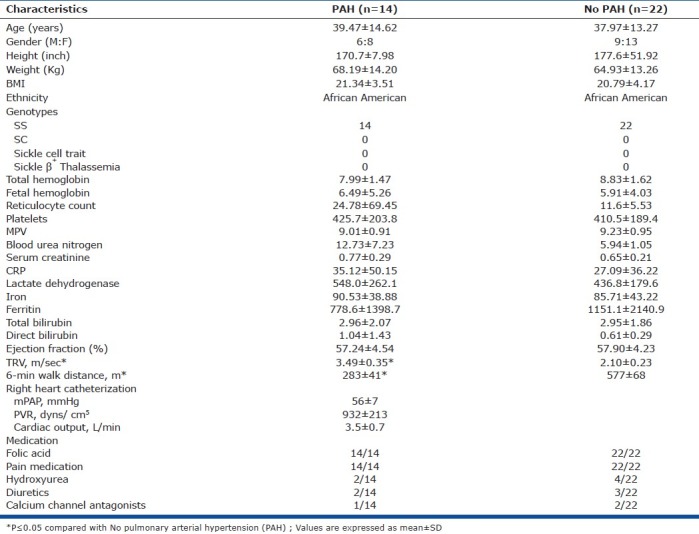
Table 1.
Patient characteristics in sickle cell disease, PAH vs. No PAH
Evaluation for PAH
Two-dimensional and Doppler echocardiographic studies (Phillips iE33 and 5500, Andover, Massachusetts) were performed by an experienced technician according to standard American Society of Echocardiography protocol. A 2.5 MHz ultrasound transducer (model S5-1 and S3) recorded continuous wave signals using multiple views to obtain peak TRV. Right ventricular systolic pressure (RVSP) was calculated using modified Bernoulli equation [4v2 + right atrial pressure (RAP), v=peak TRV meter/second] and was considered to be equal to the PASP in the absence of RV outflow obstruction. We defined TRV≥2.5 m/s as a marker of PAH in SCD. The presence of PAH was confirmed via right heart catheterization (RHC) defined as mPAP≥25 mmHg. Hemodynamic measurements were performed in a recumbent position. Cardiac output was obtained, using triplicate measurements with the thermodilution method (Agilent, Boeblingen, Germany). Pulmonary vascular resistance (PVR) was calculated using the standard formula PVR=80 × (mean pulmonary artery pressure [mmHg] − pulmonary capillary wedge pressure [mmHg])/cardiac output [l/min]. Fourteen patients were found to have PAH while 22 patients were found to lack PAH. Exercise capacity was assessed by a 6-minute walk distance.
Flow cytometry
Flow cytometry was used to quantify EPCs in peripheral blood by identifying eight distinctive cell markers (CD34, CD14, CD106, CD105, CD31, CD133, KDR, CD146) in 20 different combinations in both groups of patients. One milliliter of venous blood (anticoagulant: EDTA) was prepared by lysing red cells with 10 ml of FACS lysing solution (10×, diluted 1:10) for 10 minutes. The white cells (WBC) were then blocked with 20 μl of specific Fc-receptor antibodies (Octagam; Octapharma) and 200 μl of mouse serum (Sigma) for a minimum of 20 minutes at room temperature. Next, the cells were incubated with monoclonal anti-human mouse antibodies, namely PE-CD146, and PE-Cy 5-CD34 (Becton Dickinson) CD31- FITC, CD106-PE, CD14-FITC (eBiosciences), CD105-PE, VEGFR2 antibody [KDR/EIC] (Abcam) and PE-CD133 (Miltenyi Biotec) for 20 minutes at room temperature, washed with cell buffer solution (PBS + 1% bovine serum albumin + 0.05% sodium azide), and centrifuged at 500 g to repellet the cells. The cells were then fixed with 200 μl of 2% paraformaldehyde for 20 minutes at 4°C and made up to a final volume of 1 ml with cell buffer solution ready for analysis. All samples were analyzed using a 3-color FAC scan flow cytometer (Becton Dickinson). At least 10,000 events were acquired and analyzed using Cell-Quest 3.3 software. Isotype-matched irrelevant antibodies were used as negative controls for nonspecific binding.
EPCs were predefined as cells expressing the following combinations of surface markers: CD34+/CD14−/CD106+ and CD31+/CD133+/CD146+ cells. In general, CD34+ is a marker of bone marrow origin while CD14− excludes monocytic origin and CD106+ denotes endothelial origin, labeling these cells as EPCs. Whereas in another subset, CD133+ indicates immaturity/bone marrow origin and CD31+ and CD146+ indicate endothelial origin, marking these cells as a different subpopulation of EPCs. Circulating endothelial cells (CECs) were defined as CD14−/KDR+ or CD31+/CD146+ cells.
Statistical analysis
A statistical software package (SAS, version 9.2; SAS institute Inc, Cary, N.C., USA) was applied for data management. Results were expressed as mean ± standard deviation (SD). Statistical analyses were performed by a Wilcoxon t test and Spearman correlation. Multivariate regression analysis was applied to determine independent relations of all clinical variables with progenitor cells. A P value < 0.05 was considered statistically significant.
RESULTS
Numbers of circulating CD34+/CD14−/CD106+ (EPC Group-1) progenitor cells were found to be significantly lower in SCD patients with PAH than those in the non-PAH group (P=0.025) (Fig. 1). However, CD31+/CD133+/CD146+ cells (EPC Group-2) did not show a significant difference in PAH versus non-PAH SCD patients (Fig. 1). In addition, the number of CD34+/CD14−/CD106+ cells (EPC Group-1) was also found to be significantly correlated with TRV on Doppler echo (r=−0.44, P=0.033), 6MWD (r=0.72, P=0.001), mPAP (r=−0.43, P=0.05), and PVR (r=−0.45, P=0.05) on RHC (Fig. 2). The EPC number did not correlate with age, sex, body mass index (BMI), hemoglobin, WBC count, reticulocyte count, lactate dehydrogenase (LDH), iron/ferritin levels, BUN, and serum creatinine.
Figure 1.
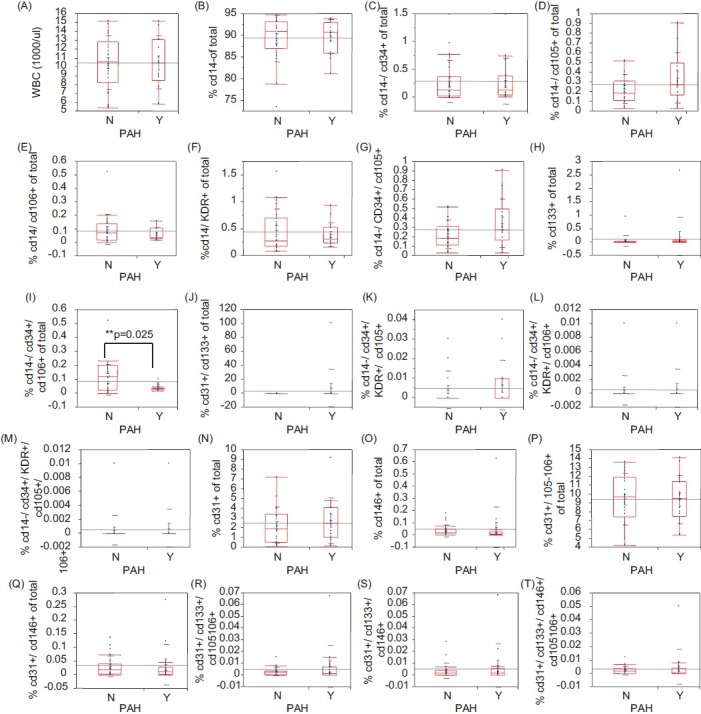
Flow cytometric analysis and characterization of cell surface markers on EPC from peripheral blood. Using several[8] distinctive cell markers (CD34, CD14, CD106, CD105, CD31, CD133, KDR, CD146) in 20 different combinations in both groups of patients flow cytometric analysis was performed as described in the “Methods” section. EPCs with CD14−/CD34+/CD106+ markers showed a significant difference between SCD patients with PAH versus without PAH (P=0.025), while EPCs with CD31+/CD34+/CD146+ markers and CECs did not show any significant difference between the two groups of patients. Statistical analysis was performed as described in the methods section.
Figure 2.
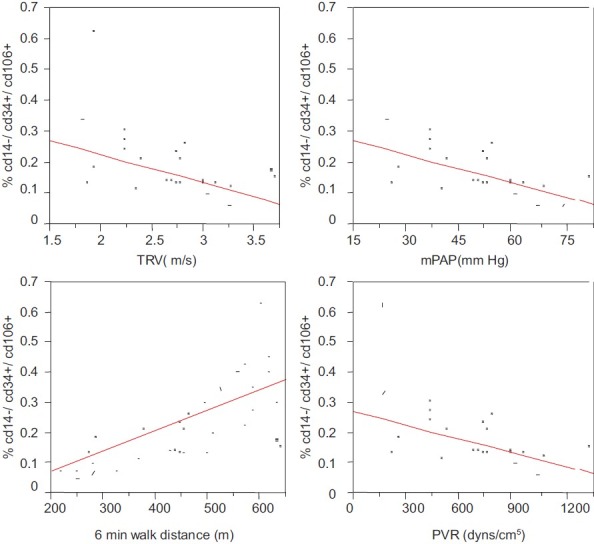
Correlation of EPC’ with various PAH parameters. EPCs with CD34+/CD14−/CD106+markers (EPC Group-1) were found to be to be significantly correlated with TRV on Doppler echo (r=−0.44, P=0.033), 6MWD (r=0.72, P=0.001), mPAP (r=−0.43, P=0.05), and PVR (r=−0.45, P=0.05) on RHC. Multivariate regression analysis was applied to determine independent relations of all clinical variables with EPCs. A P value ≤0.05 was considered statistically significant.
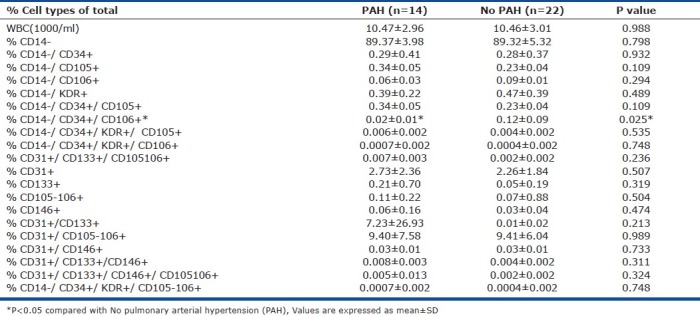
Percentage cell types of the total for various marker combinations are shown in (Table 2). Other than CD34+/CD14−/CD106+ (EPC Group-1) we did not find significant differences in cells with other combinations of the endothelial markers including circulating endothelial cells (CECs) that are mature endothelial cells resulting partially from normal shedding of endothelial lining. Characterization of these cells by flow cytometry showed that CECs, namely CD14−/KDR+ and CD31+/CD146+ cells, were similar among PAH versus non-PAH SCD patients (Fig. 1). Patient demographics of SCD patients with PAH versus non-PAH are shown in (Table 1).
Table 2.
Numbers of different progenitor and circulating endothelial cells (percentage of total cells) in SCD patients with PAH vs. No PAH
DISCUSSION
Endothelial dysfunction is a hallmark of PAH. Although EPCs have been largely implicated in endothelial dysfunction in PAH, none of the studies have directly assessed their significance in setting of SCD-related PAH. We are unaware of a prior study that has characterized EPC subtypes in the setting of SCD-related PAH. Analysis of data from 20 different cell marker combinations revealed two significant subpopulations of EPCs, CD34+/CD14−/CD106+ cells (EPC Group-1) and CD31+/CD133+/CD146+ cells (EPC Group-2). The number of circulating EPC Group-1 was significantly lower in SCD patients with PAH than that in the non-PAH group. In addition, these cell numbers were found to be significantly correlated with TRV on 2D-echo, 6MWD, mPAP and PVR on RHC, indicating that SCD-related PAH is associated with lower number of EPCs.
These findings are consistent with prior studies that showed a decrease in the number of EPCs in other forms of PH. One such study found deficiency of bone marrow-derived progenitor cells (CD34+, CD133+/VEGFR2+, CD34+/CD133+/KDR+ cells) in patients with PH secondary to idiopathic pulmonary fibrosis.[20] Other studies have described a reduction in bone marrow-derived progenitor cells in idiopathic pulmonary hypertension (IPAH) patients versus healthy controls.[21,22] Depletion of circulating EPCs has also been found in patients with COPD, who are at risk of developing associated PH[16] . In contrast, some have shown an increase in number of circulating EPCs in IPAH patients versus healthy controls,[23,24] whereas others have found no difference.[25] The reason for this disparity is not fully clear, but may reflect differences in the markers used to identify and quantify EPCs.
The exact mechanism underlying the association between low numbers of EPCs and SCD-related PAH is not fully understood. One possibility is decreased mobilization of EPCs from the bone marrow. The fact that EPC mobilization is a protective compensatory mechanism during PH rather than a pathogenic event is supported by several observational studies.[15,22,26,27] One of the studies showed that the deletion of erythropoietin receptor in nonerythroid cells worsens histopathological features of hypoxia-induced PH compared to wild-type mice.[26] Indeed, more severe PH in EPOR-/- mice was associated with complete inhibition of CD133+KDR+ EPC mobilization. Similarly, chronic treatment of IPAH patients with sildenafil is associated with a dose-dependent increase in circulating EPC numbers.[22] Moreover, potential new therapies for PAH, such as statins and PPAR-γ agonists, also induce the mobilization and differentiation of EPCs.[15,27,28] These findings suggest that decreased mobilization of EPCs from bone marrow in PAH might contribute to the pulmonary vascular pathology.
Alternatively, fewer peripheral EPCs might be a consequence of greater homing of EPCs in pulmonary vasculature at the site of endothelial injury. The homing of CD34+KDR+EPCs to diseased pulmonary arteries has been demonstrated in a study of endarterectomised tissue from patients with chronic thromboembolic PH.[29] Davie et al. reported that hypoxia-induced PH in calves is characterized by increased development of perfused vasa vasorum that are c-kit positive (a marker of bone marrow-derived cells).[30] Using a GFP-positive bone marrow chimeric mouse model, Hayashida et al. confirmed that bone marrow cells are located at sites of remodeling pulmonary arteries in hypoxia-induced PH, but that they coexpress markers of smooth muscle cells rather than endothelial markers.[31] Interestingly, another study found that early EPCs isolated from mice with hypoxia-induced PH show defective pro-angiogenic activities in comparison to EPCs from control mice when transplanted into nude mouse ischemic hind limbs.[32] The in vitro functions of mononuclear cells (e.g., colony forming capacity, adherence, migration and sensitivity to apoptosis) isolated from the peripheral blood of IPAH patients have also been found to vary when compared with cells from healthy controls.[21–23,33] Therefore, it appears that compensatory EPC mobilization and recruitment occurs in the setting of PAH, which in turn may cause a relative deficiency of EPCs in peripheral blood due to increased homing of cells to pulmonary vasculature. Whether the response is insufficient to prevent disease or detrimental because of EPC dysfunctionality is unknown.
In the current study, we did not find a significant difference among other subgroups of progenitor cells such as CD31+/CD133+/CD146+ cells (EPC Group-2) in PAH versus non-PAH SCD patients. As shown by Diller et al.,[22] circulating EPC numbers (CD34+/KDR+, CD34+/CD133+/KDR+ cells) were significantly lower in patients with Eisenmenger's syndrome or IPAH than healthy controls. However, numbers of CD34+/CD45−/low and CD34+/CD133+ progenitor cells were not significantly different from control in IPAH population versus Eisenmenger's syndrome. These findings suggest that subpopulations of progenitor cells may be differentially affected in these two well-known forms of PH and support our observation of two significant subpopulations of EPCs in SCD. Although in this prior study by Diller et al. observations were based on subpopulations of progenitor cells in general rather than EPCs alone, it nonetheless appears that different subsets of EPCs might be involved in vascular repair and hence differentially affected in SCD-related PAH.
Circulating endothelial cells (CECs) are mature endothelial cells that can result either from conversion of circulating EPCs into mature endothelial cells or from normal shedding of endothelial lining into circulation.[7] Increased concentrations of CECs are thought to be a marker of vascular injury in patients with cardiovascular disease, neoplasia, vasculitis, shock, and sepsis.[34] In the current study, characterization of different types of CECs by flow cytometry revealed that CD14−/KDR+ and CD31+/CD146+ cell numbers were similar among PAH versus non-PAH SCD patients. These findings differ from those of a prior study showing higher numbers of CECs in 15 SCD patients than in healthy controls. Of note, CECs were significantly higher in patients with PAH.[35] This disparity in results may be due to the difference in patient characteristics. All participants in our study were free of wounds, ulcers, and retinopathy that may influence CECs kinetics. Our findings suggest that endothelial cell turn over might not be different among SCD patients with and without PAH.
In conclusion, our study was limited, as we did right-heart catheterization in patients with TRV≥2.5 only to confirm PAH, but not in patients with normal TRV. Therefore, patients with PAH but normal TRV on echo may have been misclassified. Furthermore, the term EPC has been widely used and comprises a heterogeneous population of mononuclear cells. Further studies are needed to precisely identify their origins, different cell phenotypes, and the terminology to be used when describing them.[36] In summary, we show here for the first time that subpopulations of EPCs are differentially affected in patients with SCD-related PAH. Relative deficiency of these EPCs subgroups in PAH patients may contribute to the pulmonary vascular pathology. These findings will provide potential insight into the pathophysiological mechanisms and can be useful for identifying suitable therapeutic targets in these patients.
Footnotes
Source of Support: Nil,
Conflict of Interest: None declared.
REFERENCES
- 1.Sutton LL, Castro O, Cross DJ, Spencer JE, Lewis JF. Pulmonary hypertension in sickle cell disease. Am J Cardiol. 1994;74:626–8. doi: 10.1016/0002-9149(94)90760-9. [DOI] [PubMed] [Google Scholar]
- 2.Castro O, Hoque M, Brown BD. Pulmonary hypertension in sickle cell disease: cardiac catheterization results and survival. Blood. 2003;101:1257–61. doi: 10.1182/blood-2002-03-0948. [DOI] [PubMed] [Google Scholar]
- 3.Haque AK, Gokhale S, Rampy BA, Adegboyega P, Duarte A, Saldana MJ. Pulmonary hypertension in sickle cell hemoglobinopathy: A clinicopathologic study of 20 cases. Hum Pathol. 2002;33:1037–43. doi: 10.1053/hupa.2002.128059. [DOI] [PubMed] [Google Scholar]
- 4.Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159–65. doi: 10.1161/01.CIR.0000102381.57477.50. [DOI] [PubMed] [Google Scholar]
- 5.Dorfmüller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J. 2003;22:358–63. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi T, Kalka C, Masuda H, Chen D, Silver M, Kearney M, et al. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med. 1999;5:434–8. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 7.Steurer M, Kern J, Zitt M, Amberger A, Bauer M, Gastl G, et al. Quantification of circulating endothelial and progenitor cells: comparison of quantitative PCR and four-channel flow cytometry. BMC Res Notes. 2008;1:71. doi: 10.1186/1756-0500-1-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petit I, Jin D, Rafii S. The SDF-1-CXCR4 signaling pathway: A molecular hub modulating neo-angiogenesis. Trends Immunol. 2007;28:299–307. doi: 10.1016/j.it.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hur J, Yoon CH, Kim HS, Choi JH, Kang HJ, Hwang KK, et al. Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arterioscler Thromb Vasc Biol. 2004;24:288–93. doi: 10.1161/01.ATV.0000114236.77009.06. [DOI] [PubMed] [Google Scholar]
- 10.Ingram DA, Mead LE, Tanaka H, Meade V, Fenoglio A, Mortell K, et al. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. 2004;104:2752–60. doi: 10.1182/blood-2004-04-1396. [DOI] [PubMed] [Google Scholar]
- 11.Hristov M, Erl W, Weber PC. Endothelial progenitor cells: mobilization, differentiation, and homing. Arterioscler Thromb Vasc Biol. 2003;23:1185–9. doi: 10.1161/01.ATV.0000073832.49290.B5. [DOI] [PubMed] [Google Scholar]
- 12.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 13.Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 14.Werner N, Nickenig G. Influence of cardiovascular risk factors on endothelial progenitor cells: limitations for therapy? Arterioscler Thromb Vasc Biol. 2006;26:257–66. doi: 10.1161/01.ATV.0000198239.41189.5d. [DOI] [PubMed] [Google Scholar]
- 15.Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, et al. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89:E1–7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- 16.Peinado VI, Ramírez J, Roca J, Rodriguez-Roisin R, Barberà JA. Identification of vascular progenitor cells in pulmonary arteries of patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2006;34:257–63. doi: 10.1165/rcmb.2005-0255OC. [DOI] [PubMed] [Google Scholar]
- 17.Weiss DJ. Stem cells and cell therapies for cystic fibrosis and other lung diseases. Pulm Pharmacol Ther. 2008;21:588–94. doi: 10.1016/j.pupt.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khakoo AY, Finkel T. Endothelial progenitor cells. Annu Rev Med. 2005;56:79–101. doi: 10.1146/annurev.med.56.090203.104149. [DOI] [PubMed] [Google Scholar]
- 19.Liew A, Barry F, O’Brien T. Endothelial progenitor cells: diagnostic and therapeutic considerations. Bioessays. 2006;28:261–70. doi: 10.1002/bies.20372. [DOI] [PubMed] [Google Scholar]
- 20.Fadini GP, Schiavon M, Rea F, Avogaro A, Agostini C. Depletion of endothelial progenitor cells may link pulmonary fibrosis and pulmonary hypertension. Am J Respir Crit Care Med. 2007;176:724–5. doi: 10.1164/ajrccm.176.7.724a. [DOI] [PubMed] [Google Scholar]
- 21.Junhui Z, Xingxiang W, Guosheng F, Yunpeng S, Furong Z, Junzhu C. Reduced number and activity of circulating endothelial progenitor cells in patients with idiopathic pulmonary arterial hypertension. Respir Med. 2008;102:1073–9. doi: 10.1016/j.rmed.2007.12.030. [DOI] [PubMed] [Google Scholar]
- 22.Diller GP, van Eijl S, Okonko DO, Howard LS, Ali O, Thum T, et al. Circulating endothelial progenitor cells in patients with Eisenmenger syndrome and idiopathic pulmonary arterial hypertension. Circulation. 2008;117:3020–30. doi: 10.1161/CIRCULATIONAHA.108.769646. [DOI] [PubMed] [Google Scholar]
- 23.Asosingh K, Aldred MA, Vasanji A, Drazba J, Sharp J, Farver C, et al. Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am J Pathol. 2008;172:615–27. doi: 10.2353/ajpath.2008.070705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toshner M, Voswinckel R, Southwood M, Al-Lamki R, Howard LS, Marchesan D, et al. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2009;180:780–7. doi: 10.1164/rccm.200810-1662OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smadja DM, Gaussem P, Mauge L, Israël-Biet D, Dignat-George F, Peyrard S, et al. Circulating endothelial cells: A new candidate biomarker of irreversible pulmonary hypertension secondary to congenital heart disease. Circulation. 2009;119:374–81. doi: 10.1161/CIRCULATIONAHA.108.808246. [DOI] [PubMed] [Google Scholar]
- 26.Satoh K, Kagaya Y, Nakano M, Ito Y, Ohta J, Tada H, et al. Important role of endogenous erythropoietin system in recruitment of endothelial progenitor cells in hypoxia-induced pulmonary hypertension in mice. Circulation. 2006;113:1442–50. doi: 10.1161/CIRCULATIONAHA.105.583732. [DOI] [PubMed] [Google Scholar]
- 27.Dimmeler S, Aicher A, Vasa M, Mildner-Rihm C, Adler K, Tiemann M, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391–7. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang CH, Ciliberti N, Li SH, Szmitko PE, Weisel RD, Fedak PW, et al. Rosiglitazone facilitates angiogenic progenitor cell differentiation toward endothelial lineage: a new paradigm in glitazone pleiotropy. Circulation. 2004;109:1392–400. doi: 10.1161/01.CIR.0000123231.49594.21. [DOI] [PubMed] [Google Scholar]
- 29.Yao W, Firth AL, Sacks RS, Ogawa A, Auger WR, Fedullo PF, et al. Identification of putative endothelial progenitor cells (CD34+CD133+Flk-1+) in endarterectomized tissue of patients with chronic thromboembolic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2009;296:L870–8. doi: 10.1152/ajplung.90413.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davie NJ, Crossno JT, Jr, Frid MG, Hofmeister SE, Reeves JT, Hyde DM, et al. Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: contribution of progenitor cells. Am J Physiol Lung Cell Mol Physiol. 2004;286:L668–78. doi: 10.1152/ajplung.00108.2003. [DOI] [PubMed] [Google Scholar]
- 31.Hayashida K, Fujita J, Miyake Y, Kawada H, Ando K, Ogawa S, et al. Bone marrow-derived cells contribute to pulmonary vascular remodeling in hypoxia-induced pulmonary hypertension. Chest. 2005;127:1793–8. doi: 10.1378/chest.127.5.1793. [DOI] [PubMed] [Google Scholar]
- 32.Marsboom G, Pokreisz P, Gheysens O, Vermeersch P, Gillijns H, Pellens M, et al. Sustained endothelial progenitor cell dysfunction after chronic hypoxia-induced pulmonary hypertension. Stem Cells. 2008;26:1017–26. doi: 10.1634/stemcells.2007-0562. [DOI] [PubMed] [Google Scholar]
- 33.Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, et al. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res. 2006;98:209–17. doi: 10.1161/01.RES.0000200180.01710.e6. [DOI] [PubMed] [Google Scholar]
- 34.Wu H, Chen H, Hu PC. Circulating endothelial cells and endothelial progenitors as surrogate biomarkers in vascular dysfunction. Clin Lab. 2007;53:285–95. [PubMed] [Google Scholar]
- 35.Strijbos MH, Landburg PP, Nur E, Teerlink T, Leebeek FW, Rijneveld AW, et al. CURAMA study group.Circulating endothelial cells: A potential parameter of organ damage in sickle cell anemia? Blood Cells Mol Dis. 2009;43:63–7. doi: 10.1016/j.bcmd.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 36.Zampetaki A, Kirton JP, Xu Q. Vascular repair by endothelial progenitor cells. Cardiovasc Res. 2008;78:413–21. doi: 10.1093/cvr/cvn081. [DOI] [PubMed] [Google Scholar]