

Abstract
We aimed to investigate the role of endothelin-mediated vasoconstriction following acute pulmonary thromboembolism (APTE). Thirteen anesthetized piglets (~25 kg) were ventilated with 0 PEEP. Cardiac output (Qt) and wedge pressure (Pw) were measured by a Swan Ganz catheter, along with arterial and venous blood gases. APTE was induced by autologous blood clots (~0.8 g/kg, 12-16 pieces) via a jugular venous catheter at time = 0 minutes until the mean pulmonary arterial pressure (Ppa) was about 2.5 times the baseline at 30 minutes. Eight control animals (Group 1) received only normal saline afterward, while the remaining five (Group 2) received at time = 40-minute saline plus Tezosentan, a nonspecific endothelin antagonist. The drug was initially given as an intravenous bolus (10 mg/kg), followed by an infusion (2 mg/min) until the end of the experiment at 2 hours. Hemodynamic data were measured before APTE and then at 30-minute intervals. Pulmonary vascular resistance index (PVRI) was calculated as (Ppa-Pw)/CI, where CI was cardiac index or Qt/W (body weight). Fluorescent microspheres (FMS) were used to mark regional blood flows and ventilation for cluster analysis. PVRI acutely increased within minutes and remained high despite some recovery over time. With Tezosentan treatment, the results showed that endothelin-mediated vasoconstriction persisted significantly up to 2 hours and accounted for about 25% of the increase in PVRI while clot obstruction accounted for the remaining 75%. CI remained relatively constant throughout. Tezosentan also affected PVRI indirectly by mitigating the shift of regional blood flow back to the embolized areas over time, possibly by attenuating vasoconstriction in the nonembolized areas. We conclude that following APTE, although the increased PVRI is mostly due to mechanical embolic obstruction, secondary factors such as vasoconstriction and pattern of regional blood flow over time also play important roles.
Keywords: endothelin antagonist, pulmonary embolism, pulmonary vascular resistance, pulmonary vasoconstriction, smooth muscle contraction
INTRODUCTION
Following acute pulmonary thromboembolism (APTE), the increase in pulmonary vasculature resistance (PVR) arises not only from vascular obstruction but also from vasoconstriction. However, there are still some controversial opinions with regard to the importance of the latter mechanism.[1,2]
Some suggested that vasoconstriction existed but soon became insignificant afterward,[3,4] while others showed that its magnitude depended on the embolic size, with the smaller ones causing more vasoconstriction than the larger ones.[5] In a standardized, angiographic obstruction model using embolic beads, some investigators concluded that there was no significant component of vascular tone.[1] Thus, it remained unclear whether these disagreements could be accounted for by the differences in their experimental preparations, animal species, sizes of embolic materials, timing of hemodynamic measurements or even interpretation of data.
In order to resolve these issues, we aimed to use a novel approach to study the impact of pulmonary vasoconstriction after APTE by examining regional blood flow after APTE using fluorescent microspheres (FMS) instead of MIGET or pressure flow plots. Specifically, the degree of vasoconstriction mediated by endothelins after APTE was studied, focusing on the first 2 hours when the mortality among these patients was the highest.[6] This pathway was chosen because endothelins, which were found omnipresent in the lung along with their ubiquitous receptors, have been considered to be one of the key mediators in the pathogenesis.[7,8] Lee et al. reported that endothelin-1 was expressed locally in the embolized lung at the tissue level and its antagonist mitigated the subsequent pulmonary hypertension.[9] Similarly, others have confirmed its role in pulmonary air embolism.[10] It has also been established that they play important parts in many other pulmonary vascular diseases, such as primary or secondary pulmonary hypertension[11] as well as different forms of acute lung injury.[12] Finally, Bosentan, an oral endothelin antagonist, has been used successfully for some time for patients with chronic thromboembolic pulmonary hypertension,[13] implying that this same vasoconstrictor is likely to be also relevant in the acute phase.
We hypothesize that endothelin-mediated vasoconstriction persists to a significant degree after APTE in the first 2 hours and that it affects the redistribution of regional blood flow, which could also impact pulmonary hemodynamics. The data will then serve to quantify its relative importance.
MATERIALS AND METHODS
Surgical preparations and physiological measurements
The experimental protocol was approved by the University of Washington Animal Care and Use Committee. Thirteen piglets (25±5 kg) were premedicated with ketamine 20 mg/ kg i.m. and xylazine 2 mg/kg i.m. They were then maintained under a general anesthetic using intravenous pentothal, initially set at 100 mg/h and the dose occasionally titrated afterward.
One femoral arterial line was inserted for the purpose of monitoring systemic blood pressure (BP) and blood gases. Two femoral venous lines allowed for fluid infusion and fluorescent microspheres (FMS) injection. A Swan Ganz catheter (Edwards Lab, CA) was inserted in the right external jugular vein for measuring pulmonary arterial pressure (Ppa), pulmonary capillary wedge pressure (Pw), and cardiac ouput (Qt) by thermodilution technique. A large bore catheter (5 mm internal diameter) was inserted in the left external jugular vein for the rapid infusion of preformed blood clots (see below). No heparin was used.
Eighty ml of blood were withdrawn from the femoral arterial line and mixed with 2500 units of Thrombin - JMI at room temperature. Clots were allowed to form and fibrinize over the next 2 hours.
Upon completion of the surgical procedures, animals were placed in the prone posture and received at least three consecutive stacked breaths to peak airway pressures (Paw) ~25 cm H2O to remove residual atelectasis. Their ventilatory settings were adjusted to maintain PaCO2 at 35±2 Torr, PEEP at 0, tidal volume (TV) at 12-15 ml/kg, respiratory rate (RR) at 18–20/min and FIO2 at 21% or room air at sea level. No further adjustments of these settings occurred afterward. At each of the subsequent data collection times, hemodynamic parameters such as BP, Ppa, Pw, heart rate, and Qt were measured, along with hemoglobin (Hb), arterial and venous blood gases, Fowler dead space (MacLab at 100 mm/s), TV, RR, and Paw. Each measurement of Qt was the mean of at least three consistent readings at the same experimental setting.
FMS of 10 different colors and 2 different sizes (Molecular Probes, Eugene, OR) were used to mark both regional blood flow (15 μm) and regional lung ventilation (1 μm), respectively, in random order at five time points, beginning at t = – 30 minutes. The details of the FMS techniques were as previously described.[14]
After control runs at –30 and –5 minutes and all physiological measurements had been recorded, APTE was induced for all 13 animals at time = 0 minutes. All other previous and subsequent events would be recorded relative to that time. Preformed fibrinized clots, 12–16 pieces (1.5×0.5×0.5 cm3), were cut into uniform size[5,15] , suspended in normal saline in a large catheter tip syringe, and injected into the left external jugular vein over the next 10–15 minutes until Ppa was stabilized at about 2.5 times the baseline value at 30 minutes. Immediately after initial clot infusion, Ppa often reached over 40 mm Hg temporarily. At t = 30 minutes after achieving a stable Ppa, we repeated physiological measurements, blood sampling, and FMS mapping as in the control runs.
After time = 30 minutes, the animals were randomly divided into two groups. In group 1 (control, n=8) the animals continued to receive only normal saline at 100 ml/h for the remainder of 2 hours. In group 2 (n=5), the animals received similar volumes of normal saline but also Tezosentan, a nonspecific endothelin antagonist[16] (Courtesy Actelion Ltd., Switzerland) at t=40 minutes, initially as an intravenous bolus of 10 mg/kg over 20 minutes, followed by 2 mg/min infusion until t=120 minutes.
This approach was preferred because it would allow us to establish a steady level of Tezosentan prior to the hemodynamic measurements in our interested time frame (i.e., between 60 and 120 minutes). On the other hand, pretreatment of Tezosentan before APTE would not allow standardization of similar embolic load in both groups to the same Ppa end point, about 2.5 times the baseline. The size of the emboli was quite uniform, rarely exceeding 50% from each other.
At t=60, 90 and 120 minutes, we repeated all the measurements done at t=30 minutes except that FMS mappings were omitted at t=90 minutes. There were no data for 90 minutes because the maximum number of FMS available for the same experiment was reached at 10. Any additional color would compromise the accuracy of spectrum analysis.[17] No other vasoactive substance was administered.
Post mortem lung preparation
After t=120 minutes, the animals from both groups were treated identically. They were all deeply anesthetized with intravenous pentothal, heparinized with 5000 units and exsanguinated. The lungs were removed after a gentle saline vascular flush, suspended vertically and inflated to no more than 25 cm H2O. The lobes were kept in their in vivo anatomical positions by a few spot adhesions using cyanoacrylate glue. Lungs were then dried for 72 hours by blowing warm, dry air at Paw ~20 cm H2O into the trachea, exiting via 10-20 small puncture holes.
Dried lungs were sectioned into cubes (~2 cm3), with each sample carefully assigned to a three-dimensional (3D) coordinate according to a pre-established grid pattern. The injected thromboemboli, readily seen in the major pulmonary arteries, had not been macerated. Approximately 1000 pieces were analyzed per animal (see the “Results” section). For each piece, we recorded its spatial location, dry weight, estimated airway tissue, and the presence or absence of blood clots in arteries >1 mm.
The fluorescent intensities of FMS, which were used to mark both the regional blood flow and ventilation, were each measured at five time points (–30, –5, 30, 60, and 120 minutes) as previously described.[14] Briefly, the FMS were mixed with saline and either injected via the femoral vein to mark regional blood flow, or nebulized into the lung to mark regional ventilation. The FMS signal per piece, reflecting the number of trapped microspheres, was determined by measuring its intensity in a spectrofluorometer (Perkin-Elmer LS-50B), following elution after 4 days of soaking in 2 ml of organic solvent (Cellosolve, Sigma-Aldrich, Mo.). Overlaps from adjacent colors were then corrected using a matrix inversion method.[17]
Data analysis
For both groups, hemodynamic and gas exchange parameters before and after APTE were recorded (i.e., Ppa, Pw, Qt, PaO2, PVO2, and PaCO2). Averages of Qt prior to APTE at -30 and -5 minutes were plotted against the corresponding body weight (W) in kg for the 13 animals to establish a linear correlation. Once confirmed, each Qt was then normalized by W of the same animal to derive cardiac indices or CI=Qt/W. W was also used to normalize PVR to the pulmonary vascular resistance index[18] (PVRI) and calculated as (Ppa – Pw)/CI. We quantified pulmonary vasoconstriction after APTE from the differences in PVRI between the two groups at 60, 90 and 120 minutes. This consideration was intended to reduce the confounding effects of the variations in Qt with body size among animals and also to provide a better estimate of the vasoactive component of vascular resistance.
Cluster analysis
Hemodynamically, Ppa at 30 minutes was standardized to about 2.5 times the pre-APTE value. Furthermore, in order to ensure that the two groups were comparable after APTE, we checked the patterns of embolic obstruction beforeTezosentan by cluster analysis.
These measured regional flows in all the samples, approximately 1000 of them, were then separated into three clusters a priori, according to how they changed between –5 minutes and +30 minutes (i.e., immediately before and after APTE). After their grouping, each cluster would act as cohort and be followed in the remainder of the experiment to see their physiological behavior over time.
Weight normalized relative flow (WNRQ) was the parameter used for defining the characteristics of these 3 clusters. It was calculated in the following steps:
-
After obtaining each regional blood flow from the FMS intensity of a specific color and Qt at a given time, the flow value (Qi, ml/min) of each piece was first normalized for its size, by dividing Qiby the weight of that piece (wi). These weight normalized piece flow (WNQi) values or Qi/ wi, were then normalized a second time by dividing it by the mean of WNQi values for all pieces at that corresponding time, i.e.,.
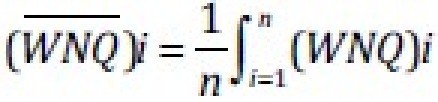
This resulted in a weight normalized relative flow, namely [
 ] was represented as either 1 or 100% respectively. WNRQicould now be fairly compared within and between clusters, despite some minor variations in Qt, at different conditions and times.
] was represented as either 1 or 100% respectively. WNRQicould now be fairly compared within and between clusters, despite some minor variations in Qt, at different conditions and times.
Subsequently, in order to identify how different areas of the lung were affected by blood clots in terms of regional blood flows, we proceeded to portion the lung pieces into three clusters according to the change in WNRQifrom the baseline condition prior to APTE (t=–5 minutes) to the first measurement after APTE (t=30 minutes) in the following manner: Cluster 1 included lung pieces that had an absolute decrease of 50% or more in WNRQifrom time = –5 minutes to +30 minutes after APTE; cluster 2 included lung pieces in which WNRQiwas relatively unchanged; cluster 3 included lung pieces that had an absolute increase of 50% or more in WNRQifrom time = –5 minutes to 30 minutes after APTE. It was important to note that these clusters were created prior to the initiation of Tezosentan at time = 40 minutes. The absolute increase or decrease of 50% in WNRQiwas chosen because this would clearly differentiate the three cluster patterns, particularly when WNRQiof most samples was about 1 before embolization.
From our current and previous studies, these subpopulations represented anatomically those regions distal to the emboli (Cluster 1), those in the central regions (Cluster 2) and those in the least embolized and more cephalad regions (Cluster 3).[19]
Weight normalized relative ventilations (WNRV)iwere similarly obtained from the aerosolized microspheres (1 μm) data, using the same clusters which were already defined according to WNRQ, at all the designated times.
Statistics
PVRI was considered as the primary variable and all other hemodynamic parameters as secondary ones. Paired and 2-sample t-tests were performed to compare PVRI within groups at different times and between groups at the same times. These tests were then similarly done for other physiological variables but Bonferroni adjustment of P values was not performed for the secondary variables. CI was compared between groups at the same time point to see if there was statistical difference. Repeated measure ANOVA was performed to assess any difference in the same group over time. Two- and 1-sample Hotelling T2 tests were used to compare the distribution of regional blood flow and regional ventilation in the three clusters, in the same time between groups and in the same group across time respectively. All data were expressed as mean±standard deviation unless indicated otherwise. P<0.05 was used to designate statistical significance.
RESULTS
Table 1 shows the hemodynamic parameters over time. During early APTE, Ppa increased quickly and reached a plateau at around 40 Torr despite some early dissipation between clot injections. It gradually stabilized prior to 30 minutes and over time it recovered gradually toward baseline. Pw itself was relatively constant at 5±2 Torr for all the animals as they were regularly hydrated during the experiments.
Table 1.
The hemodynamic parameters during the experiment
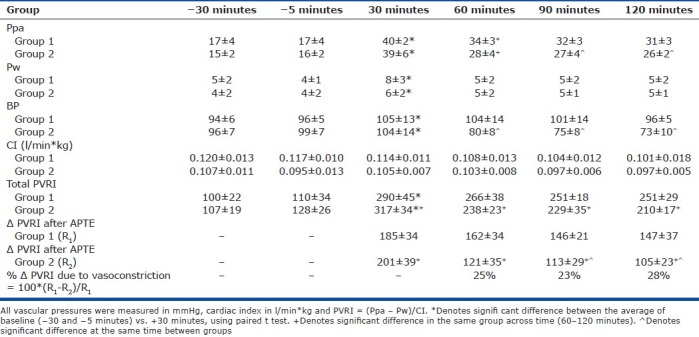
Systemic blood pressure (BP) dropped after the initiation of Tezosentan, which was given in Group 2 at 40 minutes to abolish any endothelin-mediated pulmonary vasoconstriction after APTE but its systemic effect was also seen. Sufficient dose was given.
The square of the correlation coefficient (r2) between Qt and W was 0.62, indicating a good correlation. When Qt was normalized with W in the same animal, cardiac index CI was calculated. It shows that CI remained stable and relatively constant during the experiments in both groups and there was no statistical significance between them.
Table 1 and Figure 1 show the pulmonary vascular resistance index (PVRI) during the experiment. The average total PVRI at –30 and –5 minutes for Groups 1 and 2 were calculated and used as the baseline values for both of them respectively. They were not statistically different.
Figure 1.
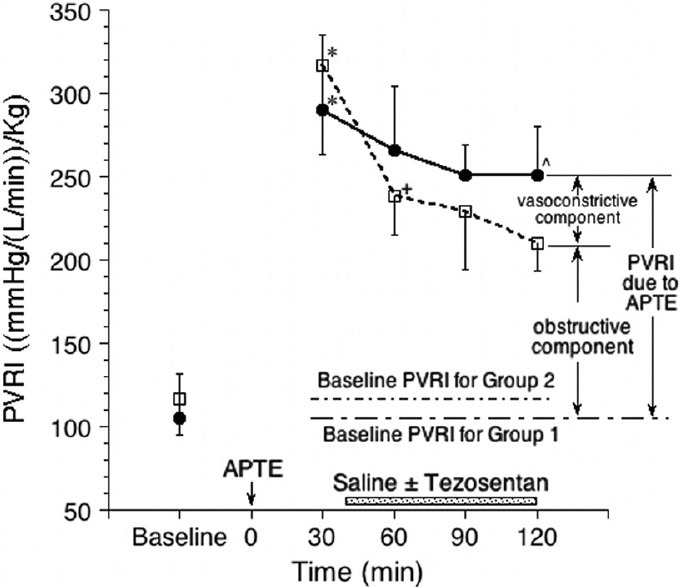
The total pulmonary vascular resistance index (PVRI) during the experiment for Group 1 (APTE only, solid circles) and Group 2 (APTE + Tezozentan, open squares). *Denotes significant difference between the average of baseline (−30 and -5 minutes) vs. +30 minutes, using paired ttest. +Denotes significant difference in the same group across time (60–120 minutes). ^Denotes significant difference at the same time between groups.
The total PVRI in Group 1 at 30 minutes and thereafter represented the total pulmonary vascular resistance index immediately after APTE due to a combination of the obstructive and vasoconstrictive components, plus the baseline value. It would remain so for Group 1 until the end of the experiment. However, for Group 2 at 60 minutes onward when Tezosentan was already started, their total PVRI represented the pulmonary vascular resistance index due to the obstructive component plus the baseline value only, without the contribution from the vasoconstriction mediated by endothelins.
By comparing the net increase (Δ) in PVRI after APTE , i.e., R1 and R2 for Groups 1 and 2 respectively, and adjusting for their slightly different baseline PVRI, the vasoconstriction component as mediated by endothelins after APTE was estimated to be about 25%, 23% and 28% at 60, 90, and 120 minutes, respectively (Table 1).
Table 2 shows that percentage of the lung samples distributed to the three clusters, assigned by WNRQ according to the definition determined a priori (see the “Methods” section). The patterns of embolization before Tezosentan at 30 minutes were similar in groups 1 and 2 and there was no statistical difference between them. For both groups, roughly 50% of the pieces were assigned to cluster 1, while about 25% of the pieces were assigned to clusters 2 and 3 each, namely half of the lung tissue received reduction of regional blood flow after APTE.
Table 2.
Cluster definition and sample distribution within lung at 30 minutes after APTE
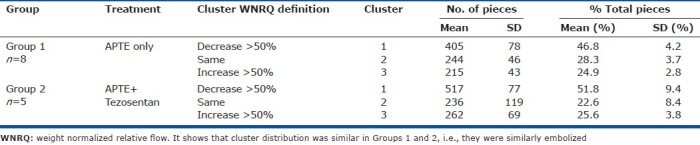
Table 3 shows the percentage of regional blood flow summed up in different clusters during the experiments. Once the clusters were defined by WNRQi, the percentage of regional blood flow in each cluster was calculated by the summation of all raw regional flow from those samples within that cluster and dividing it by the corresponding total pulmonary blood flow or Qt.
Table 3.
Regional blood flow in each cluster as a percentage of total pulmonary blood flow over time

At –30 and –5 minutes, the percentage of regional blood flow in each cluster was roughly proportional to the percentage of total pieces (Tables 2 and 3) at ~2 cm3 each, indicating their homogeneity. They were quite similar between groups. At +30 minutes, the distributions of the regional blood flow in the three clusters immediately after embolization between groups were also comparable, indicating that the measurements at these times were reproducible. Thus, these data indicated that the animals in groups 1 and 2 were similarly embolized at 30 minutes in terms of embolic load (about 20 g), Ppa end points and the resulted blood flow pattern among these clusters. It was part of our experimental goal to complete the embolization process in both groups toward similar end points.
Note that at time = +30 minutes after APTE, cluster 1 in both groups, which by definition received less than 50% of WNRQ % (Table 3), actually received only 6.4% and 6.2% of total pulmonary blood flow respectively, since some of these samples, located distal to the emboli, received almost no flow at all.
The percentage of regional blood flow in Cluster 2 at 30 minutes for both groups remained comparable to the pre-embolic state, as their WNRQ, by definition (Table 2), remained relatively unchanged.
Cluster 3 at 30 minutes, located in the least embolized regions, received disproportionately more flow after APTE, mostly diverted from Cluster 1, because their WNRQ % was at least 50% higher after embolization. However, at time = 60 and 120 minutes, the distributions of regional blood flow between Groups 1 and 2 were different. While there was progressively more percentage of regional blood flow returning to Cluster 1 from Cluster 3 over time in both groups, the recovery was sooner and more significant in Group 1 than in Group 2. The latter was treated with Tezosentan which caused relative vasodilatation in these less embolized areas (Cluster 3) and facilitated persistent recruitment there.
The data for the percentage of regional ventilation did not show any significant difference between groups or after Tezosentan and will not be presented here.
DISCUSSION AND CONCLUSIONS
In our experiments, we ensured that all the animals received comparable embolic load and similar hemodynamic profiles before those in Group 2 were given the endothelin antagonist. There was no difference in Ppa and embolic load between groups prior to Tezosentan. A pre-treatment approach was not used because the embolic load during the experiment would be different at the same hemodynamic end point at 30 minutes or the hemodynamic end point would be different at the same embolic load. Instead, a bolus of Tezosentan was given after APTE at 40 minutes, followed by an infusion. This quickly resulted in achieving a good plasma level of this drug and reached a steady state at the earliest time. Its impact was seen in their systemic blood pressure. However, the animals were never in shock with a steady CI and PvO2throughout (37±4 Torr).
We were able to maintain CI relatively constant during the experiments (Table 1). Although the CI in both groups across time was not strictly constant, because it could not be rigidly controlled as in isolated perfusion models, we considered them to be sufficiently steady for our present investigation (i.e., to fairly compare the pulmonary vascular resistance index (PVRI) between groups and over time).
Ppa acutely increased as embolization begun and did so after each additional injection of clots, sometimes exceeding 40 Torr. However, its magnitude intermittently dissipated within minutes until a sufficient embolic load was finally given. Thereafter, a more steady state was achieved. It is reasonable to suggest that these early temporaryincreases in pulmonary vascular resistance were possibly due to the more severe but transient vasoconstriction[3] at the early stage of embolic lung injury. Indeed, if the very early vasomotor response was disproportionately intense due to one large embolic load rather than the gradual accumulation of many over time, we speculated that these patients might succumb more readily[6] , accounting for their early mortality.
However, we were also interested to see if significant vasoconstriction persisted beyond the immediate phase. In our experimental model, the data showed that PVRI nearly tripled at 30 minutes after APTE. At and after 60 minutes, PVRI in Group 1 represented both obstructive and vasoconstrictive components of PVRI after APTE, plus the baseline PVRI. On the other hand, for Group 2, the PVRI represented only the obstructive component after APTE plus the baseline PVRI, but did not include the vasoconstrictive component mediated by endothelins, as they were well antagonized by Tezosentan at our chosen doses (Fig. 1).
Our data showed that the vasoconstrictive component, as mediated by the endothelins pathway in the early hours after APTE, was consistently about 25% of the total while the obstructive component accounted for the remaining 75%. We recognized that the result did not include all vasoconstrictive pathways.
After 30 minutes, Ppa and PVRI slowly recovered toward baseline condition, but more so in Group 2 than Group 1 (Table 1 and Fig. 1). The differences in their changes, even though they were moving in the same direction, could be explained by both the abolition of endothelin-mediated vasoconstriction in the entire pulmonary circulation in Group 2 and the more persistent recruitment of blood flow to the nonembolized regions (Cluster 3, Group 2) which effectively lowered the PVRI, presumably due to the relative vasodilatation there. These data were unique in that they gave insight on the relationship between regional blood flow pattern and PVRI in real time, which was measured only indirectly by MIGET[18,20,21] and not at all in pressure flow plots.
Endothelin antagonism also had some unexpected consequences beyond the pure reduction of vasomotor tone in the pulmonary vasculature. It has been previously reported that treating APTE with vasodilators, such as hydralazine, might result in more extensive pulmonary infarction.[22] This observation was compatible with our data in that vasodilatation might further reduce blood flow into the embolized areas over a longer period of time, resulting in ischemia and potential harm.
Table 1 shows that Tezosentan at sufficient dose could cause moderate systemic hypotension. It is a nonspecific endothelin antagonist that broadly abolishes vasoconstriction when it is activated. Its dosage was chosen to assert undisputed effect on the designated pulmonary vasoconstriction without detrimental changes in CI.
It was observed that among patients with prior normal cardiopulmonary condition, their Ppa rarely exceeded 40 Torr after APTE.[23] The phenomenon was usually explained by the fact that the less muscular right ventricle (RV) could not generate sufficient pressure to overcome the increased pulmonary vascular resistance,[24] while patients with previous lung diseases could, because this same cardiac chamber was chronically hypertrophied under stress. While this explanation could certainly account for the observation, we proposed another plausible consideration relating to regional blood flow.
Pulmonary vessels in an otherwise healthy subject, with their larger potential cross sectional area for recruitment, could allow for more ready accommodation of increased flow into the nonembolized regions, if APTE was not massive and central. Beyond the strength of the RV, this compensatory mechanism in Poiseuille flow might also mitigate to some extent the subsequent pulmonary hypertension at normal Qt (i.e., Ppa reached a maximum round 40 Torr).[25] On the other hand, if the patient had emphysema, with a reduction of recruitment potential from loss of vasculature, their Ppa might reach a higher level more readily after APTE, along with their stronger RV. In both cases, if the patient was in shock, Ppa would remain low as it could not be interpreted in isolation without the simultaneous CI.
In summary, our data show that endothelin-mediated vasoconstriction persists in the early hours after APTE and accounts significantly for about 25% of the increase in pulmonary vascular resistance. Tezosentan also facilitates continual redistribution of regional blood flow into the nonembolized regions, which may also mitigate pulmonary hypertension.
ACKNOWLEDGMENTS
The authors thank Dr. Jack Hildebrandt at the University of Washington for his insights and constructive criticisms in the preparation of this manuscript.
Footnotes
Source of Support: B. C. Lung Association
Conflict of Interest: None declared.
REFERENCES
- 1.Melot C, Delcroix M, Closset J, Vanderhoeft P, Lejeune P, Leeman M, et al. Starling resister vs distensible vessel models for embolic pulmonary hypertension. Am J Physiol. 1995;267:H817–27. doi: 10.1152/ajpheart.1995.268.2.H817. [DOI] [PubMed] [Google Scholar]
- 2.Smulders YM. Pathophysiology and treatment of hemodynamic instability in acute pulmonary embolism: The pivotal role of pulmonary vasoconstriction. Circ Res. 2000;48:23–33. doi: 10.1016/s0008-6363(00)00168-1. [DOI] [PubMed] [Google Scholar]
- 3.Smulders YM. Contribution of pulmonary vasoconstriction to hemodynamic instability after acute pulmonary embolism: Implications for treatment? Netherland J Med. 2001;58:241–7. doi: 10.1016/s0300-2977(01)00117-6. [DOI] [PubMed] [Google Scholar]
- 4.Torbicki A, Perrier A, Konstantinides S, Agnelli G, Gallie N, Pruszczyk P, et al. Guidelines on the diagnosis and management of acute pulmonary embolism. Eur Heart J. 2008;29:2276–315. doi: 10.1093/eurheartj/ehn310. [DOI] [PubMed] [Google Scholar]
- 5.Delcroix M, Melot C, Vachiery JL, Lejune P, Leeman M, Naeije R. Effects of embolus size on hemodynamics and gas exchange in canine embolic pulmonary hypertension. J Appl Physiol. 1990;69:2254–61. doi: 10.1152/jappl.1990.69.6.2254. [DOI] [PubMed] [Google Scholar]
- 6.Wood KE. Major pulmonary embolism. Review of a pathophysiologic approach to the golden hour of hemodynamically significant pulmonary embolism. N Eng J Med. 2002;121:877–905. doi: 10.1378/chest.121.3.877. [DOI] [PubMed] [Google Scholar]
- 7.Battistini B, Dussault P. Biosynthesis, distribution and metabolism of endothelins in the pulmonary system. Pul Pharmacol Therapeut. 1998;11:79–88. doi: 10.1006/pupt.1998.0148. [DOI] [PubMed] [Google Scholar]
- 8.Benigni A, Remuzzi G. Endothelin antagonists. Lancet. 1999;353:133–8. doi: 10.1016/S0140-6736(98)09423-9. [DOI] [PubMed] [Google Scholar]
- 9.Lee J, Chun Y, Lee I, Tudor R, Hong S, Shim T, et al. Pathogenic role of endothelin-1 in hemodynamic dysfunction in experimental acute pulmonary thromboembolism. Am J Respir Crit Care Med. 2001;164:1282–7. doi: 10.1164/ajrccm.164.7.2011011. [DOI] [PubMed] [Google Scholar]
- 10.Schmeck J, Koch T, Patt B, Heller A, Neuhof H, Ackern K. The role of endothelin-1 as a mediator of the pressure response after air embolism in blood perfused lungs. Intens Care Med. 1998;24:605–11. doi: 10.1007/s001340050622. [DOI] [PubMed] [Google Scholar]
- 11.Giaid A, Yansgisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. New Engl J Med. 1993;328:1732–9. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- 12.Michael J, Markewitz B. Endothelins and the lung. Am J Respir Crit Care Med. 1996;154:555–81. doi: 10.1164/ajrccm.154.3.8810589. [DOI] [PubMed] [Google Scholar]
- 13.Hoeper MM, Kramm T, Wilkens H, Schulze C, Schafers HJ, Welte T, et al. Bosentan therapy for inoperable chronic thromboembolic pulmonary hypertension. Chest. 2005;128:2363–7. doi: 10.1378/chest.128.4.2363. [DOI] [PubMed] [Google Scholar]
- 14.Altemeier W, Robertson H, Glenny R. Pulmonary gas exchange analysis by using simultaneous deposition of aerosolized and injected microspheres. J Appl Physiol. 1998;85:2344–51. doi: 10.1152/jappl.1998.85.6.2344. [DOI] [PubMed] [Google Scholar]
- 15.Clark AR, Burrows KS, Tawhai MH. The impact of micro-embolism size on hemodynamic changes in the pulmonary microcirculation. Respir Physiol Neurbiol. 2011;175:365–74. doi: 10.1016/j.resp.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 16.Clozel M, Ramuz H, Clozel J, Breu V, Loffler B, Coassolo P, et al. Pharmacology of Tezosentan, a new endothelin receptor antagonist designed for parenteral use. J Pharmacol Exp Ther. 1999;290:840–6. [PubMed] [Google Scholar]
- 17.Schimmel C, Frazer D, Glenny RW. Extending fluorescent microsphere methods for regional blood flow to 13 simultaneous colors. Am J Physiol Heart Circ Physiol. 2001;280:H2496–506. doi: 10.1152/ajpheart.2001.280.6.H2496. [DOI] [PubMed] [Google Scholar]
- 18.Melot C, Naeije R, Mois P, Hallemans R, Lejeune P, Jasper N. Pulmonary vascular tone improves pulmonary gas exchange in the adult respiratory distress syndrome. Am Rev Respir Dis. 1987;136:1232–6. doi: 10.1164/ajrccm/136.5.1232. [DOI] [PubMed] [Google Scholar]
- 19.Tsang J, Lamm W, Starr I, Hlastala M. Spatial pattern of ventilation-perfusion mismatch following acute pulmonary thromboembolism in pigs. J Appl Physiol. 2005;99:662–9. doi: 10.1152/japplphysiol.01018.2004. [DOI] [PubMed] [Google Scholar]
- 20.Dantzker DR, Bower JS. Pulmonary vascular tone improves V/Q matching in obliterative pulmonary hypertension. J Appl Physiol. 1981;51:607–13. doi: 10.1152/jappl.1981.51.3.607. [DOI] [PubMed] [Google Scholar]
- 21.Melot C, Hallemans R, Naeije R, Mois P, Lejeune P. Deleterious effect of nifedipine on pulmonary gas exchange in chronic obstructive pulmonary disease. Am Rev Respi Dis. 1984;130:612–6. doi: 10.1164/arrd.1984.130.4.612. [DOI] [PubMed] [Google Scholar]
- 22.Schraufnagel DE, Tsao MS, Yao YT, Wang NS. Factors associated with pulmonary infarction. Am J Clin Pathol. 1985;84:15–8. doi: 10.1093/ajcp/84.1.15. [DOI] [PubMed] [Google Scholar]
- 23.McIntyre KM, Sasahara AA. The hemodynamic response to pulmonary embolism in patients without prior cardiopulmonary disease. Am J Cardiol. 1971;28:288–94. doi: 10.1016/0002-9149(71)90116-0. [DOI] [PubMed] [Google Scholar]
- 24.Sasahara AA. Pulmonary vascular response to thromboembolism. Modern Concepts of Cardiovas Disease. 1967;36:55–60. [PubMed] [Google Scholar]
- 25.Sharma GV, McIntyre KM, Sharma S, Sasahara AA. Clinical and hemodynamic correlates in pulmonary embolism. Clinics in Chest Med. 1984;5:421–37. [PubMed] [Google Scholar]