

Abstract
The explosion of protein sequence information requires that current strategies for function assignment must evolve to complement experimental approaches with computationally-based function prediction. This necessitates the development of strategies based on the identification of sequence markers in the form of specificity determinants and a more informed definition of orthologues. Herein, we have undertaken the function assignment of the unknown Haloalkanoate Dehalogenase superfamily member BT2127 (Uniprot accession # Q8A5V9) from Bacteroides thetaiotaomicron using an integrated bioinformatics/structure/mechanism approach. The substrate specificity profile and steady-state rate constants of BT2127 (with kcat/Km value for pyrophosphate of ∼1 × 105 M−1 s−1), together with the gene context, supports the assigned in vivo function as an inorganic pyrophosphatase. The X-ray structural analysis of the wild-type BT2127 and several variants generated by site-directed mutagenesis shows that substrate discrimination is based, in part, on active site space restrictions imposed by the cap domain (specifically by residues Tyr76 and Glu47). Structure guided site directed mutagenesis coupled with kinetic analysis of the mutant enzymes identified the residues required for catalysis, substrate binding, and domain-domain association. Based on this structure-function analysis, the catalytic residues Asp11, Asp13, Thr113, and Lys147 as well the metal binding residues Asp171, Asn172 and Glu47 were used as markers to confirm BT2127 orthologues identified via sequence searches. This bioinformatic analysis demonstrated that the biological range of BT2127 orthologue is restricted to the phylum Bacteroidetes/Chlorobi. The key structural determinants in the divergence of BT2127 and its closest homologue β-phosphoglucomutase control the leaving group size (phosphate vs. glucose-phosphate) and the position of the Asp acid/base in the open vs. closed conformations. HADSF pyrophosphatases represent a third mechanistic and fold type for bacterial pyrophosphatases.
Keywords: BT2127, haloacid dehalogenase, HAD enzyme superfamily, inorganic pyrophosphatase, β-phosphoglucomutase, enzyme evolution, enzyme function assignment
Functional annotation of the rapidly growing numbers of enzyme sequences in the public databases is a challenge that should be met quickly so that the scientific community can effectively mine these databases. Ultimately, enzyme function annotation must be performed computationally because experimental-based function determinations are far too time consuming. The Enzyme Function Initiative (http://enzymefunction.org/) was established in 2010, with the long-term goal of defining an overall strategy that merges experimental approaches with complementary computationally-based function prediction. The work reported herein is an early product of this initiative that describes an integrated bioinformatic/structure/mechanism approach to define the function of a member of the Haloalkanoate Dehalogenase superfamily (HADSF). Beyond this single functional annotation, our integrated approach provides for the reliable tracking and annotation of orthologues.
The HADSF was chosen to explore methods of function assignment because of its large size, its presence in all organisms represented in the NCBI genomic database, and because of the diversity in the substrates upon which its members act(1-2). Although the HADSF members include dehalogenases, phosphonatases, and phosphomutases, the vast majority are phosphatases. HADSF phosphatases can number up to 30 in bacterial cells and up to 200 - 300 in eukaryotic cells(3). A central question to be addressed is- how many unique in vivo functions exist within this subfamily? The study of bacterial, as opposed to eukaryotic, HADSF phosphatases avoids the challenges arising from splice variants and reduces the complications associated with protein binding partners and subcellular localizations as modulators of in vivo function. Functional assignment of the bacterial HADSF phosphatases thus entails identification of the physiological substrate among the pool of organophosphate metabolites that exist in the cell, followed by defining the biological context of the catalyzed reaction.
Earlier work on the HADSF that involved the screening of 23 cytoplasmic E. coli HADSF phosphatases with an 80-compound library of known metabolites made clear the challenge of identifying the physiological substrates(4). Specifically, most of the phosphatases displayed activities towards multiple substrates, which in numerous instances were structurally dissimilar. In addition, there also existed some degree of overlap in the substrate ranges between phosphatases. Nevertheless, the physiological substrates of several of the 23 E. coli HADSF phosphatases have been convincingly identified and their in vivo functions assigned. Included in this group are phosphoserine phosphatase (SerB)(4-5), histidinol phosphate phosphatase (HisB)(6), trehalose-6-phosphate phosphatase (OtsB)(7), 3-deoxy-D-manno-octulosonate-8-phosphate phosphatase (YrbI)(8), and glycerol-manno-heptose 1,7-bisphosphate phosphatase (YaeD)(9) . Notably, each of these phosphatases functions in a biosynthetic pathway. The in vivo function assignment was facilitated by gene context as well as by a narrow substrate range coupled with a high kcat/Km value for the physiological substrate. In addition, gene knockout experiments have been used to show that the promiscuous phosphatases YniC(4) and YjjG(10) can function in vivo to remove toxic 2-deoxy-glucose 6-phosphate and noncanonical nucleoside-5′-monophosphates, respectively. Genetic experiments have also shown that YjjG performs in the thymidine salvage pathway as a dUMP nucleotidase(11).
To gain insight into the question of why many HADSF phosphatases from two different species of bacteria share common function while many do not, with the ultimate goal of facilitating orthologue assignment, we have initiated a side-by-side comparison of the HADSF phosphatases from E. coli and Bacteroides thetaiotaomicron. Even though these two species derive from different phyla, they share a common habitat: the human gut. E. coli possesses 23 HADSF phosphatases and B. thetaiotaomicron possesses 19, two of which have been experimentally characterized. One is 3-deoxy-D-manno-octulosonate-8-phosphate phosphatase (BT1677)(12), ortholog to the E. coli YrbI of the 3-deoxy-D-manno-octulosonate pathway. The second is 2-keto-3-deoxy-D-glycero-D-galactonate-9-phosphate phosphatase (BT1713) of the 2-keto-3-deoxy-D-glycero-D-galactonate pathway(13), which, notably, is not a pathway found in E. coli.
In the present study, we have undertaken the functional assignment of the HADSF member BT2127 (Uniprot accession # Q8A5V9), a member of unknown function from B. thetaiotaomicron. We report the function of BT2127 as an inorganic pyrophosphatase, and detail the X-ray structure and biological range, in contrast to those of the closest homologue, β-phosphoglucomutase (β-PGM) and to those of a HADSF Archeal inorganic pyrophosphatase.
Materials and Methods
General
All chemicals and buffers were purchased from Sigma-Aldrich. The sources of the gene cloning materials are as follows: primers, T4 DNA ligase, restriction enzymes (Invitrogen); E. coli BL21 (DE3) competent cells and Pfu, Turbo polymerases (Strategene); pET14b, pET23a and pET28a vector kits (Novagen); Qiaprep Spin Miniprep Kit (Qiagen). DEAE Sepharose was from Amersham Biosciences. Butyl and Phenyl-Sepharose resins were purchased from Sigma-Aldrich, and the Ni-NTA resin was from Qiagen. Snakeskin pleated dialysis tubing was purchased from Thermo Scientific. SDS-PAGE analysis was performed with a 12% acrylamide running gel and a 4% stacking gel (BioRad, Hercules, CA). Protein solutions were concentrated using a 10K Amicon Ultra Centrifugal filter (Millipore). The nucleotide sequence of each cloned gene or site-directed mutation bearing gene was determined by the Center for Genetics in Medicine, University of New Mexico. Electro-spray mass-spectrometry (ES-MS) determinations were carried out by the University of the New Mexico Mass Spectrometry Facility. Protein concentrations were determined using the Bradford assay kit from Sigma-Aldrich with BSA standards.
Preparation of recombinant wild-type and mutant BT2127
The DNA encoding the gene NP_811040 from B. thetaiotaomicron was amplified by PCR using the genomic DNA from B. thetaiotaomicron (ATCC 29148D), Pfu Turbo DNA polymerase and oligonucleotide primers (5′-GATTCCATCTAACCCACATATGAGAAAGAAAC) and (5′-CTTTTGCATAGTAGGATCCGTATTTATAGGT) containing restriction endonuclease cleavage sites NdeI and BamHI. The pET-28A vector, cut by restriction enzymes NdeI and BamHI, was ligated to the PCR product that had been purified and digested with the same restriction enzymes. The ligation product was used to transform E. coli BL21(DE3) competent cells which were then grown on a Kanamycin-containing agar plate. A selected colony was checked for BT2127 expression and the isolated plasmid was sequenced to verify the correct gene sequence. For BT2127 preparation, the transformed cells (9 L) were grown at 25 °C with agitation at 200 rpm in Luria broth containing 40 μg/mL Kanamycin to an OD600 of 0.6-0.7, and then induced for 12 h at 20 °C with 0.4 mM isopropyl α-D-thiogalactopyranoside. The cells were harvested by centrifugation (7,855g for 15 min at 4 °C) to yield 2.2 g/L of culture medium. The cell pellet was suspended (1 g wet cells/10 mL) in ice-cold buffer A (50 mM Tris (pH 7.6), 5 mM MgCl2 and 1 mM DTT). The cell suspension was passed through a French press at 1,200 PSIG before centrifugation at 48,384g and 4°C for 45 min. The supernatant was loaded onto a 40 × 5 cm DEAE-Sepharose 50-120 column, which was eluted with a 2 L linear gradient of NaCl (from 0 to 0.5 M) in buffer A. The column fractions were analyzed by SDS-PAGE. The desired fractions were combined and loaded onto 10 mL Ni-NTA Agarose column at 4 °C. After washing the column with 100 mL of buffer B (50 mM NaH2PO4, 300 mM NaCl, 20 mM imidazole (pH 8.0)), the enzyme was eluted with 200 mL elution buffer C (50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole (pH 8.0)). The column fractions were analyzed by SDS-PAGE, and the desired fractions were combined and concentrated with an Amicon Ultrafiltration apparatus (PM10) before dialysis at 4 °C against buffer A. The final yield was 11 mg BT2127/g wet cells.
Site directed mutagenesis was performed with a PCR-based strategy, using the BT2127-pET-28A clone as template and commercial primers. The protein variants were purified in the same manner as described for the wild-type BT2127.
BT2127 molecular weight and quaternary structure determination
The theoretical subunit molecular mass of recombinant BT2127 was calculated by using the amino acid composition, derived from the gene sequence, and the ExPASy Molecular Biology Server program Compute pI/MW. The subunit size of recombinant BT2127 was estimated by SDS-PAGE analysis, which included the molecular weight standards from New England Biolabs. The exact subunit mass was determined by MS-ES mass spectrometry. The molecular weight of native BT2127 was estimated by FPLC gel filtration column chromatography against protein standards (13.7-220 kDa from GE Healthcare). Elution of protein from the 1.6 cm × 60 cm Sephacryl S-200HR column (GE Healthcare) was performed at 4 °C with buffer D (50 mM HEPES, 100 mM NaCl (pH 7.5)) at a flow rate of 1 mL/min. The BT2127 molecular weight was derived from the measured elution volume by extrapolation of the plot of the elution volume of the molecular weight standard versus log molecular weight. BT2127 native mass was also analyzed at the HHMI Biopolymer/Keck Foundation Biotechnology Resource Laboratory at Yale University by size exclusion chromatography coupled with on-line laser scattering, refractive index, and ultraviolet detection.
Kinetic assay for β-phosphoglucomutase activity
Reaction solutions initially contained 7 μM wild-type BT2127, 200 μM β-glucose 1-phosphate, 10 μM β-glucose1,6-bisphosphate, 2 mM MgCl2, 0.4 mM NADP and 5.4 units/ml glucose-6-phosphate dehydrogenase in 50 mM K+-HEPES (pH 7.5). The formation of NADPH from reduction of glucose-6-phosphate was monitored at 340 nm (ε = 6.2 mM−1 cm−1).
Steady-state kinetic constant determinations
Initial velocities for BT2127 catalyzed hydrolysis of phosphate esters and anhydrides were measured at 25 °C using assay solutions that contained 1 mM MgCl2, 1.0 unit/ml purine nucleoside phosphorylase, and 0.2 mM MESG in 50 mM Tris (pH 7.5). The reactions were monitored at 360 nm (Δε = 9.8 mM−1 cm−1). The steady-state kinetic parameters (Km and kcat) were determined by fitting the initial velocity data measured at varying substrate concentrations (ranging from 0.5Km to 5Km) to equation 1 using the program KinetAsyst I,
| (1) |
where V0 is the initial velocity, Vmax the maximum velocity, [S] the substrate concentration, and Km the Michaelis constant for the substrate. The kcat value was calculated from Vmax and [E] according to the equation kcat=Vmax/[E], where [E] is the enzyme concentration.
The steady-state competitive inhibition constant Ki was determined for imidodiphosphate by fitting the initial velocity data, measured as a function of pyrophosphate (3, 5, 8, 10, 15, 20 μM) and imidodiphosphate (0, 10, 35, 50 μM) to equation 2 using KinetAsyst I.
| (2) |
Where [I] is the inhibitor concentration and Ki is the inhibition constant.
pH rate profile determination
The steady-state kinetic constants kcat and Km for BT2127-catalyzed hydrolysis of pyrophosphate were measured at 25 °C as a function of reaction solution pH. Reaction solutions initially contained pyrophosphate (0.5Km to 10Km), 1 mM MgCl2, 1.0 unit/ml purine nucleoside phosphorylase, 0.2 mM MESG in 50 mM buffer (MES, pH 5.0-6.0; HEPES, pH 6.5-7.5; Tris, pH 8.0-8.5). The kcat/Km data were fitted with equation 3 and the kcat data were fitted with equation 4.
| (3) |
| (4) |
where Y is the kcat or kcat/Km, [H] is the hydrogen ion concentration of the reaction solution, Ka and Kb are the apparent ionization constants, and C is the constant value of Y.
BT2127 crystallization and structure determination
Proteins were crystallized by the sitting-drop vapor diffusion method. In brief, the protein solutions (usually 0.3 or 1 μL) were mixed with an equal volume of a precipitant solution and equilibrated at room temperature (294 K) against the same precipitant solution in clear tape-sealed 96-well INTELLI-plates (Art Robbins Instruments, Sunnyvale, CA, USA). Crystallization was performed using either a TECAN crystallization robot (TECAN US, Research Triangle Park, NC, USA) or a PHOENIX crystallization robot (Art Robbins Instruments) and three types of commercial crystallization screens: the WIZARD screen (Emerald BioSystems, Bainbridge Island, WA, USA), the INDEX and the CRYSTAL SCREEN I and II (both from Hampton Research, Aliso Viejo, CA, USA). A number of conditions produced diffraction-quality protein crystals starting within 24-72 h of incubation. The crystals were harvested using cryogenic loops (Hampton Research), quickly transferred into liquid nitrogen, and stored frozen in liquid nitrogen until X-ray analysis and/or data collection. Only in one case (PDB ID 3QU2) was the mother liquor supplemented with the cryoprotectant glycerol. Where necessary, the crystallization conditions were optimized manually using 24-well Cryschem sitting drop plates (Hampton Research). The final crystallization conditions for each X-ray crystal structure are listed in Table S1 (Supporting Information)
The X-ray diffraction data for the majority of frozen crystals were collected at 100 K on the Beamline X29A (NSLS, Brookhaven National Laboratory) using a wavelength of either 0.93 or 1.08 Å (see Table S1). The X-ray diffraction data for the D13A variant were collected at 100 K using an RU-200 rotating-anode X-ray generator (λ = 1.5418 Å) coupled to a Rigaku R-AXIS IV area detector. All diffraction data were processed and scaled with the HKL2000 software package(14). The first of 13 crystal structures (PDB ID 3QU2) reported here was determined by molecular replacement using coordinates for the PDB structure 3DV9 (68% sequence identity) as the search model and the program MOLREP (the CCP4 program package suite(15)). The other 12 structures were determined by molecular replacement using coordinates of the 3QU2 structure as the search model. Each structure was refined using REFMAC 5.03 (CCP4 suit(16)) and the resulting models were rebuilt manually using COOT visualization and refinement software (17). The data collection and refinement statistics for these structures are shown in the Tables S2-S5.
Bioinformatic Analysis
Putative orthologues of BT2127 and the pyrophosphatase from Thermococcus onnurineus TON0002 were identified using BLAST searches of the NCBI microbial genome bank (http://www.ncbi.nlm.nih.gov/sutils/genom_table.cgi) and selecting those sequences in which the key marker residues are conserved. The multiple sequence alignments were made in Cobalt (http://www.ncbi.nlm.nih.gov/tools/cobalt/) and displayed in ESPript (http://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi).
In order to compare biological ranges of BT2127 and β-PGM a more automated approach was employed. BLAST searches were performed for BT2127, β-PGM, maltose phosphorylase, and trehalose phosphorylase using a locally installed copy of BLAST against the NCBI non-redundant protein database (ftp://ftp.ncbi.nih.gov/blast/db/) with default parameters. The resulting hits were filtered for query coverage >90% and >30% sequence identity. A reciprocal BLAST search, using an e-value threshold of 1e−10, was performed for each filtered hit. A hit was considered successful if the reciprocal BLAST search identified the initial query sequence. For each successful hit, taxonomic information was downloaded from NCBI taxonomy website (http://www.ncbi.nlm.nih.gov/Taxonomy/taxonomyhome.html/). For the selection of β-PGM orthologues, only those species with a predicted maltose or trehalose phosphorylase gene were considered as positive hits. A phylogenetic tree for the orthologues was generated using iTOL (interactive Tree Of Life (http://itol.embl.de/)) and visualized using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Results and Discussion
BT2127 Substrate Specificity Profile
Because the closest characterized structural homolog of BT2127 is β-phosphoglucomutase (β-PGM), the ability of purified recombinant BT2127 (see SDS/PAGE gel Figure S1) to catalyze the conversion of β-glucose1-phosphate to glucose 6-phosphate in the presence of the cofactor β-glucose1, 6-(bis)phosphate was tested. No activity was observed above the detection limit of one catalytic turnover per hour.
To determine the substrate range of BT2127, phosphatase activity was tested using a structurally diverse chemical library of 21 organophosphate metabolites as substrate (Table S6 and S7). The most active substrates identified by this screen, pyrophosphate, glycerol 1-phosphate, D-ribose 5-phosphate, fructose 6-phosphate, uridine 5-phosphate and pNPP, were analyzed to determine kcat and kcat/Km values (Table 1) at the pH optimum of 7.5 (pH/rate profile shown in Figure S2). HADSF phosphatases are known to utilize a two-step reaction entailing phosphoryl transfer from the bound substrate to the Asp nucleophile followed by hydrolysis of the aspartyl-phosphate intermediate (Scheme 1). Variation in the kcat value with substrate structure indicates that the first step is rate-limiting (or at least partially rate-limiting) and that some substrates bind more productively than others. The largest kcat value was measured for the substrate inorganic pyrophosphate (0.3 s−1). Inorganic pyrophosphate exists in the cell as a magnesium complex that undergoes spontaneous hydrolysis to two molecules of orthophosphate at an uncatalyzed rate in solution of 2.8 × 10−10 s−1 (at 25 °C, pH 8.5)(18). BT2127 thus increases the hydrolysis rate of pyrophosphate ∼1 × 109-fold (a transition state stabilization of greater than 12 kcal/mol).
Table 1.
Steady-state kinetic constants for BT2127-catalyzed hydrolysis of pyrophosphate and selected phosphate monoesters at 25 °C and pH 7.5 (see Materials and Methods for details).
| Substrate | kcat (s−1) | Km (mM) | kcat/Km (M−1s−1) |
|---|---|---|---|
| pyrophosphate | 0.32 ± 0.01 | 0.0036 ± 0.0004 | 9 × 104 |
| glycerol 1-phosphate | 0.35 ± 0.02 | 4.8 ± 0.3 | 7 × 101 |
| D-ribose 5-phosphate | 0.20 ± 0.02 | 3.8 ± 0.3 | 6 × 101 |
| fructose 6-phosphate | 0.066 ± 0.002 | 6.4 ± 0.2 | 1 × 101 |
| uridine 5-phosphate | 0.15 ± 0.01 | 2.6 ± 0.4 | 6 × 101 |
| p-nitrophenylphosphate | 0.0019 ± 0.0004 | 0.05 ± 0.01 | 4 × 101 |
Scheme 1.

The HADSF phosphatase chemical pathway.
The kcat/Km value (also known as the substrate specificity constant) is determined by the substrate binding affinity as well as by the efficiency with which the bound substrate is converted to product. Because the concentration of the substrate in the cell is likely to be sub-saturating, the kcat/Km value is most useful for identifying substrates that have physiologically relevant activities. HADSF phosphatases that target a single physiological substrate typically display a kcat/Km value in the range of 1 × 105 to 1 × 107 M−1 s−1(8-9, 13), whereas those that function as regulators of the level and composition of organophosphate metabolite pools typically have Km values in the milli-molar range and kcat/Km values in the 1 × 103 to 1 × 104 M−1 s−1 range(19-20). The kcat/Km value of ∼1 × 105 M−1 s−1 measured for inorganic pyrophosphate is consistent with its assignment as a physiological substrate. Furthermore, the fact that the screen did not identify other substrates with activities in a range to be considered physiologically relevant suggests that BT2127 might indeed function in vivo as an inorganic pyrophosphatase.
BT2127 Structure Determination
The structure of BT2127 was examined in order to identify the structural determinants of substrate recognition. A total of 13 X-ray structure determinations were carried out with wild-type enzyme and the D11N, D13A, D13N, E47A, E47D, E47N variants unliganded and bound to ligands. The crystallization conditions (Table S1) and the crystallographic data collection and refinement statistics are reported in Supporting Information (Tables S2-S5). The various crystallization conditions led to different crystal packing and to different ligands observed in the active site. All crystallizations were performed in the presence of 5 mM MgCl2; however, only six of the structures (PDB ID 3QU2, 3QU4, 3QU9, 3QUQ, 3QUT and 3QX7) contained Mg2+ within the active site. In two of the structures (PDB ID 3QU7 and 3QYP), Ca2+ derived from the crystallization solution replaced Mg2+. This replacement has previously been observed in HADSF enzymes(21). Co-crystallization and crystal soaking of each enzyme were attempted with pyrophosphate as well as with its analog imidodiphosphate (a competitive inhibitor with a Ki = 13 ± 1 μM (Figure S3)). No ligand density was observed in the imidodiphosphate-soaked crystals. Three structures derived from the pyrophosphate-soaked crystals (PDB IDs 3QYP, QX7, and 3QU7) contained the hydrolysis product phosphate, showing the enzyme is active under the crystallization conditions utilized.
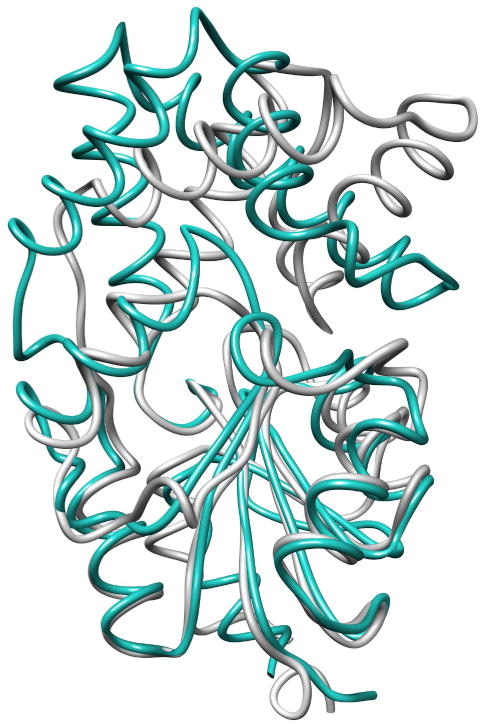
The BT2127 protomer possesses the conserved HADSF α/β Rossmann-like catalytic domain composed of a six-stranded parallel β sheet (β1, and β4-β8) surrounded by six helices (α1-α3, and α5-α7) and a four helix cap domain (residues 19 - 89) inserted between β1 and α1 (denoted type C1 cap domain). Well-characterized members of the C1 class include Lactobacillus lactis β-PGM (Z-score 10.2)(22) and Bacillus cereus phosphonatase (Z-score 7.8)(23) (Z-scores calculated by DALI(24). The C1 type cap domains of BT2127 and β-PGM superimpose (Figure 1) with a RMSD of 2.4 Å (13.4% identity in cap domain), whereas the two catalytic domains superimpose with a RMSD of 1.9 Å (26.7% identity in core domain). HADSF members with similar cap domain folds were determined using the program DALI and the cap domain (Table S8). The closest homolog (Z-score 14.7) is the HADSF member from Bacteroides vulgatis, which was previously annotated “putative β-PGM” (PDB ID 3DV9). As detailed below, we posit that this B. vulgatis protein is not β-PGM but a pyrophosphatase and an orthologue of BT2127.
Figure 1.
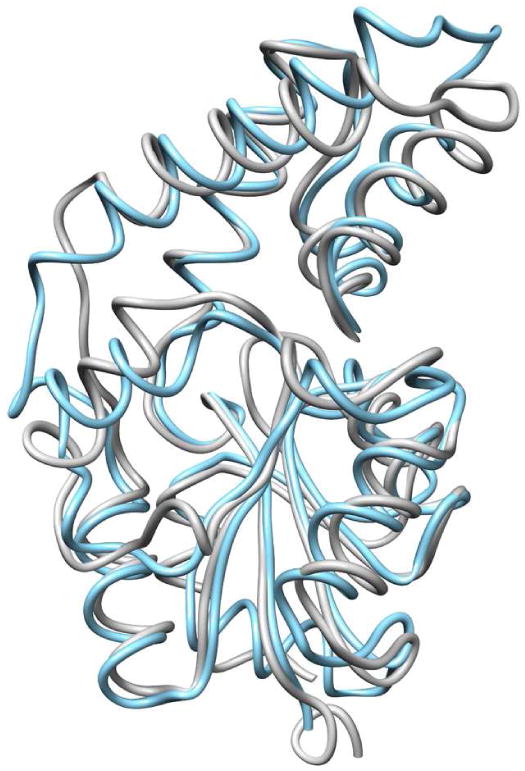
Superposition of BT2127 (PDB ID 3QX7) (gray) and L. lactis β-PGM (PDB ID 1O08) (cyan).
BT2127 is a monomer in solution, as the molecular weight determined by molecular size gel filtration chromatography (27-28 kDa) matches the protomer molecular weight of 26,970 Da determined by mass spectrometry (theoretical mass of BT2127 minus the N-terminal Met is 26,983 Da) and of 27 kDa determined by SDS-PAGE (Figure S1).
Association of the Cap and Catalytic Domains
CA1 cap motion has been most thoroughly studied in L. lactis β-PGM for which crystallographic snapshots of the enzyme in the open and closed conformations have been obtained (Figure 2)(25-26). The structures of BT2127 (PDB ID 3QX7; wild-type bound to phosphate and Mg2+) reveals a cap-closed conformation that is similar to that of β-PGM (Figure 1) and a cap-open conformation (3QUT; D13N variant with Mg2+ and malate bound to the transferring phosphate site) that is not as open as that observed for β-PGM (Figure 2A and 2B).
Figure 2.
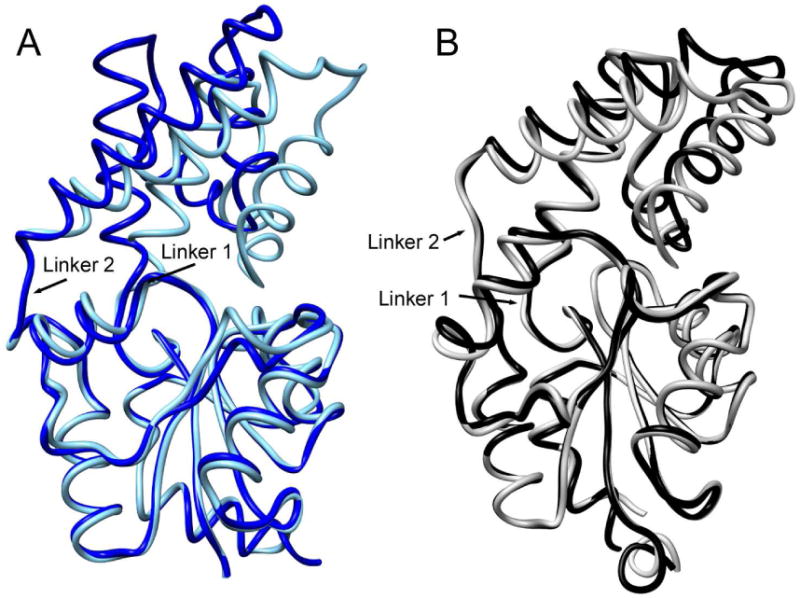
Backbone coil depictions of the superposition of the (A) cap-open (royal blue) and cap-closed (cyan) conformations of L. lactis β-PGM (PDB ID 1ZOL and 1O08) and (B)the cap-open (black) and cap-closed (gray) conformations of BT2127 (PDB ID 3QUQ and 3QX7).
In the case of β-PGM, the cap and catalytic domains associate for catalysis and dissociate to allow ligand exchange with solvent. An earlier crystallographic based analysis of the “clam-like” domain movement in β-PGM using DynDom(27-28) indicated that the cap and catalytic domains move as rigid bodies with a large relative rotation of 26°; this movement is primarily the result of changes in backbone conformations (psi and phi angles) at domain-domain linker 1 hinge residues Thr114-Asp15-Thr16-Ala17 (Dai 2009). A similar analysis shows that the more modest domain movement observed in BT2127 is due to changes in phi and psi angles at linker 1 hinge residues Ser19-Met20-Pro21 and linker 2 hinge residues Pro87-Glu88-Ala89-Glu90-Arg91. The rotation angle between the cap and catalytic domains in BT2127 is 16.3°, with a translation of 0.6 Å. As seen with β-PGM, the BT2127 cap and core domains must dissociate in order for pyrophosphate to bind to the active site and phosphate to dissociate because only in the cap-open conformation is there bulk solvent access to the active site.
In order to assess the relative sizes of the active site cavities of the two enzymes, the program Voidoo (http://xray.bmc.uu.se/usf/voidoo.html) was used to calculate the volume of the active sites of BT2127 (189.8 Å3) and β-PGM (285.1 Å3) in their cap-closed conformations. Next, the molecular volumes of pyrophosphate (99.4 Å3) and β-glucose1,6-(bis)phosphate (242.5 Å3) were calculated using the molinspiration server (http://www.molinspiration.com/). Whereas pyrophosphate is small enough to fit in the active site of BT2127, β-glucose1,6-(bis)phosphate is not. Models of BT2127 bound to pyrophosphate and to β-glucose1,6-(bis)phosphate were generated to show the fit of the ligands in the active site cavity (Figure 3). Whereas the pyrophosphate can be easily docked in a productive binding mode without steric clash, the larger β-glucose-1,6-bisphosphate ligand cannot fit without the cap dissociating to some extent from the core domain. The low level activity that is observed with the larger substrates (Table 1 and Table S6) might be rationalized by catalytic turnover in a conformer in which the cap domain is partially dissociated to enlarge the active-site cavity.
Figure 3.
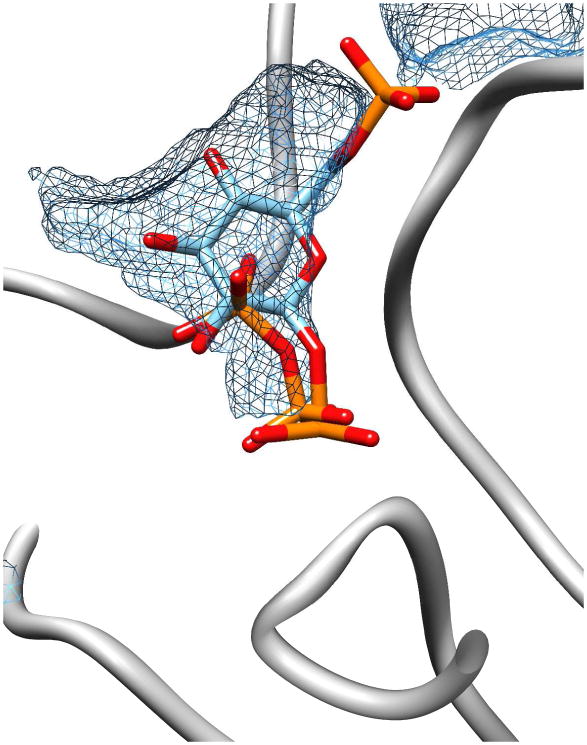
The active site of BT2127 in the cap-closed conformation (PDB ID 3QX7) showing the active site volume calculated in Voidoo (mesh) and the pyrophosphate and β-glucose-1,6-bisphosphate ligands (shown as sticks, phosphorus atoms orange) modeled in COOT.
The Active Site Scaffold
The active site scaffold of the HADSF phosphotransferase catalytic domain consists of a 4-loop platform on which the conserved catalytic residues are located: the Asp nucleophile and Asp acid/base on loop 1, the Thr/Ser and Lys hydrogen bond donors to the transferring phosphoryl group on loop 2 and 3, respectively, and the Asp/Glu Mg2+ ligand on loop 4(1). Figure 4 shows the residues surrounding the Mg2+ cofactor in the BT2127-Mg complex and the interacting residues in the BT2127 E47N variant complexed with Ca2+ and phosphate. As observed with other HADSF phosphotransferases, the Mg2+ cofactor is coordinated to the loop 1 Asp nucleophile (Asp11) carboxylate group and the Asp acid/base (Asp13) backbone C=O (2.2 Å). The loop 4 Asn172 forms a coordination bond with Mg2+ (2.3 Å) and loop 4 Asp171 forms a hydrogen bond (2.7 Å) to one of two waters that coordinate the Mg2+. A deviation from the classical HADSF Mg2+ binding site(29) is the identity of the sixth ligand, Glu47 (2.0 Å), which is located on the specificity loop of the cap domain (Glu47-Gly48-Arg49-Thr50).2
Figure 4.
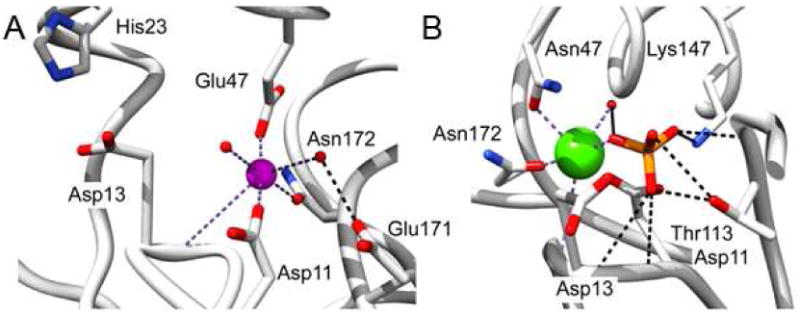
(A) The Mg2+ binding site observed structure of wild-type BT2127 bound with Mg2+ (magenta sphere) (PDB ID 3QUQ). (B) The phosphate binding site observed in the structure of the BT2127 E47N mutant bound with phosphate (phosphorous in orange) and Ca2+ (green sphere) (PDB ID 3QYP). Water molecules are represented as red spheres, coordination bonds as dashed purple lines, and hydrogen bonds as dashed black lines.
From the structure of the E47N variant bound to phosphate and Ca2+ at the catalytic site, we observe that an oxygen atom of the phosphate ligand replaces a water ligand (Figure 4), as is usually observed in the HADSF(12). The phosphate ligand also is engaged in numerous hydrogen-bond interactions with the loop 2 Thr113 and loop 3 Lys147 side chains, and loop 1 Asp13, loop 2 Gly114 and Ser115 backbone amide N atoms. In order to best examine the key interactions in the context of the pyrophosphate substrate, a model was made of the BT2127 active site (cap-closed conformer) with pyrophosphate bound in an orientation that superimposes the transferring phosphoryl group on the phosphate ligand of the BT2127 E74N/Ca2+/phosphate structure (Figure 5). Only one P-O-P conformer placed the phosphate-leaving group within the vacant space defined by the Voidoo analysis (vide supra). Here it can engage in hydrogen bond formation with a water molecule that is bound to cap domain Trp27 and with the backbone amide NH of cap domain residue Gly48 and catalytic domain Ser115. Moreover, the Asp13 acid/base is positioned to donate a hydrogen bond to a nonbridging oxygen atom, and to the bridging oxygen atom of the phosphate-leaving group. Notably, at pH 7.5 the pyrophosphate is mono-protonated (pKa = 9.3) and if it were to bind with the protonated phosphate group in the leaving-group position there would be no need for acid catalysis. On the other hand, we have proposed that the Asp acid/base of the HADSF phosphotransferases is protonated by the substrate phosphate ester, which binds to the active site in the mono-protonated form and upon coordination with the Mg2+ the phosphate substituent loses its proton(30). Thus, in order for the Asp13 to be protonated the pyrophosphate would need to be fully ionized.
Figure 5.
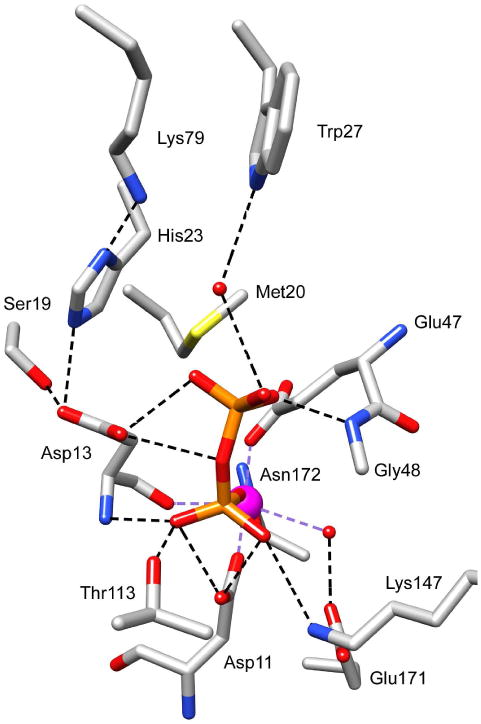
The BT2127 active site with pyrophosphate (manually docked). Atom coloring and bonds shown as in Figure 4.
Notably, by replacing the pyrophosphate bridging oxygen atom with an NH group, as in imidodiphosphate, tight binding to BT2127 is retained (Ki = 13 ± 1 μM) but catalytic turnover is greatly diminished (Table S2). We infer from this observation that the Asp13 is not able to align with the nonbonding electron pair of the bridging nitrogen atom for proton transfer. In contrast, several of the 23 E. coli HADSF phosphatases examined by Kuznetsova et al. (4) showed significant hydrolase activity with imidodiphosphate but curiously, not with pyrophosphate.
An interesting finding is presented by the position that the phosphate ligand assumes in the structure of the wild-type BT2127/Mg2+/phosphate complex (PDB ID 3QX7). Superposition of this structure with the structure of L. lactis β-PGM bound to β-glucose1,6-(bis)phosphate places the phosphate ligand at the same position that the non-transferring phosphoryl group (i.e., C6-phosphate) is found (Figure S4). It is plausible that once formed, the phosphate product in the BT2127 active site might diffuse into this vacant space to make room for the water nucleophile, which will perform in-line attack at the phosphorus atom of the aspartylphosphate intermediate in the second step of the reaction (Scheme 1). Alternatively, the movement of the phosphate into the cap domain might trigger cap dissociation.
Site Directed Mutagenesis of BT2127 Active Site and Domain-Domain Interface Residues
The individual contributions that the active site residues make to catalysis were evaluated by site-directed mutagenesis coupled with steady-state kinetic analysis of the mutant enzymes in catalysis of pyrophosphate hydrolysis. The structures of the BT2127 mutants showed that the wild-type native conformation, including the active site residue side chains, was retained. Therefore, we interpret the changes in catalytic activity in the mutants as resulting from changes of side chain function and not alterations of the native conformation. The mutants that did not display activity above background are listed in Table 3 as not active (“NA”). The mutants that displayed detectable yet very low activity were evaluated by measuring the initial velocity of the reaction using a concentration of pyrophosphate (300 μM) that we assume to be saturating. The ratio of the initial velocity and the enzyme concentration gives an approximate kcat value (i.e., turnover rate). For the mutants that displayed higher activity, kcat and Km values were measured.
First, the impact of Ala and/or Asn replacement of the key catalytic residues Asp11 nucleophile and Asp13 acid/base, both of which are invariant was tested (see multiple sequence alignment of putative BT2127 orthologues in Figure S5). Not surprisingly, Ala and/or Asn replacement of BT2127 Asp11 or Asp13 resulted in the loss of all detectable activity (Table 3). However, the observation of a phosphate ligand in the active sites of pyrophosphate-soaked crystals of BT2127 D13N indicates that a single-turnover reaction had most likely occurred during the 15 min soaking time.
The replacement with Ala of the residues that contribute to the positioning of the Asp13 acid-base, namely, Ser19, His23, Ser115, or Met20 (each of which is invariant) reduced the turnover rate 950, 60, 230, and 370-fold, respectively. The Met20 was also replaced by Leu and by Lys. The M20L and M20K mutants were found to be 330 and 80-fold lower in turnover rate compared to that of the wild-type BT2127. Because the Met20 side chain restricts both the volume and the polarity of the region available to accommodate the substrate leaving-group we tested the activities of M20A, M20L and M20K towards the same panel of phosphate esters that were used to screen the wild-type enzyme. The results (Table S7) indicate that the substrate specificity of the mutants was not significantly different than that of wild-type BT2127.
The coordination of the Mg2+ by the loop 1 Asp nucleophile and by the backbone amide C=O of the Asp acid-base is observed in all structurally characterized Mg2+ bound HADSF phosphatases(29). Likewise, an Asp or Glu residue located on loop 4 invariably adds a third metal contact. In some cases, an Asp or Glu also located on loop 4 forms a water-mediated contact with the Mg2+ cofactor. We have not previously encountered the collaboration between a core domain loop 4 amide (Asn172) and cap domain specificity loop carboxylate (Glu47) to contribute to the Mg2+ binding. The replacement by Ala of the stringently conserved Asn172 resulted in only a 26-fold reduction in turnover rate. In contrast, Ala replacement of the conserved Glu47 resulted in ∼6000-fold decrease in turnover rate and the same reduction is observed with the E47D and E47N mutants. Glu47 might be required for Mg2+ binding. We note that in the four structures of the three E47 mutants crystallized in the presence of Mg2+ the positions of the Asp11 and Asn172 side chains and the Asp13 backbone C=O are the same as in wild-type, yet the Mg2+ is absent. In contrast, the E47N variant crystallized in the presence of Ca2+ (PDB ID 3QYP) contains Ca2+ which is coordinated to the loop 1 and 4 ligands as well as to the Asn47. The larger ionic radius of Ca2+ apparently compensates for the shortened reach of the Asn side chain (Figure 4). Accordingly, the replacement of Glu47 with Gln should be conservative as it would allow Mg2+ coordination; this is reflected in the activity of the E47Q mutant which has a turnover rate only 40-fold lower than the wild-type enzyme (Table 2). These findings suggest that E47 rather than the loop 4 Asn172 plays the key role in Mg2+ binding. Furthermore, the fact that the Glu47 is coordinated to the Mg2+ in the cap-open as well as the cap-closed conformation might explain why the BT2127 cap domain does not dissociate from the catalytic domain to the extent observed with β-PGM.
Table 2.
Steady-state kinetic constants for wild-type and mutant BT2127-catalyzed hydrolysis of pyrophosphate at 25 °C and pH 7.5. See Materials and Methods for Details.
| BT2127 | kcat (min−1) | Km (μM) | kcat/Km (M−1s−1) |
|---|---|---|---|
| Wild-type | 1.9 (± 0.06) × 101 | 3.6 ± 0.4 | 8.9 × 104 |
| D11N | NAa | ------ | ------ |
| D13A | NAa | ------- | ------ |
| D13N | NAa | ------- | ------ |
| K147A | ∼2 × 10−3b | ------- | ------ |
| N172A | 7.4 (± 1.8) × 10−1 | 4.5 ± 0.3 | 3 × 103 |
| T113A | 4.0 (± 0.03) × 10−1 | 5.9 ± 0.2 | 1 × 103 |
| S115A | 8.4 (± 0.12) ×10−2 | 3.6 ± 0.4 | 4 × 102 |
| T50A | 4.1 (± 0.18) × 10−1 | 2.7 ± 0.7 | 3 × 103 |
| R49A | ∼3 × 10−2b | ------- | ------- |
| K79A | ∼2 × 10−2b | ------- | ------- |
| H23A | 3.3 (± 0.06) × 10−1 | 1.2 (± 0.08) × 101 | 5 × 102 |
| M20A | ∼5.1 × 10−2b | ------- | ------- |
| M20K | 2.4 (± 0.04) × 10−1 | 2.5 (±0.3) × 101 | 2 × 102 |
| M20L | 5.8 (± 0.04) × 10−2 | 3.5 ± 0.1 | 3 × 102 |
| E47A | ∼3 × 10−3 | ------- | ------- |
| E47Q | 4.8 (± 0.4) × 10−1 | 6 ± 1 | 1 × 103 |
| E47N | ∼3 × 10−3b | ------- | ------- |
| E47D | ∼3 × 10−3b | ------- | ------- |
| S80A | 1.7 (± 0.04) × 10−1 | 7.1 ± 0.9 | 4.1 × 102 |
| Q117A | ∼3 × 10−2b | ------- | ------- |
| Y76F | ∼2 × 10−2b | ------- | ------- |
| S19A | ∼2 × 1 0−2b | ------- | ------- |
NA represents no activity detected above the detection limit kcat < 0.001 min−1.
The rate was determined by measuring the initial velocity of orthophosphate formation (μM/min) and dividing that value by the enzyme concentration. Reaction solutions initially contained 10 μM enzyme, 300 μM pyrophosphate, 1 mM MgCl2, 1.0 unit/ml purine nucleoside phosphorylase, and 0.2 mM MESG in 50 mM Tris (pH 7.5).
In examining the ligands to the substrate, the Thr113 and Lys147 activate the transferring phosphoryl group via hydrogen bond formation (Figures 4 and 5). Both are stringently conserved, and we found that the turnover rate was reduced 42-fold in the case of the T113A mutant and 9,500-fold in the case of the K147A mutant. The stringently conserved cap domain residue Lys49 is not close enough to the catalytic site for direct interaction with pyrophosphate. It does however, form a hydrogen bond with the stringently conserved cap domain residue His23, which in turn forms a hydrogen bond with the catalytic domain Asp13 acid/base (Figure 5). Whereas the turnover rate of the K49A mutant was found to be 950-fold slower than that of the wild-type BT2127, that of the H23A mutant was reduced only 60-fold. Thus, Lys49 appears to contributing to catalysis in some way beyond simply serving as a hydrogen bond partner to His23 (vide infra).
Additional mutants were examined to evaluate the contributions made by residues located at the domain-domain interface, which appear from the structures to stabilize the cap-closed conformation through hydrogen bond formation with a partner located on the opposing domain (Figure S6). First, the stringently conserved Gln117 of the catalytic domain forms a hydrogen bond with cap domain residue Ser80 (replaced with Thr, Ala, or Arg in 8 out of 33 orthologous sequences). Ala replacement of Gln117 reduced the turnover rate 630-fold whereas Ala replacement of Ser80 reduced the turnover rate only 110-fold. Second, the stringently conserved Tyr76 located on the inter-domain linker 2 is close enough (3.7 Å) to make hydrogen bond formation possible with the backbone amide NH of catalytic domain Gln117 and Gly116 (highly conserved; replaced with Ala in 1 out of 33 sequences). The conservative replacement of Tyr76 with Phe reduced the turnover rate 950-fold. Third, the highly conserved Arg49 (31 out of 33 sequences) and Thr50 (32 out of 33 sequences) are located on the cap domain specificity loop (composed of Glu47-Gly48-Arg49-Thr50). The Arg49 side chain projects into solvent where the nonpolar hydrocarbon region of the side chain packs against the side chains of the catalytic domain Pro148 (stringently conserved) and Leu175 (conservatively replaced by Met in 5 out of 33 sequences), an interaction that might facilitate the proper positioning of Glu47 within the active and/or influence the association of the cap and core domains. Ala replacement of Arg49 reduced the turnover rate 630-fold. Thr50 is seen in the crystal structures to be too far (4.3 Å) from the stringently conserved catalytic domain Gly114 C=O for hydrogen bond formation. Nevertheless, Ala replacement of Thr50 reduced catalytic turnover 50-fold, suggesting that the solution conformation might allow hydrogen bond formation or alternatively, that the Thr50 contributes to the conformation of the specificity loop.
BT2127 Biological Range
Based on the structure-function analysis described in the previous sections we selected the catalytic residues Asp11, Asp13, Thr113, and Lys147 as well the metal binding residues Asp171, Asn172 and Glu47 to curate the list of BT2127 homologues identified by BLAST searches. An alignment of 33 putative homologues was generated (Figure S5), which highlighted the invariant conservation of 46 residues (including the 7 marker residues) out of 244 residues total (19% conservation). The lowest pair-wise sequence identity between BT2127 and any putative sequence orthologue is 47%. Many of these sequences have N-terminal extensions (∼20 amino acids) when compared to the BT2127 sequence. One of these is the orthologue from Bacteroides vulgatis (Uniprot accession code A6L7P8) that was also identified as the most similar structure in the DALI search for structural homologues (vide supra). The superposition of the X-ray structure (PDB ID 3DV9) with that of BT2127 (not shown) reveals that N-terminal extension forms an α-helix at the surface of the catalytic domain located at the edge of the β-sheet, which is distant from the active site and is not expected to impact catalysis.
Although the marker residues used in curating the BT2127 homologues are by definition invariant, 14 additional sequences could be identified that replaced a marker residue, Asn172 with Asp (multiple sequence alignment shown in Figure S7). These sequences are more divergent (28-30% pairwise sequence identity with BT2127) and all but one of them also possesses the conservative replacement of the Met12 in BT2127 by Leu in the Asp11-Met12-Asp13 loop 1 catalytic motif. Although the residue that separates the Asp nucleophile and Asp acid/base on loop 1 is not known to have a direct catalytic role, it has been our observation that it is typically conserved within function families that have clear sequence homology boundaries. Moreover, the residues of the specificity loop and/or the residues supporting domain-domain interaction (such as Tyr76, Thr50, Gln117, Ser19, Met20 and Lys49) are not conserved. Nevertheless, the 14 sequences might in fact be pyrophosphatases, but such annotation should be first supported by an in vitro activity assay of a representative member, and as such these proteins will be referred to as “BT2127-like” homologues.
The biological range of the putative BT2127 orthologues is restricted to the phylum Bacteroidetes/Chlorobi (Figure 6). BT2127 orthologues are found in all Bacteroides species/strains represented in the gene databases (with the exception of B. pectinophilus that instead possesses a BT2127-like homologue), as well as in the Parabacteroides species/strains and Prevotella species/strains. Bacteroides, Parabacteroides, and Prevotella colonize the human cavity from mouth to colon, yet occupy different regions along this environmentally stratified track. The BT2127-like homologues are also contained in the Bacteroidetes/Chlorobi phylum where they are found in select species that occupy a diverse range of habitats: Capnocytophaga ochracea (human mouth); Chlorobium luteolum and Pelodictyon phaeoclathratiforme (sulfide rich aquatic environments), Croceibacter atlanticus and Flavobacteria bacterium BAL38, BBFL7, ALC-1 (sea water); Listeria innocua, Listeria marthii, Listeria monocytogenes and Listeria seeligeri (ubiquitous in the environment), Paenibacillus sp. Y412MC10 and Paenibacillus vortex (heterogeneous and complex environments such as soil and rhizosphere).
Figure 6.

A phylogenic representation of the biological range of the putative orthologues of BT2127 (blue), β-PGM (brown) and the Archeal pyrophosphatase TON0002 (purple).
BT2127 In Vivo Function
The substrate-specificity profile and the active-site structure strongly indicate that inorganic pyrophosphate is the physiological substrate for BT2127. Independent supportive evidence for the in vivo function of BT2127 as an inorganic pyrophosphatase derives from gene context analysis. Pyrophosphate is derived from nucleoside 5′-triphosphates as they undergo nucleotidyl transfer reactions catalyzed by ligases, synthases, nucleotidyl transferases and DNA and RNA polymerases. The hydrolysis of the pyrophosphate product drives these reactions forward and at the same time replenishes the orthophosphate pool.
The gene neighborhoods of BT2127 and the 33 putative orthologues were examined for the bacterial genomes that have been mapped. The BT2127 gene overlaps the fkp gene that encodes the bifunctional L-fucose kinase/L-fucose-1-phosphate guanyltransferase. The products of the kinase-catalyzed reaction are β-L-fucose-1-phosphate (a poor substrate for BT2127 with kcat ∼ 0.1 min−1) and ADP whereas the products of the transferase-catalyzed reaction are inorganic pyrophosphate and β-L-fucose-GDP. The β-L-fucose-GDP is in turn used as a precursor for the decoration of surface capsular polysaccharides and glycoproteins with L-fucose for colonizing the mammalian intestine(31). Thus, we posit that the biochemical function of BT2127 is inorganic pyrophosphate hydrolysis, which supports the biological function of host colonization. Intriguingly, several genes upstream and on the same DNA strand as BT2127 is the lysyl-tRNA synthase gene lysS (BT2122). Thus, BT2127 might also assist protein synthesis by removal of the synthase pyrophosphate product.
Structural Determinants in the Divergence of Function in BT2127 and β-PGM
β-PGM and maltose phosphorylase or trehalose phosphorylase collaborate in the utilization of maltose or trehalose as a carbon and energy source. β-PGM and BT2127 share 30% sequence identity and a common cap domain fold (four α-helices) in addition to the highly conserved catalytic domain fold. Most significantly, they share the cap His20-Lys76 hydrogen bond diad motif that in β-PGM is responsible for the discrimination between β-glucose-1-phosphate and solvent as reactants with the aspartyl phosphate intermediate (Figure S8)(30). Briefly, in the cap domain-open conformation the β-PGM Asp acid/base side chain assumes a rotomer conformation stabilized by hydrogen bond formation with the backbone amide NH and side chain of the linker hinge residue Thr117, which places it outside of the active site(25). Upon substrate-induced cap closure the His20 of the His20-Lys76 diad and the β-glucose 1,6-(bis)phosphate bridging oxygen atom form a hydrogen bond to the Asp acid/base, placing it into position for catalysis (Figure S8)(26). BT2127 conserves the His23-Lys79 diad, and in both the cap domain-open and closed conformations the Asp13 side is in the same conformation, engaged in a hydrogen bond with linker residue Ser15. In the cap-closed conformation, the His23-Lys79 diad is close enough to the Asp13 carboxylate for hydrogen bond formation with the His23, however in the cap-open conformation it is too far away. Thus, the cap domain His-Lys diad appears to be a vestige of the common ancestry between BT2127 and β-PGM.
Examination of the biological range of β-PGM using the same approach applied to BT2127 showed that the putative β-PGM orthologues are primarily found in the phylum Firmicutes and especially in the genera Bacilli (as represented in Figure 6). However, they can also be found in certain species of the phyla Proteobacteria, Actinobacteria and Bactoidetes/Chlorobi (not represented in Figure 6). Notably, we did not uncover a bacterial species that contain orthologues of both BT2127 and β-PGM. A detailed analysis of β-PGM orthologues and their biological range will be published separately.
Evolution of Pyrophosphatases Within the HADSF and Other Superfamilies
Inorganic pyrophosphatases are ubiquitous and essential to cellular function. The large (660-770 amino acid) membrane-bound pyrophosphatases are limited in range to plants and certain bacteria, where they function as proton pumps. The soluble pyrophosphatases are ubiquitous and are found in two distinct fold families (type I and type II). The type I pyrophosphatases are widely distributed in all three kingdoms of life. This is an ancient family with high sequence divergence. The more recently characterized family is the type II family. Inorganic pyrophosphatases of the type II family are restricted in range to select lineages of bacteria(32).
The type I pyrophosphatases are single domain, β-barrel proteins whereas the type II pyrophosphatases are two-domain α/β-proteins in which the active site is located at the domain-domain interface. Type I and II pyrophosphatases employ three or four Mg2+ cofactors to activate the transferring phosphoryl group and water nucleophile and to stabilize the phosphate-leaving group(32-34). HADSF pyrophosphatases represent a third mechanistic and fold type for bacterial pyrophosphatases.
Recently, a novel pyrophosphatase (TON0002) from Thermococcus onnurineus was discovered(35); this Archeal pyrophosphatase is a member of the HADSF. Like BT2127, TON0002 possesses a C1 four-a-helix cap domain, yet this enzyme has evolved unique structural features as inferred from the structure of the close sequence homolog (60% identity) from Pyrococcus horikoshii (PDB ID 2OM6). The superposition of the structures of BT2127 and the P. horikoshii enzyme is shown in Figure 7. The two sets of a-helices that form the TON0002 cap domain are splayed apart thereby expanding the area above the catalytic site. The active site is protected from bulk solvent by: (1) the insertion of a hydrophobic loop (residues 123-128; viz. loop 2) from the catalytic domain between two cap a-helices and (2) the long side chains of Arg48, Lys52, and Arg55 from the cap.
Figure 7.
Superposition of the structures of BT2127 (gray) (PDB ID 3QX7) and the putative pyrophosphatase from P. horikoshii (teal) (PDB ID 2OM6).
A model of the P. horikoshii pyrophosphatase of the active site with Mg2+ and pyrophosphate bound was made by using the positions of the Mg2+ and the phosphate (to define the cofactor and transferring phosphoryl position) in the BT2127 structure and the position of the sulfate ligand in the P. horikoshii enzyme structure to define the position of the phosphate leaving group (Figure 8). Remarkably, the loop 1 Asp acid/base and loop 2 Thr/Ser are not conserved and instead Trp12 and Gly122, respectively, are observed at these positions. The loop 1 Asp nucleophile is Asp10 and the loop 3 Lys is Lys158. The counterparts to the BT2127 loop 4 Glu171 and Asn172 are Gly182 and Asp183, respectively. In the model, the phosphate leaving group is positioned for favorable interaction with Arg48, Lys52, and Arg55 of the cap domain whereas the transferring phosphoryl group is positioned to coordinate Mg2+ and to favorably interact with catalytic domain residues Asn123 (loop 2) and Lys158 (loop 3).
Figure 8.
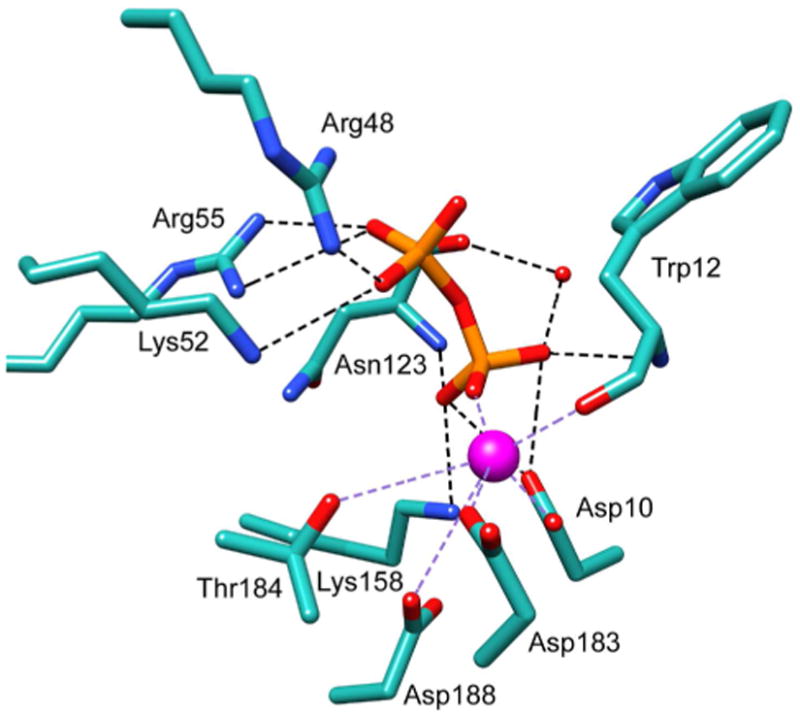
The active site of the putative pyrophosphatase from T. onnurineus BT2127 modeled with pyrophosphate (manually docked) and Mg2+ (derived from the superposition of BT2127 PDB ID 3QUQ). The Mg2+ is shown as a magenta sphere and the water molecules are represented as red spheres. Atom coloring and bonds shown as in Figure 4.
Because the sequence identity is high (60%) and because the key active site residues identified in the P. horikoshii enzyme structure are conserved in the T. onnurineus pyrophosphatase it is reasonable to conclude that the P. horikoshii enzyme is also a pyrophosphatase. The active site model suggests that the backbone amide NH of Asn123 substitutes for the conserved loop 2 Thr/Ser in activating the transferring leaving group and that cap domain residues Arg48, Lys52, and Arg55 stabilize the phosphate leaving group. In the second partial reaction (Scheme 1), the loop 1 Asp acid/base activates the water nucleophile for attack on the aspartylphosphate. If it were to assume a slightly altered side chain rotomer conformation from that observed in the structure, the Asn123 could orient (but not deprotonate) the water nucleophile.
The biological range of the Archeal pyrophosphatase orthologues was compared to that of the BT2127 orthologues. A BLAST search carried out using the T. onnurineus pyrophosphatase sequence identified three orders of Archea with Archeal pyrophosphatase orthologues: Thermococcales (53-80% sequence identity), Desulfurococcales (31-41% sequence identity) and Thermoproteales (29-33% sequence identity). A sequence alignment of all homologues (all sequences conserved Trp12) was made and those sequences that did not conserve the marker residues Arg48, Lys52, Arg55 and Asn123 were omitted, producing an alignment of orthologues (orders Thermococcales and Thermoproteales; Figure S9). Inspection of the gene contexts of these putative orthologues revealed that in some of the thermophiles a gene encoding DNA polymerase was found either on the same DNA strand next to pyrophosphates gene, or across from it on the opposite strand. This suggests a possible biochemical context for the pyrophosphatase, i.e. aiding the polymerase by removing its product, inorganic pyrophosphate. On the other hand, the TON0002 kcat/Km ∼ 4 × 102 M−1 s−1 (35) is so low one might question the physiologically relevance especially given that the T. onnurineus genome encodes a type I inorganic pyrophosphatase.
Conclusion
A BLAST search of the B. thetaiotaomicron genome using the E. coli type I inorganic pyrophosphatase and the Bacillus subtilus type II inorganic pyrophosphatase sequences as query failed to identify a homologue. Thus, the C1-type HADSF member BT2127 appears to be the only known cytoplasmic inorganic pyrophosphatase in B. thetaiotaomicron. The C1-type class appears to be the most prevalent and functionally versatile of the three HADSF classes (C0, C1, C2)(2). Aside from the many phosphatases that have evolved within this class, the dehalogenase, phosphonatase, β-PGM and the newly identified pyrophosphatase activities have evolved as well. Through the comparison of these structures we gain insight into the key structural changes that might be responsible for the change in function. In the case of the inorganic pyrophosphatase BT2127 and its closest relative β-PGM the key structural changes are seemingly directed at how large the leaving group can be (phosphate vs. glucose-phosphate) and the position of the Asp acid/base in the cap-open vs. cap-closed conformation. The BT2127 pyrophosphatase active site volume precludes binding of β-glucose 1, 6-(bis)phosphate. The two key residues that restrict the volume of the catalytic site are the cap domain residues Tyr76 and Glu47. The β-PGM activity requires the movement of the Asp acid/base out of the active site when the cap opens to allow the association of the β-glucose1-phosphate or glucose 6-phosphate for reaction with the aspartylphosphate. In both BT2127 and β-PGM the Asp acid/base forms a hydrogen bond with the linker hinge residue. In β-PGM this hydrogen bond only occurs in the cap-open conformation and it positions the Asp acid/base outside of the active site. In the BT2127 pyrophosphatase this hydrogen bond is present in both the cap-open and cap-closed conformations and the Asp acid/base remains in the active site. When the β-PGM Asp acid/base swings into the active site upon cap domain closure it forms a hydrogen bond with the His20 of the cap domain His20-Lys76 diad. Ala replacement of β-PGM His20 reduces the kcat 7000-fold whereas Ala replacement of the Lys76 reduces the kcat only 100-fold(36) . Ala replacement of the BT2127 counterparts His23 and Lys49 indicates that the hydrogen bond formation with the Asp13 acid/base makes a minor contribution to the turnover rate (kcat reduced 60-fold) compared to the contribution made by the Lys79 (kcat reduced 950-fold), which we posit might function as a docking site for the displaced phosphate as needed for the hydrolysis of the aspartyl phosphate intermediate in the second partial reaction. The pathway followed in the evolution of β-PGM and BT2127 is not clear. One might have evolved from the other or they might have evolved separately from a common ancestor. The fact that they have assumed very specific and separate biological niches is intriguing.
From a mechanistic enzymology perspective the function of BT2127 identifies the HADSF as a third protein fold family that has evolved a bona fide inorganic pyrophosphatase. Moreover, the catalytic mechanism employed by BT2127 is unlike the Mg2+-centered catalytic mechanisms used by the type I and type II inorganic pyrophosphatases. From an orthologue tracking perspective, the Bacteroides inorganic pyrophosphatase BT2127 is a HADSF member that extends the chemistries beyond those of the HADSF phosphatase activities found in E. coli
Supplementary Material
1. Abbreviations used are
- DTT
dithiothreitol
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- Tris
tris(hydroxymethyl)aminomethane
- MESG
2-amino-6-mercapto-7-methylpurine ribonucleoside
- MES
2-(N-morpholino)ethanesulfonic acid
- β-PGM
β-phosphoglucomutase
Footnotes
This work was supported by NIH grants U54 GM093342 (K.N.A., D.D.-M., and S. A.), N.I.H. grant GM61099
The coordinates for the BT2127 X-ray crystal structures are deposited in the protein database under the accession codes 3QU2 (wild-type), 3QXG (wild-type), 3QX7 (wild-type), 3QUQ (wild-type), 3QU5 (D11N mutant), 3QU4 (D13A mutant), 3QU7 (D13N mutant), 3QU9 (D13N mutant), 3QUT (D13N mutant), 3QU7 (D13N mutant), 3R9K (E47D mutant), 3QYP (E47N mutant), 3QUC (E47N mutant) and 3QUB (E47A mutant).
The “substrate specificity loop” (first identified in phosphonatase and β-PGM) is the region of the cap domain that places one or more amino-acid side chains at the catalytic site. In the case of phosphonatase and β-PGM the key loop residue is a Lys which in the former forms a Schiff base adduct with the substrate and in the latter binds the substrate leaving group. In BT2127 the substrate specificity loop Glu47 forms a coordination bond with the Mg2+ cofactor. We do not believe that this interaction is an artifact of crystallization because it is observed in all BT2127 structures that possess Glu47. Moreover, superposition of the X-ray structure of the BT2127 orthologue from Bacteroides vulgatus (PDB ID 3DV9) (vide infra) shows that the homologous residue (Glu62) is likewise coordinated to the Mg2+. The BT2127 Glu47-Mg2+ coordination is not essential for cap domain closure as is indicated by the structures of the BT2127 E47 variants (PDB ID 3QUB (E47A), 3R9K (E47D), 3QUC (E47N)), which are all observed in the cap-closed conformation.
Supporting Information Available: The supporting information includes Tables S1-S8 and Figures S1-S9. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Allen KN, Dunaway-Mariano D. Phosphoryl group transfer: evolution of a catalytic scaffold. Trends Biochem Sci. 2004;29:495–503. doi: 10.1016/j.tibs.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Burroughs AM, Allen KN, Dunaway-Mariano D, Aravind L. Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J Mol Biol. 2006;361:1003–1034. doi: 10.1016/j.jmb.2006.06.049. [DOI] [PubMed] [Google Scholar]
- 3.Allen KN, Dunaway-Mariano D. Markers of fitness in a successful enzyme superfamily. Curr Opin Struct Biol. 2009;19:658–665. doi: 10.1016/j.sbi.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuznetsova E, Proudfoot M, Gonzalez CF, Brown G, Omelchenko MV, Borozan I, Carmel L, Wolf YI, Mori H, Savchenko AV, Arrowsmith CH, Koonin EV, Edwards AM, Yakunin AF. Genome-wide analysis of substrate specificities of the Escherichia coli haloacid dehalogenase-like phosphatase family. J Biol Chem. 2006;281:36149–36161. doi: 10.1074/jbc.M605449200. [DOI] [PubMed] [Google Scholar]
- 5.Wang W, Cho HS, Kim R, Jancarik J, Yokota H, Nguyen HH, Grigoriev IV, Wemmer DE, Kim SH. Structural characterization of the reaction pathway in phosphoserine phosphatase: crystallographic “snapshots” of intermediate states. J Mol Biol. 2002;319:421–431. doi: 10.1016/S0022-2836(02)00324-8. [DOI] [PubMed] [Google Scholar]
- 6.Rangarajan ES, Proteau A, Wagner J, Hung MN, Matte A, Cygler M. Structural snapshots of Escherichia coli histidinol phosphate phosphatase along the reaction pathway. J Biol Chem. 2006;281:37930–37941. doi: 10.1074/jbc.M604916200. [DOI] [PubMed] [Google Scholar]
- 7.Joseph TC, Rajan LA, Thampuran N, James R. Functional characterization of trehalose biosynthesis genes from E. coli: an osmolyte involved in stress tolerance. Mol Biotechnol. 2010;46:20–25. doi: 10.1007/s12033-010-9259-4. [DOI] [PubMed] [Google Scholar]
- 8.Wu J, Woodard RW. Escherichia coli YrbI is 3-deoxy-D-manno-octulosonate 8-phosphate phosphatase. J Biol Chem. 2003;278:18117–18123. doi: 10.1074/jbc.M301983200. [DOI] [PubMed] [Google Scholar]
- 9.Wang L, Huang H, Nguyen HH, Allen KN, Mariano PS, Dunaway-Mariano D. Divergence of biochemical function in the HAD superfamily: D-glycero-D-manno-heptose-1,7-bisphosphate phosphatase (GmhB) Biochemistry. 2010;49:1072–1081. doi: 10.1021/bi902018y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Titz B, Hauser R, Engelbrecher A, Uetz P. The Escherichia coli protein YjjG is a house-cleaning nucleotidase in vivo. FEMS Microbiol Lett. 2007;270:49–57. doi: 10.1111/j.1574-6968.2007.00646.x. [DOI] [PubMed] [Google Scholar]
- 11.Weiss B. YjjG, a dUMP phosphatase, is critical for thymine utilization by Escherichia coli K-12. J Bacteriol. 2007;189:2186–2189. doi: 10.1128/JB.01645-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu Z, Wang L, Dunaway-Mariano D, Allen KN. Structure-function analysis of 2-keto-3-deoxy-D-glycero-D-galactonononate-9-phosphate phosphatase defines specificity elements in type C0 haloalkanoate dehalogenase family members. J Biol Chem. 2009;284:1224–1233. doi: 10.1074/jbc.M807056200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, Lu Z, Allen KN, Mariano PS, Dunaway-Mariano D. Human symbiont Bacteroides thetaiotaomicron synthesizes 2-keto-3-deoxy-D-glycero-D- galacto-nononic acid (KDN) Chem Biol. 2008;15:893–897. doi: 10.1016/j.chembiol.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 15.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 16.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 17.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 18.Stockbridge RB, Wolfenden R. Enhancement of the rate of pyrophosphate hydrolysis by nonenzymatic catalysts and by inorganic pyrophosphatase. J Biol Chem. 2011;286:18538–18546. doi: 10.1074/jbc.M110.214510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu Z, Dunaway-Mariano D, Allen KN. HAD superfamily phosphotransferase substrate diversification: structure and function analysis of HAD subclass IIB sugar phosphatase BT4131. Biochemistry. 2005;44:8684–8696. doi: 10.1021/bi050009j. [DOI] [PubMed] [Google Scholar]
- 20.Tremblay L, Zhang G, Dai J, Dunaway-Mariano D, Allen KN. Structure and Activity Analyses of E. coli K-12 NagD Provide Insight Into the Evolution of Biochemical Function in the HAD Enzyme Superfamily. Biochemistry. 2005 doi: 10.1021/bi051842j. in press. [DOI] [PubMed] [Google Scholar]
- 21.Peeraer Y, Rabijns A, Verboven C, Collet JF, Van Schaftingen E, De Ranter C. High-resolution structure of human phosphoserine phosphatase in open conformation. Acta Crystallogr D Biol Crystallogr. 2003;59:971–977. doi: 10.1107/s0907444903005407. [DOI] [PubMed] [Google Scholar]
- 22.Lahiri SD, Zhang G, Dunaway-Mariano D, Allen KN. Caught in the act: the structure of phosphorylated beta-phosphoglucomutase from Lactococcus lactis. Biochemistry. 2002;41:8351–8359. doi: 10.1021/bi0202373. [DOI] [PubMed] [Google Scholar]
- 23.Morais MC, Zhang W, Baker AS, Zhang G, Dunaway-Mariano D, Allen KN. The crystal structure of Bacillus cereus phosphonoacetaldehyde hydrolase: insight into catalysis of phosphorus bond cleavage and catalytic diversification within the HAD enzyme superfamily. Biochemistry. 2000;39:10385–10396. doi: 10.1021/bi001171j. [DOI] [PubMed] [Google Scholar]
- 24.Holm L, Sander C. Dali/FSSP classification of three-dimensional protein folds. Nucleic Acids Res. 1997;25:231–234. doi: 10.1093/nar/25.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang G, Tremblay L, Dai J, Wang L, Allen KN, Dunaway-Mariano D. Catalytic Cycling in β-Phosphoglucomutase: Coupled X-ray Structure and Kinetic Analysis. Biochemistry submitted. 2005 doi: 10.1021/bi050558p. [DOI] [PubMed] [Google Scholar]
- 26.Lahiri SD, Zhang G, Dunaway-Mariano D, Allen KN. The pentacovalent phosphorus intermediate of a phosphoryl transfer reaction. Science. 2003;299:2067–2071. doi: 10.1126/science.1082710. [DOI] [PubMed] [Google Scholar]
- 27.Hayward S, Kitao A, Berendsen HJ. Model-free methods of analyzing domain motions in proteins from simulation: a comparison of normal mode analysis and molecular dynamics simulation of lysozyme. Proteins. 1997;27:425–437. doi: 10.1002/(sici)1097-0134(199703)27:3<425::aid-prot10>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 28.Hayward S, Lee RA. Improvements in the analysis of domain motions in proteins from conformational change: DynDom version 1.50. J Mol Graph Model. 2002;21:181–183. doi: 10.1016/s1093-3263(02)00140-7. [DOI] [PubMed] [Google Scholar]
- 29.Zhang G, Morais MC, Dai J, Zhang W, Dunaway-Mariano D, Allen KN. Investigation of Metal Ion Binding in Phosphonoacetaldehyde Hydrolase Identifies Sequence Markers for Metal-Activated Enzymes of the HAD Enzyme Superfamily. Biochemistry. 2004;43:4990–4997. doi: 10.1021/bi036309n. [DOI] [PubMed] [Google Scholar]
- 30.Dai J, Finci L, Zhang C, Lahiri S, Zhang G, Peisach E, Allen KN, Dunaway-Mariano D. Analysis of the structural determinants underlying discrimination between substrate and solvent in beta-phosphoglucomutase catalysis. Biochemistry. 2009;48:1984–1995. doi: 10.1021/bi801653r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coyne MJ, Reinap B, Lee MM, Comstock LE. Human symbionts use a host-like pathway for surface fucosylation. Science. 2005;307:1778–1781. doi: 10.1126/science.1106469. [DOI] [PubMed] [Google Scholar]
- 32.Ahn S, Milner AJ, Futterer K, Konopka M, Ilias M, Young TW, White SA. The “open” and “closed” structures of the type-C inorganic pyrophosphatases from Bacillus subtilis and Streptococcus gordonii. J Mol Biol. 2001;313:797–811. doi: 10.1006/jmbi.2001.5070. [DOI] [PubMed] [Google Scholar]
- 33.Oksanen E, Ahonen AK, Tuominen H, Tuominen V, Lahti R, Goldman A, Heikinheimo P. A complete structural description of the catalytic cycle of yeast pyrophosphatase. Biochemistry. 2007;46:1228–1239. doi: 10.1021/bi0619977. [DOI] [PubMed] [Google Scholar]
- 34.Fabrichniy IP, Lehtio L, Tammenkoski M, Zyryanov AB, Oksanen E, Baykov AA, Lahti R, Goldman A. A trimetal site and substrate distortion in a family II inorganic pyrophosphatase. J Biol Chem. 2007;282:1422–1431. doi: 10.1074/jbc.M513161200. [DOI] [PubMed] [Google Scholar]
- 35.Lee HS, Cho Y, Kim YJ, Lho TO, Cha SS, Lee JH, Kang SG. A novel inorganic pyrophosphatase in Thermococcus onnurineus NA1. FEMS Microbiol Lett. 2009;300:68–74. doi: 10.1111/j.1574-6968.2009.01766.x. [DOI] [PubMed] [Google Scholar]
- 36.Dai J. Department of Chemistry. University of New Mexico; Albuquerque: 2005. Mechanism of β-phosphoglucomutase catalysis. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.